

Abstract
The rat protein tyrosine phosphatase η, rPTPη, is a class I “classical” transmembrane RPTP, with an intracellular portion composed of a unique catalytic region. The rPTPη and the human homolog DEP-1 are downregulated in rat and human neoplastic cells, respectively. However, the malignant phenotype is reverted after exogenous reconstitution of rPTPη, suggesting that its function restoration could be an important tool for gene therapy of human cancers. Using small-angle x-ray scattering (SAXS) and biophysical techniques, we characterized the intracellular catalytic domain of rat protein tyrosine phosphatase η (rPTPηCD) in solution. The protein forms dimers in solution as confirmed by SAXS data analysis. The SAXS data also indicated that rPTPηCD dimers are elongated and have an average radius of gyration of 2.65 nm and a Dmax of 8.5 nm. To further study the rPTPηCD conformation in solution, we built rPTPηCD homology models using as scaffolds the crystallographic structures of RPTPα-D1 and RPTPμ-D1 dimers. These models were, then, superimposed onto ab initio low-resolution SAXS structures. The structural comparisons and sequence alignment analysis of the putative dimerization interfaces provide support to the notion that the rPTPηCD dimer architecture is more closely related to the crystal structure of autoinhibitory RPTPα-D1 dimer than to the dimeric arrangement exemplified by RPTPμ-D1. Finally, the characterization of rPTPηCD by fluorescence anisotropy measurements demonstrates that the dimer dissociation is concentration dependent with a dissociation constant of 21.6 ± 2.0 μM.
INTRODUCTION
Protein tyrosine phosphorylation is a reversible posttranslational modification involved in regulation of physiological processes as well as human health and disease. The antagonistic activities of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) dictate the intracellular levels of protein tyrosine phosphorylation, one of the most important eukaryotic cell signaling mechanisms (1–4). The increasing number and diversity of protein tyrosine kinases and phosphatases has been directly related to the increase in eukaryotic complexity. In recent years it became evident that, like PTKs, the PTPs play important roles in many cellular processes such as embryogenesis, organ development, tissue homeostasis, the immune system, fertilization, cell migration, and proliferation (5–7). The protein tyrosine phosphatases belong to a large group of enzymes that are subdivided into four main families based on the amino acid sequence of their catalytic domains and substrate specificity (8). The class I cysteine-based PTPs constitute by far the largest group and contain the 38 strictly tyrosine-specific PTPs. The “classical PTPs” can be further divided into transmembrane, receptor-like enzymes (RPTPs), and the intracellular, nonreceptor PTPs (NRPTPs), all sharing an intracellular catalytic domain of ∼240 residues. Its active site bears a signature motif (PIVVHCSAGvGRTG) where the catalytically essential cysteine and arginine are separated by five residues (9,10). The crystal structures of all known RPTP catalytic domains revealed a common architecture comprising: a globular fold that consists of an eight-stranded twisted β-sheet flanked by four α-helices on one side and another one on the opposite side; the PTP signature motif located at the bottom of the catalytic site cleft; and four loops, three of which provide residues needed for catalysis and substrate specificity (11–13).
The rat protein tyrosine phosphatase η, rPTPη, is a class I “classical” transmembrane RPTP, and like its human counterpart (also known as DEP-1, PTPRJ, RPTPη, and CD148) it is a ubiquitous gene. In fact, rPTPη is expressed in all studied tissues, including brain, liver, spleen, thyroid, and endothelial cells. The receptor-like rPTPη consists of an extended extracellular region containing eight fibronectin type III motifs, a short transmembrane domain, and an intracellular portion composed of a unique catalytic region (14). Soon after its discovery, it was reported that rPTPη expression is regulated by the two main thyroid regulatory pathways, suggesting it involvement in both growth and differentiation of thyroid cells (15). Further observations demonstrated that rPTPη and the human homolog DEP-1 are downregulated in rat and human neoplastic cells, respectively. However, the malignant phenotype can be reverted after exogenous reconstitution of rPTPη through increasing levels of cell cycle inhibitor p27Kip1 and dephosphorylation of PLCγ1, a substrate of DEP-1 (16–19). It was also demonstrated that the PTPη protein is capable of binding to c-Src in living cells. The dephosphorylation of the negative regulatory tyrosine (Tyr-529 of the c-Src family protein tyrosine kinases) increases c-Src tyrosine kinase activity in malignant rat thyroid cells stably transfected with rPTPη (20). Besides, the implication of PTPη as a possible candidate for the mouse colon-cancer susceptibility locus, Scc1, came to reinforce the idea that restoration of rPTPη function could be a useful tool for gene therapy of human cancers (21–23).
