

Abstract
Reflexes from the carotid body have been implicated in cardiorespiratory
disorders associated with chronic intermittent hypoxia (CIH). To investigate
whether CIH causes functional and/or structural plasticity in the carotid
body, rats were subjected to 10 days of recurrent hypoxia or normoxia. Acute
exposures to 10 episodes of hypoxia evoked long-term facilitation (LTF) of
carotid body sensory activity in CIH-conditioned but not in control animals.
The magnitude of sensory LTF depended on the length of CIH conditioning and
was completely reversible and unique to CIH, because conditioning with a
comparable duration of sustained hypoxia was ineffective. Histological
analysis revealed no differences in carotid body morphology between control
and CIH animals. Previous treatment with superoxide anion
(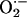 ) scavenger prevented
sensory LTF. In the CIH-conditioned animals, carotid body aconitase enzyme
activity decreased compared with controls. These observations suggest that
increased generation of reactive oxygen species contribute to sensory LTF. In
CIH animals, carotid body complex I activity of the mitochondrial electron
transport is inhibited, suggesting mitochondria as one source of
) scavenger prevented
sensory LTF. In the CIH-conditioned animals, carotid body aconitase enzyme
activity decreased compared with controls. These observations suggest that
increased generation of reactive oxygen species contribute to sensory LTF. In
CIH animals, carotid body complex I activity of the mitochondrial electron
transport is inhibited, suggesting mitochondria as one source of
 generation. These
observations demonstrate that CIH induces a previously uncharacterized form of
reactive oxygen species-dependent, reversible, functional plasticity in
carotid body sensory activity. The sensory LTF may contribute to persistent
reflex activation of sympathetic nerve activity and blood pressure in
recurrent apnea patients experiencing CIH.
generation. These
observations demonstrate that CIH induces a previously uncharacterized form of
reactive oxygen species-dependent, reversible, functional plasticity in
carotid body sensory activity. The sensory LTF may contribute to persistent
reflex activation of sympathetic nerve activity and blood pressure in
recurrent apnea patients experiencing CIH.
Hypoxia is a pervasive physiological stimulus. Changes in arterial blood oxygen levels are monitored continuously by peripheral chemoreceptors, especially the carotid bodies (reviewed in ref. 1). The ensuing reflexes are critical for maintaining homeostasis during hypoxia. Hypoxia is encountered under many different circumstances. For instance, sojourn to high altitude exposes individuals to sustained hypoxia (SH). On the other hand, people living at sea level experience intermittent hypoxia more often in life than SH. Many pathophysiological situations including recurrent apnea syndromes (central or obstructive sleep apneas), apneas in premature infants, and asthmatic attacks are associated with intermittent hypoxia. Decreases in arterial blood oxygen occur in both types of hypoxia. However, cardiorespiratory systems adapt to chronic SH to maintain adequate oxygen delivery to tissues (reviewed in refs. 2 and 3), whereas chronic intermittent hypoxia (CIH) leads to serious pathophysiological consequences such as hypertension, myocardial infarcts, and even stroke (4, 5).
Reflexes from the carotid body have been proposed to play a critical role
in ventilatory and circulatory adaptations to chronic SH and are associated
with structural changes in the carotid body including hypertrophy of the
organ, increased vascularization, and increased number of glomus cells (the
putative oxygen-sensing cells; reviewed in refs.
2 and
3). There is evidence that
reflexes arising from the carotid body also play an important role in the
development of the pathophysiology associated with CIH. Evidence includes the
findings that glomectomized subjects (removal of carotid bodies) with
recurrent apneas do not develop hypertension
(6), and chronic ablation of
the sinus nerves that innervate the carotid bodies prevents development of
hypertension as well as increased sympathetic nerve activity in rats exposed
to CIH (7). It is not known,
however, whether the effects of CIH are due to a direct effect on the carotid
body and/or enhanced processing of sensory information from the carotid body
at the central nervous system. Given its clinical significance, it is of
considerable importance to understand whether CIH results in functional and/or
structural plasticity of the carotid body and, if so, by what mechanisms.
Consequently, we examined the sensory activity of the carotid body in a rat
model of CIH. Because we are interested in examining the effects of
intermittent hypoxia similar to that seen in recurrent apneas, the duration of
individual hypoxic episodes was kept close to that seen in recurrent apneas.
Our results show that CIH induces a previously uncharacterized form of
functional plasticity in the carotid body manifested as long-lasting
activation of sensory discharge that persists for 1 h after termination of
acute intermittent hypoxia (AIH). In sharp contrast, such long-lasting
activation of sensory discharge was not elicited in animals previously exposed
to either acute or multiple exposures to continuous hypoxia. Further, our data
also show that CIH-induced functional plasticity is associated with increased
superoxide anion (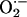 )
generation in the carotid body.
)
generation in the carotid body.
Methods
Experiments were performed on male Sprague–Dawley rats weighing 250–350 g. The Institutional Animal Care and Use Committee of Case Western Reserve University approved the experimental protocols.