Although the process of RPTP activity regulation remains elusive, one of the proposed structural mechanisms involves formation of an autoinhibitory dimeric quaternary structure. Structural evidence supporting such a mechanism came from the crystal structure of RPTPα-D1 (24). RPTPα-D1 was crystallized as a dimer with two active sites facing each other and the amino-terminal helix-turn-helix wedge-like segment of each monomer blocking the opposing active site (24). In addition, functional inhibition of RPTPs was observed in ligand-induced dimerization of EGFR/CD45, a chimeric protein containing the extracellular domain of the epidermal growth factor receptor (EGFR), and the intracellular domain of the RPTP, CD45 (25), as well as in the full-length mutants of RPTPα assembled as stable disulphide-bonded homodimers (26,27). By contrast, the crystal structures of both RPTPμ-D1 that shares 46% sequence identity with RPTPα-D1 reveal completely different dimer formation. The RPTPμ-D1 dimer has neither the catalytic sites nor the amino-terminal helix-turn-helix segment participating in protein-protein interactions (28). Taking these results together one can conclude that although dimerization seems likely to be of physiological relevance in blocking the biological activity of RPTPα and CD45, it might not be a general mechanism of RPTP inhibition.
In this study, we conducted a structural and biophysical characterization of rPTPη catalytic domain with use of small angle x-ray scattering (SAXS), homology modeling, amino acid sequence alignment, and fluorescence anisotropy techniques, aiming to determine the protein molecular shape and its oligomeric state in solution and to shed more light onto the putative autoinhibitory mechanism that might control rPTPη enzymatic activity.
EXPERIMENTAL PROCEDURES
Protein expression and purification
The catalytic domain of rPTPη has been subcloned, expressed, and purified as described in Santos et al. (29). Briefly, the cloned construct contained the open reading frame of the intracellular region of rPTPη (W875-A1216) fused at its N-terminus with the 34 additional amino acid residues (MGSSHHHHHHSSGLVPRGSHMASMTGGQQMGRGS) derived from pET-28a(+) vector that included the cluster of six histidine residues for protein purification by metal affinity chromatography. BL21 (DE3) cells harboring the plasmid containing rPTPηCD insert were grown at 30°C in 2× YT plus 50 μg/ml kanamycin with shaking until the absorbance at 600 nm reached 0.6–0.8. At this point, 0.5 mM isopropyl-β-d-thiogalactopyranoside was added to induce His6-rPTPηCD expression and cells were incubated at 30°C for 4 h. The induced bacteria were harvested by centrifugation at 6000 × g in a Sorvall RC-5C Plus centrifuge at 4°C for 20 min. Bacterial pellets were ressuspended in lysis buffer (50 mM sodium phosphate buffer, pH 7.8; 100 mM NaCl; 10% glycerol; 10 mM imidazol; 2 mM β-mercaptoethanol) containing 1 mM phenylmethanesulfonyl fluoride and 0.5 mg/ml of lysozyme (Sigma, St. Louis, MO). The suspension was incubated on ice for 30 min to lyse cells. The lysate was further disrupted by sonication to reduce viscosity. Centrifugation was done at 14,000 × g for 1 h to obtain the clear crude protein preparation. Talon Superflow resin (Clontech, Palo Alto, CA), preequilibrated with equilibration buffer (50 mM sodium phosphate buffer, pH 7.8; 300 mM NaCl; 10% glycerol; 10 mM imidazol; 2 mM β-mercaptoethanol) was mixed with the clear lysate and left rotating at 4°C for 1 h. The mixture of resin and supernatant was mounted into a c16/10 glass column (Amersham Biosciences, Uppsala, Sweden) and connected to a high-performance liquid chromatography ÄKTA purifier (Amersham Biosciences). The tightly bound proteins were eluted with buffer containing 50 mM sodium phosphate buffer, pH 7.8; 50 mM NaCl; 10% glycerol; 300 mM imidazol; 2 mM β-mercaptoethanol. The His6-rPTPηCD was further purified by size exclusion chromatography on a Superdex 200 HL 26/60 column (Amersham Biosciences) using as eluent Hepes buffer (20 mM Hepes, pH 7.8; 200 mM NaCl; 5% glycerol; 1 mM dithiothreitol). All purification procedures were carried out at 4°C. Soluble His6-rPTPηCD (molecular weight of 43,000) was concentrated to 1 mg/ml and incubated with 0.5 units/ml bovine thrombin protease for 18 h at 18°C followed by dialysis against Hepes buffer. The thrombin cleaved rPTPηCD (molecular weight of 41,000) was, then, frozen in liquid nitrogen and stored at −80°C (29).
Protein concentration measurements
Protein concentration was determined by the method of Bradford (30) with bovine serum albumin (Sigma) as reference standard, using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA).
Small-angle x-ray scattering measurements and scattering data analysis
Small-angle x-ray scattering experiments were carried out at the SAS beam line on the Brazilian National Synchrotron Light Laboratory (Campinas, Sao Paulo, Brazil), using a one-dimensional position-sensitive detector (31). SAXS data were collected at a wavelength 0.148 nm for sample-detector distance of 1046.3 mm to give a q-range from 0.28 nm−1 to 3.40 nm−1 (q = 4*sinθ/λ, where 2θ is the scattering angle). rPTPηCD concentrations between 3 and 13 mg/ml were prepared in Hepes containing 200 mM NaCl and maintained at a constant temperature of 4°C during the measurements. The scattering curves of the protein solutions and buffers were collected in several successive frames of 100 s each to avoid radiation-induced protein damage. The data reduction included normalization of the one-dimension scattered data to the intensity of the transmitted incident beam; correction for the detector response; and subtraction of the scattering of the buffers. After scaling all scattered curves for protein concentration, the composed scattering curves were constructed by merging the low angle data (<0.85 nm−1) obtained at 3 mg/ml with the high angle data, measured at 13 mg/ml, which were, then, used in ab initio model reconstruction.
Guinier analysis (32) was applied to determine the radii of gyration (Rg) of rPTPηCD in solution. The Rg and the scattered intensity, I(q), were inferred, respectively, from the slope and the intercept of the linear fit of ln[I(q)] vs. q2 in the q-range q × Rg < 1.0. The same parameter was also obtained from the theoretical fit of the merged curve by the indirect Fourier transform program GNOM (33), which also evaluated the distance distribution function, p(r), of rPTPηCD. The maximum dimension, Dmax, was estimated from the p(r) function as the distance, r, where p(r) goes down to zero.
Ab initio molecular shape determination and DAMAVER program
Ab initio models were calculated using the well-established reconstruction method DAMMIN, to determine the conformation in solution of rPTPηCD (34). Using the simulated annealing method, the program searches for the best dummy atom model (DAM) that fits the experimental data through minimizing the discrepancy function, f(X), between the calculated and experimental curves. A looseness penalty ensures the compactness and connectivity of the solution, Eq. 1, where α > 0 is the positive weight of the looseness penalty, αP(X).
 |
(1) |
The overall shape of the protein was restored from the complete range of q (0.28 nm−1 < q < 3.40 nm−1), which allowed a reasonable resolution for the discrete DAM. In each of 20 independent low-resolution reconstructions of molecular shape, a sequence of 87 ± 3 independent runs was carried out to obtain the most probable model without imposing any symmetry constrains. During this procedure, a constant was subtracted from the experimental data to ensure that the intensity at higher angles decays as q−4 following Porod's law for the homogeneous particles (35). The value of the constant was derived automatically from the outer part of the curve by linear fitting in coordinates q4I(0) versus q4 by the program DAMMIN.
DAMAVER was used for automated analysis and averaging of multiple reconstructions, permitting both to analyze the stability of the reconstruction convergence and to yield the most probable particle model (36,37).
Homology model building of rPTPηCD
The coordinate sets for the monomeric and dimeric crystallographic structures of the catalytic domain (D1) of mouse RPTPα and human RPTPμ were obtained from the Protein Data Bank ((PDB) codes 1YFO and 1RPM, respectively) (24,28). The Rg, Dmax, and scattering curves were calculated from the dimeric structures by the program CRYSOL (38), taking into account the influence of the hydration shell.