General Preparation of the Animals. Acute experiments were performed on rats anesthetized with urethane (1.2 g/kg, i.p.) and supplemented hourly with 15% of the initial dose. After tracheal intubation, a femoral artery and vein were cannulated for measuring arterial blood pressure (P122, Grass Instruments, Quincy, MA) and for i.v. administration of fluids and drugs, respectively. Animals were paralyzed with pancuronium bromide (2.5 mg/kg per h, i.v.) to prevent spontaneous breathing and ventilated with a respirator (Harvard Apparatus). Bilateral vagotomy was performed in the midcervical region. Arterial blood samples were collected from the femoral arterial catheter to determine blood gases (PaO2 and PaCO2) and pH (ABL-5, Radiometer, Copenhagen). The rectal temperature of the animals was maintained at 38 ± 1°C by means of a heating pad. At the end of the experiments, animals were killed with i.v. administration of euthanasia solution.
Exposure to CIH. The CIH paradigm consisted of 15 sec of hypoxia followed by 5 min of normoxia, nine episodes per h, 8 h/day for 10 days. This was accomplished by placing unrestrained, freely mobile animals housed in feeding cages (dimensions 0.48 × 0.26 × 0.22 m, two cages per session with up to three animals per cage) in a special chamber (dimensions 0.62 × 0.55 × 0.29 m) for exposure to intermittent hypoxia. The animals were fed ad libitum. The chamber was flushed with alternating cycles of pure nitrogen and compressed air such that inspired O2 levels reached 5% during hypoxia within 68–75 sec and 21% during normoxia within 70–85 sec. Ambient oxygen levels in the chamber were monitored continuously by using an oxygen analyzer (OM-11, Beckman, Schiller Park, IL) to sample the air in the chamber. A continuous vacuum was created within the chamber to balance the pressure between in- and outflow of the gases. Inspired CO2 levels were maintained at 0.2–0.5% and were monitored continuously by an infrared analyzer (LB-2, Beckman). In control experiments, animals were exposed to alternating cycles of normoxia instead of hypoxia while keeping the rate of gas flow similar to that described above. The duration of the gas flow during each hypoxic and normoxic episode was regulated by timed solenoid valves. Animals were subjected to intermittent hypoxia between 9:00 a.m. and 5:00 p.m. for 10 consecutive days. Acute experiments were performed on the morning after the 10th day of CIH exposure.
Exposure to SH. To examine the effects of a cumulative, comparable duration of SH, awake, unrestrained rats were exposed to isobaric hypoxia (5% O2) for 4 h by placing them in a Lucite chamber containing an inlet port for administering 5% O2 and an outlet port connected to a vacuum sufficient to create a flow of 600 ml/min through the chamber, as measured by a rate meter. Acute experiments were performed immediately after terminating SH. To assess the effect of multiple SH exposures, awake, unrestrained animals were exposed to 4 h of SH per day for 10 days. Acute experiments were performed on the morning after the 10th day of multiple SH exposures.
In Vivo Carotid Body Sensory Activity. Sensory activity of the carotid body was recorded in anesthetized rats as described (8). Briefly, carotid bifurcation was isolated, and the carotid sinus nerve was transected where it joins the glossopharyngeal nerve. Afferent activity was recorded by using a monopolar platinum–iridium wire electrode with a reference electrode placed in a nearby neck muscle. Electrical activity was amplified by an AC amplifier (P511, Grass Instruments), with a bandwidth of 100–3,000 Hz and displayed on an oscilloscope (5B12N, Tektronix). The action potentials above the baseline noise were converted to standardized pulses by using a window discriminator (RAD II-A, Winston, San Francisco) and fed into a rate meter (RIC-830, CWE, Ardmore, PA) to display the magnitude of the discharge. Signals from the rate meter, raw action potentials from the amplifier, and blood-pressure signals were recorded continuously on a chart recorder (Astro-Med, West Warwick, RI). Carotid body activity was identified by prompt augmentation of sensory discharge in response to 30 sec of asphyxia and prompt decrease in response to 100% O2. Reducing the pressure in the carotid sinus by occluding the common carotid artery for 10 sec caused no change or an increase in sinus nerve activity but never a decrease, indicating that the sensory activity is of carotid body rather than baroreceptor origin.
Ex Vivo Carotid Body Sensory Activity. Carotid bodies along with the sinus nerves were harvested from anesthetized animals and placed in ice-cold physiological saline. The carotid body along with the sinus nerve was placed in a recording chamber and superfused with warm physiological saline (36.5°C) at a rate of 2 ml/min. The composition of the medium was 125 mM NaCl/5.3 mM KCl/1.8 mM CaCl2/2 mM MgSO4/1.2 mM NaH2PO4/25 mM NaHCO3/10 mM d-glucose/5 mM sucrose, pH 7.4. The medium was bubbled continuously with either 21% O2 + 5% CO2 (normoxia) or 1% O2 + 5% CO2 (hypoxia). Afferent activity from the sinus nerve was recorded with a suction electrode by using the amplification protocols described above.