Computer models for the three-dimensional (3D) structure of rPTPηCD were constructed using homology modeling, which requires available 3D structures as templates. The mouse protein tyrosine phosphatase α has 43.21% identity to the rPTPηCD sequence, and it was selected as the template for homology model generation with the Modeller program (39). We produced 100 models, each of which was validated using the programs Procheck (40,41), Whatcheck (42), and Verify3D (43,44). The best model was chosen using as criteria the energy function calculated by the Modeller program, the Ramachandran plot, and absence of structural anomalies. The best homology model was superimposed with the dimeric crystallographic structures of RPTPα-D1 and RPTPμ-D1 using SUPCOMB (36), which computes and carries out best-matching alignment of the 3D models. The same program was used for superpositions of high-resolution models with ab initio low-resolution structures derived from experimental solution-scattering data.
Fluorescence anisotropy measurements
Fluorescence anisotropy experiments were carried on the ISS PC1 spectrofluorimeter equipped with Glan-Thompson polarizers (ISS, Champaign, IL). rPTPηCD at a concentration of 0.22 mM was labeled by incubation with 1 mM fluorescein isothiocyanate ((FITC) Sigma) in Hepes buffer (20 mM Hepes, pH 7.8; 200 mM NaCl; 5% glycerol; 1 mM DTT), at 4°C for 3 h. Free FITC was separated from labeled rPTPηCD using Hitrap desalting column (GE Healthscience, Madison, WI) equilibrated with Hepes buffer. Protein labeling was confirmed by absorbance spectrophotometry, showing both protein and fluorescein contributions in the absorption spectra. The labeling efficiency was of ∼0.24 mol fluorescein per mol protein, as calculated from
 |
(2) |
where A280 and A494 are the absorption at 280 and 494 nm, respectively, 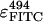 = 68,000 M−1cm−1, and
= 68,000 M−1cm−1, and  = 20,000 M−1cm−1 are the extinction coefficient for protein-bound FITC at 280 and 494 nm, respectively (45,46).
= 20,000 M−1cm−1 are the extinction coefficient for protein-bound FITC at 280 and 494 nm, respectively (45,46). 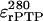 = 63,050 M−1cm−1 is the extinction coefficient for rPTPη catalytic domain at 280 nm calculated on the basis of its amino acid sequence (residues W875-A1216 of rPTPη; “Protein expression and purification” section) assuming six tryptophan residues, 20 tyrosine residues, and at most two cystines (47). The low efficiency of labeling, probably due to the pH and temperature conditions used in the reactions, is advantageous because it diminishes the chances of self-transfer between fluorophores on the same protein molecule.
= 63,050 M−1cm−1 is the extinction coefficient for rPTPη catalytic domain at 280 nm calculated on the basis of its amino acid sequence (residues W875-A1216 of rPTPη; “Protein expression and purification” section) assuming six tryptophan residues, 20 tyrosine residues, and at most two cystines (47). The low efficiency of labeling, probably due to the pH and temperature conditions used in the reactions, is advantageous because it diminishes the chances of self-transfer between fluorophores on the same protein molecule.
Fluorescein-rPTPηCD at a concentration of 10 nM was titrated with unlabeled rPTPηCD at 20°C in Hepes buffer. Fluorescence anisotropy of the labeled solution of rPTPηCD was excited at 480 nm and emission measured through a band-pass filter with a cutoff of 50% at 515 nm, as previously described (48). For each concentration of unlabeled protein, the data point represents the mean of at least five measurements after stabilization. The resulting anisotropy change was used to calculate the dissociation equilibrium constant by adjusting Eq. 3 to the experimental data:
 |
(3) |
where fd is the fraction of protein dimers and Kd is the dissociation constant. As the association led to a discrete increase in fluorescence intensity (∼40%), we applied correction:
 |
(4) |
where Aobs, Am, and Ad are, respectively, the rPTPηCD-labeled anisotropy values observed at a given concentration of unlabeled rPTPηCD, for the monomeric rPTPηCD and dimeric rPTPηCD (49). The corrections performed on changes in fluorescence intensity make use of the total fluorescence intensity IT directly provided by the instrument software, according to:
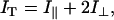 |
(5) |
where I‖ and I⊥ are, respectively, the time-independent steady-state values for fluorescent intensity polarized parallel and perpendicular to that of the excitation beam. Binding data were analyzed using SigmaPlot 2002 Windows Version 8.0.
RESULTS AND DISCUSSION
Small-angle x-ray scattering measurements demonstrate that rPTPηCD is capable of dimer formation
The biological activity of the purified rPTPηCD was tested in vitro by hydrolysis assay. The results demonstrated that rPTPηCD was active and able to hydrolyze p-nitrophenyl phosphate (29). To obtain information about the size, molecular shape, and oligomeric state of rPTPηCD in solution, we submitted the protein to small-angle x-ray scattering analysis. rPTPηCD x-ray scattering curve is shown in Fig. 1 A. The Guinier analysis, in the q2-range from 0.08 to 0.24 nm−2, reproducibly gave estimates of 2.59 nm for the radius of gyration of rPTPηCD. The linearity of the Guinier plot indicated that rPTPηCD preparation was monodisperse and constituted mainly by unique oligomeric species (Fig. 1 A, inset).
FIGURE 1.

Small angle x-ray scattering curves. (A) Experimental solution scattering curve of rPTPηCD and results of the fitting procedures. Intensity curves from rPTPηCD are shown as logarithm (log I) versus the momentum transfer q scale, with an inset containing the correspondent Guinier plots (log I vs. q2). The desmeared experimental curve (open squares) denotes the SAXS data and the error bars indicate the standard uncertainty in the measurement. The dashed curve corresponds to the best-fit model produced by DAMMIN (34). The black solid line is the theoretical scattering intensity from the dimer homology model based on the RPTPα-D1 (PDB code, 1YFO). The gray solid line is the scattering intensity computed from the dimer homology model based on the RPTPμ-D1 crystal structure (PDB code, 1RPM). (B) Distance distribution function of rPTPηCD. The p(r) values of rPTPηCD (open squares) computed from the experimental scattering curves by the indirect Fourier transform program GNOM (33). The maximum dimension (Dmax) is equal to 8.50 ± 1.00 nm. The black solid curve corresponds to p(r) derived from dimer homology model based on the RPTPα-D1 dimer. The p(r) derived from dimer homology model based on the dimer of RPTPμ-D1 is given as a gray solid line. The distance distribution function for RPTPα-D1 dimer is clearly more similar to the experimentally derived p(r) for rPTPηCD than the one calculated on the basis of RPTPμ-D1 dimer model.
The pair-distance distribution function p(r) was obtained by the inverse Fourier transform of the rPTPηCD scattering data, using the program GNOM (33). Analysis of the p(r) function led us to conclude that the protein has an elongated form with a Dmax of 8.50 ± 1.00 nm (Fig. 1 B). The value of 2.65 ± 0.02 nm for Rg calculated from the distance distribution function is in good agreement with the estimate derived from the Guinier analysis (Table 1). To compare rPTPηCD with the existing high-resolution structures, we also computed the theoretical scattering curves and the pair-distance distribution function for the crystal structures of RPTPα-D1 and RPTPμ-D1 dimers by using the program CRYSOL (38) (Fig. 1, A and B). The radii of gyration observed for RPTPα-D1 and RPTPμ-D1 were 2.72 and 2.82 nm, and the Dmax values were 9.95 and 9.50 nm, respectively. Comparison of these simulated SAXS curves with the experimental data for rPTPηCD revealed discrepancy values for RPTPα-D1 and RPTPμ-D1 of 1.47 and 2.22, respectively, for RPTPα-D1 and RPTPμ-D1. SAXS results reveal that in solution at the concentrations studied the rPTPηCD appears predominantly as dimers with a molecular shape similar to that of RPTPα-D1 (Fig. 1, A and B, and Table 1). The dimeric state of rPTPηCD also was confirmed by native gel electrophoresis (not shown).
TABLE 1.
Structural parameters
| rPTPηCD
|
||||
|---|---|---|---|---|
| Parameter/sample | Exp* | rPTPα | rPTPμ | DAM† |
| Dmax (nm) | 8.50 ± 1.00 | 9.95 | 9.5 | 8.72 |
| Rg (nm) | 2.65 ± 0.02 | 2.72 | 2.82 | 3.35 |
| Free parameters | 8‡ | – | – | 1926§ |
| Discrepancy χ | – | 1.47 | 2.22 | 1.06 |
| Resolution (nm) | 1.85¶ | – | – | 1.85¶ |
Exp, calculated from the experimental data.