Carotid Body Morphology. Morphology of carotid bodies was analyzed as described (9). Briefly, anesthetized rats were perfused at 20 ml/min with heparinized PBS, pH 7.4, followed by 4% (wt/vol) paraformaldehyde/PBS (10 min each). Carotid artery bifurcations were placed in 4% paraformaldehyde/PBS for 1 h at 4°C, washed in PBS, and cryoprotected in 30% sucrose/PBS at 4°C for 24 h. Specimens were frozen in Tissue Tek (OCT compound, VWR Scientific), serially sectioned at a thickness of 10 μm, washed three times in PBS, and exposed to 20% normal goat serum and 0.2% Triton X-100 in PBS for 2 h. Endogenous biotinylated proteins were blocked with avidin (Vector Laboratories). Sections were incubated at 4°C for 16 h with anti-chromogranin A (1:1,500, Instar, Stillwater, MN) antibody in PBS with 1% normal goat serum and 0.2% Triton X-100. After washing in PBS, sections were incubated for 2 h with biotinylated anti-rabbit IgG (1:200, Vector Laboratories) in PBS with 1% normal goat serum and 0.2% Triton X-100. Immunostaining was visualized by the Vectastain Elite avidinbiotinylated enzyme complex method (Vector Laboratories) by using diaminobenzidine. Carotid body morphology, glomic volume, and glomus cell numbers were analyzed in adjacent sections by using image software (Scion, Frederick, MD).
Mitochondrial Complex Activities. The NADH–ubiquinone reductase (complex I) activity was analyzed as described (10) except that mitochondria were not isolated because of the small size of the carotid bodies (wet weight ≈ 60 μg). Briefly, carotid bodies (n = 16) were homogenized in 60 μl of buffer containing 50 mM Tris·Cl, 1 mM DTT, 0.2 mM EGTA, 0.05 mM leupeptin, and 0.4% (vol/vol) Triton X-100, pH 7.2. The cell lysates were centrifuged for 3 min at 14,000 × g at 4°C, and the supernatants were used for analysis. The reaction mixture (300 μl) contained 1 mM KCN, 0.2 mM NADH, and 20 μg of protein. The reaction was initiated by the addition of 150 μM decylubiquinone and read at 340 nm against a blank containing all the components except decylubiquinone. After measuring for 5 min, the reaction was arrested by the addition of 2 μM rotenone, and the rate was measured for an additional 5 min. The complex I activity, measured as rotenone-sensitive rate of NADH oxidation, was expressed in micromoles of NADH oxidized per minute per milligram of protein. Ubiquinol–ferricytochrome c oxidoreductase (complex III) activity was measured by a modification of the method described previously (10). The assay mixture contained 20 μg of carotid body protein (from 16 carotid bodies) and 0.1 mM EDTA, 0.06 mM oxidized cytochrome c, 3 mM NaN3, 50 mM KH2PO4, and 0.1% BSA, pH 7.4. The reaction was initiated by the addition of 100 μM decylubiquinone, and the increase in absorbance at 550 nm was monitored every 5 sec for 1 min. The activity was measured in the presence and absence of antimycin A. The complex III activity measured as antimycin A-sensitive reaction was expressed in micromoles of cytochrome c reduced per minute per milligram of protein. Protein concentration was determined by the bicinchoninic acid method (11) by using BSA as the standard.
Aconitase Activity. For the analysis of aconitase activity, carotid bodies (n = 16) were homogenized by sonication (10 sec, three times) at 4°Cin60 μl of buffer containing 50 mM Tris·Cl, 0.6 mM MnCl2, and 20 mM (±)-fluorocitrate, pH 7.4. The aconitase activity was measured as described (12) by using a spectrophotometer (UV-2401PC, Shimadzu). Briefly, the cell lysate (10–15 μg of protein) was added to a buffer, pH 7.4, containing 50 mM Tris·Cl, 5 mM sodium citrate, 0.6 mM MnCl2, 0.2 mM NAD+, and 2 units of isocitrate dehydrogenase. The final volume of the reaction mixture was 300 μl. The change in absorbance of the reaction mixture at 340 nm was recorded for 30–60 min, and the linear rate during the last 30 min of the reaction was used for the determination of aconitase activity. The aconitase activity was expressed in micromoles of isocitrate formed per minute per milligram of protein. All enzyme assays were performed in triplicate.
Data Analysis. In anesthetized animals the variables analyzed were (i) chemoreceptor activity (impulses per sec), (ii) mean arterial blood pressure (mmHg, 1 mmHg = 133 Pa), and (iii) arterial PaO2, PaCO2, and pH. The sensory activity and blood pressure were quantified over a 1-min period before AIH, during each episode of hypoxia, and every 5 min over the 60-min post-AIH period. The results are expressed as mean ± SEM. One-way ANOVA with repeated measures, or an unpaired t test when appropriate, was used for assessing statistical significance. P values of <0.05 were considered significant.
Results
CIH Induces Sensory Long-Term Facilitation (LTF) in the Carotid Body. The effects of AIH (10 episodes of 15 sec of 12% O2 interspersed with 5 min of 95% O2, hyperoxia) were examined on carotid body sensory activity in control and CIH animals (CIH for 10 days). In control animals, sensory activity increased with each hypoxic episode, returned promptly to baseline after terminating each hypoxic challenge, and remained at this level for 60 min after terminating AIH (Fig. 1). In sharp contrast, in CIH animals, baseline sensory activity progressively increased with each successive hypoxic challenge, and this increase persisted for at least 60 min after terminating AIH. On average, in CIH animals, baseline sensory activity increased from 91 ± 10 to 228 ± 43 impulses per sec (P < 0.01, ANOVA) in the pre- to post-AIH periods, respectively (Fig. 1). This increase in baseline sensory activity was not secondary to changes in arterial O2 or blood pressure, because changes in these variables were comparable in pre- and post-AIH periods (Table 1). The time course of the persistent elevation in sensory activity resembled LTF of respiratory motor activity evoked by episodic hypoxia described previously (13). Therefore, for simplicity, the long-lasting increase in baseline carotid body activity will be referred to hereafter as sensory LTF.
Fig. 1.

AIH induces sensory LTF in the carotid body in CIH animals. (A) Carotid body sensory activity in a control (Upper) and CIH-conditioned (Lower) animal. Pre-AIH is baseline activity; AIH #1 and AIH #10 represent the first and 10th episodes of AIH; impulses (Imp) per sec, integrated sensory discharge; A.P., action potentials. (B) Average changes in the sensory activity during AIH and during every 5 min of the post-AIH period. Average data represent mean ± SEM from control (n = 7) and 10 days CIH-conditioned (n = 7) animals. The shaded area represents the difference in baseline activity in CIH and control animals during the post-AIH period.
Table 1. Changes in arterial blood gases and blood pressure.
|
Pre-AIH
|
Post-AIH
|
|||||
|---|---|---|---|---|---|---|
| MBP, mmHg | PaCO2, mmHg | PaO2, mmHg | MBP, mmHg | PaCO2, mmHg | PaO2, mmHg | |
| Control | 96 ± 5 | 34 ± 2 | 338 ± 19 | 86 ± 4n.s. | 33 ± 2n.s. | 333 ± 17n.s. |
| CIH | 106 ± 7 | 32 ± 1 | 304 ± 16 | 99 ± 6n.s. | 32 ± 1n.s. | 283 ± 13n.s. |
Data are mean ± SEM from control and CIH animals. n.s., not significant (P > 0.05 compared with pre-AIH, n = 7 in each group).
To determine whether sensory LTF can also be evoked by stimuli other than repetitive hypoxia, control and CIH animals (three each) were challenged with acute, repetitive, hyperoxic hypercapnia (10 episodes of 15 sec of 7% CO2 + 93% O2 interspersed with 5 min of 100% O2), another physiological stimulus to the carotid body. Hypercapnia augmented sensory activity in CIH-conditioned animals, and the magnitude of the response was comparable with control animals (+35 ± 13% and +29 ± 11% increase in CIH and controls, respectively). However, acute repetitive hypercapnia failed to elicit sensory LTF in either group of animals.
Characterization of Sensory LTF. Rats were exposed to 1 or 3 days of CIH to determine whether the development of LTF depends on the duration of conditioning with CIH. LTF was not apparent after 1 day, began after 3 days of conditioning, and was increased further after 10 days of CIH (Fig. 2A). Animals conditioned with 10 days of CIH were placed in room air for 3 or 10 days before testing the effects of AIH to determine whether CIH-induced sensory LTF is reversible. Sensory LTF was reduced by 39% at 3 days and completely reversed after 10 days in room air (Fig. 2B). Another group of animals was conditioned for 10 days with 10% O2 to determine whether the effect of CIH depends on the severity of hypoxia. Conditioning with 10% O2 induced a similar degree of sensory LTF to 5% O2 (Fig. 2C).
Fig. 2.

Characterization of CIH-induced sensory LTF in the carotid body. (A) Animals were conditioned with 0 (control, n = 7), 1 (1D, n = 5), 3 (3D, n = 6), and 10 days (10D, n = 7) of CIH. (B) Reversibility of LTF. Animals conditioned with 10 days of CIH were placed in room air for 3 (3D, n = 5) or 10 (10D, n = 6) days, and LTF was determined in response to AIH. (C) The effect of severity of hypoxia during CIH conditioning on the magnitude of sensory LTF. Animals were conditioned with either 5% (5% O2, n = 7) or 10% (10% O2, n = 6) inspired oxygen. From A to C, LTF was determined and expressed as the average percent of baseline activity during 60 min of post-AIH. Data are presented as mean ± SEM. **, P < 0.01; *, P < 0.05.
Pattern of CIH Conditioning Is Critical for Eliciting Sensory LTF. Animals were conditioned with 4 h of SH, a duration of hypoxia equivalent to that accumulated during 10 days of CIH. Four hours of SH did not elicit sensory LTF in the carotid body (70 ± 10 and 66 ± 12 impulses per sec in pre- and post-AIH periods, respectively; P > 0.05, ANOVA) (Fig. 3). To determine whether multiple exposures to SH result in sensory LTF, another group of animals was conditioned with 4 h of SH per day for 10 days. Conditioning with 10 days of SH showed a tendency for sensory LTF up to the 25th min of post-AIH (Fig. 3). However, changes in sensory activity in post-AIH were not significant (P = 0.172; n = 7).