DAM, parameters of the dummy atoms models averaged over 10 models.
Number of Shannon channels is given Ns = [Dmax(qmax − qmin)]/π.
Symmetry not imposed.
Resolution is calculated as 2π/qmax.
Ab initio molecular shapes of rPTPηCD and its high-resolution models
The ab initio low-resolution structure of rPTPηCD was determined from the SAXS data using the program DAMMIN (34) without any symmetry constraints (Table 1). An average of 10 independently generated dummy atom models produced by the DAMAVER package fitted well the experimental x-ray scattering curve (χ = 1.06; Table 1). The low-resolution rPTPηCD dimer has an elongated “cigar-like” shape with ∼3:1 ratio between its longest and shortest dimensions (Fig. 2). Given that the dimeric arrangement is important for rPTPη function, we attempted to model it using as templates the homologous RPTPα-D1 and RPTPμ-D1 dimer.
FIGURE 2.

Three orthogonal stereoviews of the ab initio low-resolution rPTPηCD structure superposed with the dimeric homology model of rPTPηCD based on the crystallographic structure of RPTPα-D1. The figure was prepared using PyMOL (DeLano Scientific, San Carlos, CA; http://www.pymol.org).
The rPTPηCD dimer homology models based on the RPTPα-D1 and RPTPμ-D1 crystal structures templates were automatically superposed, using the program SUPCOMB (36), as rigid bodies with the ab initio low-resolution structure of the protein (Figs. 2 and 3). After automated best-matching alignment procedure, visual analysis clearly indicates that RPTPα-style dimer (Fig. 2) fits the rPTPηCD low-resolution structure better than the RPTPμ-D1-based homology model (Fig. 3). Furthermore, in the case of the RPTPα-D1-type dimer, the normalized spatial discrepancy (NSD) value is equal to 0.823, whereas for the dimer of RPTPμ-D1-type the NSD value was 0.993. Because we can assume structural similarity between models with NSD values <1 (41), these results provide further support to the idea that in solution rPTPηCD forms dimers with the molecular shape resembling the dimer arrangement revealed by the crystallographic structure of RPTPα-D1.
FIGURE 3.

Three orthogonal stereoviews of the homology model of the putative rPTPηCD dimer based on the crystallographic structure of RPTPμ-D1 superposed on the low-resolution ab initio rPTPηCD molecular envelope. The figure was prepared using PyMOL (DeLano Scientific).
To gain more data with respect of rPTPηCD dimerization mode, we compared amino acid sequence composition at the putative dimer interfaces of the two proposed models with that of the dimerization interface of RPTPα-D1 and RPTPμ-D1 (Fig. 4). Although the overall sequence identity between rPTPηCD and these two phosphatases is similar (43.21% for RPTPα-D1 and 45.16% for RPTPμ-D1), the same is not true for the dimerization regions: 17 out of 30 residues (56.7%) on the dimerization interface of RPTPα-D1 are conserved in rPTPηCD, whereas only five out of 19 residues (26.3%) are conserved in rPTPηCD at the dimerization interface of RPTPμ-D1 (Fig. 5). This lends further support to the hypothesis that rPTPη dimerization might be similar to the RPTPα dimer architecture in which the N-terminal helix-turn-helix segment of the dyad-related monomer sterically blocks the opposite active site.
FIGURE 4.

Sequence alignment for RPTPα-D1, RPTPμ-D1, and PTPηCD. The sequence of PTPηCD corresponds to the intracellular domain of the protein studied in current work (residues W875–A1216). The sequences of RPTPα-D1 and RPTPμ-D1 are of the polypeptide chains structurally defined in PDBs 1YFO and 1RPM, respectively. The residues at the dimerization interfaces are marked in gray (RPTPα-D1) and black (RPTPμ-D1). Residues in rPTPηCD amino acid sequence are gray when they match the correspondent residues of the RPTPα-D1 dimerization interface; they are black when identical to residues at the dimerization interface of RPTPμ-D1.
FIGURE 5.