Fig. 3.

Conditioning with comparable cumulative duration of SH does not elicit sensory LTF. The effects of AIH were examined in control (n = 6) and animals conditioned with 4 h of SH (equivalent to the duration of hypoxia accumulated over 10 days of CIH conditioning, n = 5) as well as with multiple exposures to SH 4 h/day for 10 days (CSH, n = 7). Data represent mean ± SEM. Note the absence of significant sensory LTF in both groups of animals.
Sensory LTF in ex Vivo Carotid Bodies. Sensory LTF might be secondary to cardiovascular changes affecting vascular perfusion of the carotid bodies in CIH animals. Therefore, LTF in sensory discharge was examined in ex vivo superfused carotid bodies wherein vascular effects are effectively absent. The AIH paradigm in ex vivo experiments was the same as that used in intact animal preparations, i.e., 10 episodes of 15 sec of hypoxia interspersed with 5 min of normoxia. LTF in sensory activity was elicited in ex vivo carotid bodies from CIH but not control animals (Fig. 4). On average, sensory activity of the CIH-conditioned ex vivo carotid bodies increased from a baseline value of 31 ± 4 to 76 ± 14 impulses per sec in the post-AIH period (P < 0.01, ANOVA). The magnitude of sensory LTF in ex vivo preparations was comparable to that seen in in vivo preparations (245% versus 250%, ex vivo and in vivo, respectively).
Fig. 4.

Sensory LTF in ex vivo carotid bodies. (A) Representative tracings of ex vivo carotid body activity from control (Upper) and CIH-conditioned (Lower) animals. Pre-AIH is baseline activity, and AIH #1 and AIH #10 represent the first and 10th AIH episodes, respectively. Impulses (Imp) per sec, integrated sensory discharge; A.P., action potentials. (B) Average changes in the sensory activity during AIH and during every 5 min of post-AIH. Data represent mean ± SEM from control and CIH animals (n = 6 carotid bodies in each group). The shaded area represents the differences in baseline activity in CIH-conditioned and control animals during the post-AIH period.
Carotid Body Morphology. Morphometric analysis was performed on sections of the carotid bodies from control and CIH animals stained with chromogranin A antibody, an established marker of glomus cells (9), to determine whether sensory LTF is associated with morphological changes in the carotid body. No significant differences were found in the total volume of the carotid body, number of glomus cells, or glomus cell volume between control and CIH rats (Table 2).
Table 2. Morphometric analysis of carotid bodies from control and CIH-conditioned rats.
| Total volume, μm3 | Glomic volume, μm3 | Glomic volume/total volume | Glomus cells/section | Glomus cell volume, μm3 | |
|---|---|---|---|---|---|
| Control | (5.4 ± 0.9) × 106 | (1.7 ± 0.3) × 106 | 0.31 ± 0.01 | 1,081 ± 165 | 1,493 ± 134 |
| CIH | (5.6 ± 0.6) × 106 n.s. | (1.6 ± 0.2) × 106 n.s. | 0.27 ± 0.01n.s. | 1,232 ± 179n.s. | 1,326 ± 72n.s. |
Data are mean ± SEM. n.s., not significant (P > 0.05 compared with control, n = 4 carotid bodies from two animals and 20 sections from each group).
Mitochondrial Complex I and III Activities in the Carotid Body. Mitochondria have been proposed to be one of the oxygen sensors in the carotid body (14). Therefore, we tested whether CIH affects activities of the electron transport complexes of the mitochondria in the carotid body. The complex I activity of the mitochondrial electron transport chain decreased by 89% in carotid bodies from CIH compared with control animals (Fig. 5A). On the other hand, complex III activity was unaltered (Fig. 5B). In sharp contrast, neither complex I nor complex III activities were affected in carotid bodies harvested from SH animals (4 h of SH, six rats, 12 carotid bodies, data not shown).
Fig. 5.

Involvement of  in
CIH-induced sensory LTF. Effect of CIH conditioning on mitochondrial complex I
(A) and III (B) activities (eight rats, 16 carotid bodies in
each group). (C) Effect of CIH on aconitase activity (eight rats, 16
carotid bodies in each group). (D) Effect of superoxide dismutase
mimetic (MnTMPyP, 5 mg/kg per day for 10 days, n = 6) or vehicle
(saline, n = 6) on CIH-induced sensory LTF. MnTMPyP prevents
CIH-induced sensory LTF. Data are mean ± SEM. **, P
< 0.01.
in
CIH-induced sensory LTF. Effect of CIH conditioning on mitochondrial complex I
(A) and III (B) activities (eight rats, 16 carotid bodies in
each group). (C) Effect of CIH on aconitase activity (eight rats, 16
carotid bodies in each group). (D) Effect of superoxide dismutase
mimetic (MnTMPyP, 5 mg/kg per day for 10 days, n = 6) or vehicle
(saline, n = 6) on CIH-induced sensory LTF. MnTMPyP prevents
CIH-induced sensory LTF. Data are mean ± SEM. **, P
< 0.01.
Evidence for the Involvement of
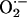 in Sensory LTF. One of
the consequences of complex I inhibition is increased generation of
in Sensory LTF. One of
the consequences of complex I inhibition is increased generation of
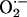 (15). Increased
(15). Increased
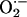 production inhibits
aconitase enzyme activity, which has been used by previous investigators as an
index of
production inhibits
aconitase enzyme activity, which has been used by previous investigators as an
index of 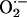 generation
(12). Therefore, we monitored
aconitase enzyme activity in carotid bodies from CIH and control animals.
Aconitase activity was 79% less in carotid bodies from CIH compared with
control animals (Fig.
5C), suggesting an increased generation of
generation
(12). Therefore, we monitored
aconitase enzyme activity in carotid bodies from CIH and control animals.
Aconitase activity was 79% less in carotid bodies from CIH compared with
control animals (Fig.
5C), suggesting an increased generation of
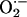 in CIH carotid bodies. To
test the contribution of
in CIH carotid bodies. To
test the contribution of  to
the development of sensory LTF, rats were treated each day for 10 days with a
stable superoxide dismutase mimetic [manganese (III)
tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP), 5 mg/kg, i.p.],
a potent scavenger of
to
the development of sensory LTF, rats were treated each day for 10 days with a
stable superoxide dismutase mimetic [manganese (III)
tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP), 5 mg/kg, i.p.],
a potent scavenger of 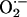 (16), or vehicle (saline)
before subjecting them to CIH. Sensory LTF was markedly attenuated in animals
treated with superoxide dismutase mimetic (P < 0.01, unpaired
t test) (Fig.
5D). On the other hand, a single application of
superoxide dismutase mimetic on the 10th day of CIH failed to prevent sensory
LTF (n = 4).
(16), or vehicle (saline)
before subjecting them to CIH. Sensory LTF was markedly attenuated in animals
treated with superoxide dismutase mimetic (P < 0.01, unpaired
t test) (Fig.
5D). On the other hand, a single application of
superoxide dismutase mimetic on the 10th day of CIH failed to prevent sensory
LTF (n = 4).
Discussion
The results described above demonstrate that conditioning with CIH induces functional plasticity in the carotid body, manifested as sensory LTF in response to AIH. Induction of sensory LTF is unique to intermittent hypoxia because it was not evident in animals exposed to acute or multiple exposures to SH. Our data further suggest that CIH-induced sensory LTF utilizes a mechanism(s) involving reactive oxygen species (ROS) and decreased activity of the mitochondrial electron transport chain at complex I.
The major finding of the present study is that CIH induces sensory LTF in the carotid body. Previous studies have documented LTF of respiratory motor output in response to repeated exposures to acute hypoxia in a number of experimental animal models (reviewed in ref. 13). However, there are some notable differences between the sensory and respiratory motor LTF. First, CIH is required for eliciting sensory LTF in the carotid body, whereas LTF in the motor activity can be elicited in control animals. Second, sensory LTF occurs at the level of the carotid body itself as evidenced by long-lasting activation in ex vivo carotid bodies, whereas LTF in motor activity involves a central mechanism (13). The contribution of sensory LTF to the LTF of respiratory motor activity, however, remains to be investigated.
Our results demonstrate that CIH-induced sensory LTF is a time-dependent and completely reversible phenomenon (Fig. 2). Carotid body sensory activity increases proportionally with increasing severity of hypoxia. Therefore, it is possible that the magnitude of sensory LTF depends on the severity of hypoxia used in CIH conditioning. However, our data revealed no significant differences in the magnitude of LTF elicited by CIH conditioning with either 10% or 5% inspired O2, suggesting that the severity of hypoxia used for CIH conditioning plays a less critical role in determining the magnitude of sensory LTF. Furthermore, a generalized increase in carotid body excitability does not seem to account for sensory LTF, because repetitive hypercapnia failed to elicit sensory LTF. However, hyperoxic hypercapnia caused relatively less increase in sensory discharge compared with hypoxia, and whether severe hypercapnia causing a comparable magnitude of sensory activity to that seen with hypoxia is capable of eliciting sensory LTF remains to be studied.
The following lines of evidence indicate that sensory LTF is intrinsic to the carotid body and not secondary to extrinsic influences such as alterations in blood pressure or gases. First, sensory LTF was elicited in ex vivo carotid bodies from CIH-conditioned animals. Second, there were no significant variations in either blood pressure or arterial blood gases (Table 1). The possible contribution of plasticizing effects of circulating vasoactive agents during the CIH-conditioning period in inducing sensory LTF, however, cannot be ruled out.
It has been well documented that chronic SH causes structural changes in the carotid body including hypertrophy of the organ and increased number and volume of glomus cells (reviewed in refs. 2 and 3). However, morphometric analysis revealed no changes in the histology of CIH carotid bodies, suggesting that alterations in gross morphology do not account for sensory LTF. These observations also demonstrate that prolonged exposures to repetitive and sustained hypoxia exert profoundly different effects on the gross morphology of the carotid body and therefore perhaps involve distinctly different mechanisms. Further experiments, however, are needed to establish whether CIH causes ultrastructural changes in the glomus tissue.
An intriguing observation of the present study is that animals exposed to 4
h of SH for a single or multiple episodes (4 h per episode, one episode per
day for 10 days) failed to evoke sensory LTF in response to AIH
(Fig. 3). These observations
suggest that induction of sensory LTF critically depends on the pattern of
hypoxia, i.e., recurrent hypoxia versus SH. What makes CIH different from SH?
The major difference between repetitive and sustained exposures to hypoxia is
the episodic oxygenation in the former. In this respect, the recurrent hypoxia
resembles ischemia reperfusion, wherein cellular generation of ROS, especially
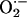 , is increased during
reperfusion (15). The
following observations are consistent with the idea that CIH leads to
increased generation of
, is increased during
reperfusion (15). The
following observations are consistent with the idea that CIH leads to
increased generation of 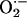 in
the carotid body that contributes to sensory LTF. First, aconitase enzyme
activity (an index of
in
the carotid body that contributes to sensory LTF. First, aconitase enzyme
activity (an index of  generation) (12) is reduced in
CIH carotid bodies, indicating increased generation of
generation) (12) is reduced in
CIH carotid bodies, indicating increased generation of
 . Second, sensory LTF was
practically absent in CIH animals pretreated with MnTMPyP, a potent,
cell-permeable
. Second, sensory LTF was
practically absent in CIH animals pretreated with MnTMPyP, a potent,
cell-permeable 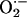 scavenger
(16). A single application of
MnTMPyP on the 10th day of CIH, on the other hand, failed to prevent sensory
LTF, indicating that it is due to long-term effects of
scavenger
(16). A single application of
MnTMPyP on the 10th day of CIH, on the other hand, failed to prevent sensory
LTF, indicating that it is due to long-term effects of
 on the sensory complex of
the carotid body.
on the sensory complex of
the carotid body.
Previous studies have suggested that mitochondria are one important source
of ROS generation (12,
15). Based on experiments with
cell cultures it has been proposed that inhibition of mitochondrial electron
transport chain at complex I as well as complex III results in increased
generation of ROS, especially
 (12,
15). Our data have shown that
complex I activity of the mitochondrial electron transport chain was markedly
down-regulated in carotid bodies from CIH compared with controls, whereas
complex III activity was unaffected. Changes in complex I activity seem unique
to CIH, because a comparable duration of SH had no effect on either complex I
or III activity. Therefore, inhibition of complex I activity of the
mitochondrial electron transport chain contributes at least in part to
increased
(12,
15). Our data have shown that
complex I activity of the mitochondrial electron transport chain was markedly
down-regulated in carotid bodies from CIH compared with controls, whereas
complex III activity was unaffected. Changes in complex I activity seem unique
to CIH, because a comparable duration of SH had no effect on either complex I
or III activity. Therefore, inhibition of complex I activity of the
mitochondrial electron transport chain contributes at least in part to
increased  in CIH carotid
bodies. Besides mitochondria, several other cellular sources such as oxidases
(e.g., NADPH oxidase) also contribute to increased generation of
in CIH carotid
bodies. Besides mitochondria, several other cellular sources such as oxidases
(e.g., NADPH oxidase) also contribute to increased generation of
 . Future studies are needed
to address the mechanism(s) by which CIH leads to inhibition of complex I
activity and whether sources other than mitochondria also contribute to
CIH-induced increases in
. Future studies are needed
to address the mechanism(s) by which CIH leads to inhibition of complex I
activity and whether sources other than mitochondria also contribute to
CIH-induced increases in
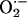 .
.
How might 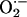 contribute to
LTF in sensory activity? One possibility is that
contribute to
LTF in sensory activity? One possibility is that
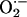 might act at the level of
O2-sensitive K+ channels and/or heme protein(s) that
regulate O2 sensing by the glomus cells (reviewed in ref.
1). Alternatively, ROS might
enhance the increase in intracellular Ca2+ in glomus
cells by affecting either voltage-gated Ca2+ channels
and/or by releasing Ca2+ from intracellular stores.
Relevant to the latter possibility is a study suggesting that ROS potentiates
the increase in intracellular Ca2+ evoked by a
depolarizing stimulus in cell cultures
(17). Finally,
might act at the level of
O2-sensitive K+ channels and/or heme protein(s) that
regulate O2 sensing by the glomus cells (reviewed in ref.
1). Alternatively, ROS might
enhance the increase in intracellular Ca2+ in glomus
cells by affecting either voltage-gated Ca2+ channels
and/or by releasing Ca2+ from intracellular stores.
Relevant to the latter possibility is a study suggesting that ROS potentiates
the increase in intracellular Ca2+ evoked by a
depolarizing stimulus in cell cultures
(17). Finally,
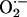 might interfere with the
release of transmitters and/or receptors in the carotid body responsible for
increasing the sensory discharge. Further experiments, however, are needed to
test these possibilities. What might be the functional significance of
CIH-induced sensory LTF in the carotid body? Human subjects experiencing
intermittent hypoxia as a consequence of recurrent apneas often exhibit
elevated blood pressures and sympathetic nerve activity during daytime even in
the absence of apneas (18).
Because increased carotid body sensory activity reflexively triggers
sympathetic nerve discharge and blood pressure, it is possible that sensory
LTF contributes to the persistent increases in sympathetic nerve activity and
blood pressure seen in recurrent apnea patients. By using a rodent model, the
present study demonstrates that CIH can directly influence the carotid body
and induces functional and reversible plasticity. However, further studies are
needed to establish whether other sensory receptors associated with regulation
of cardiorespiratory system are also affected by CIH and contribute to the
progression of pathophysiology associated with recurrent apneas.
might interfere with the
release of transmitters and/or receptors in the carotid body responsible for
increasing the sensory discharge. Further experiments, however, are needed to
test these possibilities. What might be the functional significance of
CIH-induced sensory LTF in the carotid body? Human subjects experiencing
intermittent hypoxia as a consequence of recurrent apneas often exhibit
elevated blood pressures and sympathetic nerve activity during daytime even in
the absence of apneas (18).
Because increased carotid body sensory activity reflexively triggers
sympathetic nerve discharge and blood pressure, it is possible that sensory
LTF contributes to the persistent increases in sympathetic nerve activity and
blood pressure seen in recurrent apnea patients. By using a rodent model, the
present study demonstrates that CIH can directly influence the carotid body
and induces functional and reversible plasticity. However, further studies are
needed to establish whether other sensory receptors associated with regulation
of cardiorespiratory system are also affected by CIH and contribute to the
progression of pathophysiology associated with recurrent apneas.
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute Grant HL-25830, and D.K. was supported by National Heart, Lung, and Blood Institute Training Grant T-32-HL-07887.
Abbreviations: SH, sustained hypoxia; CIH, chronic intermittent hypoxia; AIH, acute intermittent hypoxia; LTF, long-term facilitation; MnTMPyP, manganese (III) tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride; ROS, reactive oxygen species.
References
- 1.Prabhakar, N. R. (2000) J. Appl. Physiol. 88, 2287–2295. [DOI] [PubMed] [Google Scholar]
- 2.Lahiri, S., Rozanov, C. & Cherniack, N. S. (2000) High Alt. Med. Biol. 1, 63–74. [DOI] [PubMed] [Google Scholar]
- 3.Bisgard, G. E. (2000) Respir. Physiol. 121, 237–246. [DOI] [PubMed] [Google Scholar]
- 4.Nieto, F. J., Young, T. B., Lind, B. K., Shahar, E., Samet, J. M., Redline, S., D'Agostino, R. B., Newman, A. B., Lebowitz, M. D. & Pickering, T. G. (2000) J. Am. Med. Assoc. 283, 1829–1836. [DOI] [PubMed] [Google Scholar]
- 5.Shahar, E., Whitney, C. W., Redline, S., Lee, E. T., Newman, A. B., Javier, N. F., O'Connor, G. T., Boland, L. L., Schwartz, J. E. & Samet, J. M. (2001) Am. J. Respir. Crit. Care Med. 163, 19–25. [DOI] [PubMed] [Google Scholar]
- 6.Somers, V. K. & Abboud, F. M. (1993) Sleep 16, S30–S33. [PubMed] [Google Scholar]
- 7.Fletcher, E. C. (2000) Respir. Physiol. 119, 189–197. [DOI] [PubMed] [Google Scholar]
- 8.Cragg, P. A., Runold, M., Kou, Y. R. & Prabhakar, N. R. (1994) Respir. Physiol. 95, 295–310. [DOI] [PubMed] [Google Scholar]
- 9.Kline, D. D., Peng, Y. J., Manalo, D. J., Semenza, G. L. & Prabhakar, N. R. (2002) Proc. Natl. Acad. Sci. USA 99, 821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krahenbuhl, S., Schafer, T. & Wiesmann, U. (1996) Clin. Chim. Acta 253, 79–90. [DOI] [PubMed] [Google Scholar]
- 11.Redinbaugh, M. G. & Turley, R. B. (1986) Anal. Biochem. 153, 267–271. [DOI] [PubMed] [Google Scholar]
- 12.Gardner, P. R. (2002) Methods Enzymol. 349, 9–23. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell, G. S. & Johnson, S. M. (2003) J. Appl. Physiol. 94, 358–374. [DOI] [PubMed] [Google Scholar]
- 14.Rozanov, C., Roy, A., Mokashi, A, Osnai, S., Daudu, P., Storey, B. & Lahiri, S. (2000) Adv. Exp. Med. Biol. 475, 397–404. [DOI] [PubMed] [Google Scholar]
- 15.Raha, S. & Robinson, B. H. (2000) Trends Biochem. Sci. 25, 502–508. [DOI] [PubMed] [Google Scholar]
- 16.Gardner, P. R., Nguyen, D. D. & White, C. W. (1996) Arch. Biochem. Biophys. 325, 20–28. [DOI] [PubMed] [Google Scholar]
- 17.Yermolaieva, O., Brot, N., Weissbach, H., Heinemann, S. H. & Hoshi, T. (2000) Proc. Natl. Acad. Sci. USA 97, 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cistulli, P. A. & Sullivan, C. E. (1994) in Sleep and Breathing, eds. Saunders, N. A. & Sullivan, C. E. (Dekker, New York), pp. 405–448.