Crystallographic structures of RPTPα-D1 and RPTPμ-D1. The dimerization interfaces of RPTPα-D1 and RPTPμ-D1 are shown as surfaces. (A) The residues of RPTPα-D1 that are conserved in rPTPηCD are shown in black; the nonconserved residues are painted in gray. (B) The same molecule as in panel A rotated by 180° across the vertical axis. (C) The residues of RPTPμ-D1 that are conserved in rPTPηCD are depicted in black; the ones that are nonconserved are given in gray. (D) The same molecule as in panel C rotated by 180° across the vertical axis. The figure was prepared using PyMOL (DeLano Scientific).
Fluorescence anisotropy measurements provide an equilibrium dissociation constant of the rPTPηCD dimers
To determine the equilibrium dissociation constant of the rPTPηCD dimer, we labeled the recombinant protein with fluorescein isothyocianate (FITC), which reacts with amino terminus of proteins and the ɛ-amino group of lysines (50). Fluorescence spectra of both buffer and fluorescein-labeled rPTPηCD are shown in Fig. 6 A. Purified fluorescein-labeled rPTPηCD (FITC-rPTPηCD) at 10 nM shows an anisotropy value of ∼0.085 (Fig. 6 B), which is compatible with a protein-bound fluorescein value but largely different from the typical values of free fluorescein anisotropy (of the order of 0.014). Equilibration of 10 nM FITC-rPTPηCD with increasing amounts of unlabeled rPTPηCD (from 7.2 nM to 36 μM) led to an increase in fluorescein fluorescence anisotropy values, indicating multimerization (Fig. 6 B). Because we know from SAXS and native gel electrophoresis studies that at 50 μM rPTPηCD is dimeric, these changes in anisotropy values as a function of rPTPηCD concentration are indicative of a monomer-dimer equilibrium. The rPTPηCD dimer dissociation constant of 21.6 ± 2.0 μM was calculated by adjusting Eq. 3 to experimental data. This Kd corresponds to a Gibbs free-energy value of 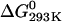 = 6.2 kcal/mol.
= 6.2 kcal/mol.
FIGURE 6.

Equilibrium fluorescence anisotropy. (A) Fluorescence spectra of buffer and fluorescein-labeled rPTPηCD. (B) Measurements of rPTPηCD dimer-monomer reaction were performed as described in experimental procedures. The results of three independent experiments are shown as raw anisotropy. (Inset) Fraction of association calculated from anisotropy values properly corrected for changes in fluorescence intensity. Solid line corresponds to the best fit of Eq. 3 to data, which yields a dissociation constant of 21.6 ± 2.0 μM (r2 = 0.988).
On the basis of theoretical evaluations, it is expected that protein dissociation (from 10 to 90%) would occur within a protein concentration log span of 2.86 (51,52). Our experimental data demonstrate that rPTPηCD dimers dissociation presents a significantly shortened log span of 1.5. A similar behavior leading to a reduced span in the dimerization isotherms has already been observed for a number of proteins, such as malate dehydrogenase (53), yeast enolase (51), arc repressor (54), and O2bZIP (55), to name a few. The deviations from the expected span in dimerization isotherms have been attributed to a conformational drift of the protein, i.e, a change in quaternary interactions affecting the structure and conformation of each subunit upon dimerization process (52). This indicates that subunits of the dimeric and monomeric species of rPTPηCD are conformationally different.
A number of RPTPs, as well as RPTKs, are known to function as transmembrane dimers and dimerization could be a common phenomenon for RPTPs in general (26,27,56). Although the dimeric interaction seen for soluble rPTPηCD is ∼22 μM, this affinity is expected to be enhanced substantially for the full-length receptors in the cell membrane. In fact, both local protein concentrations and monomer-monomer affinities could be significantly higher for the functional rPTPη dimers bound to the membrane of cell. Under these settings two extracellular domains of PTPη, each composed of eight fibronectin type 3 motifs, will also contribute to PTPη dimer formation. Membrane anchoring, which restricts both PTPη translational movement in the direction perpendicular to the membrane surface, and its rotational freedom around the axes within the membrane plane, will lead to further increase in association affinity of PTPη dimer. It has been argued that the binding affinities of the molecules in two-dimensional diffusion case are significantly (orders of magnitude) higher than observed in solution (57,58). Therefore, affinities of dimers association are expected to be substantially enhanced, and equilibrium dissociation constants significantly diminished, under conditions of the full-length rPTPηs anchored in the membrane.
CONCLUSIONS
Dimerization is a well-established regulatory mechanism for activation of RPTKs and is likely to be involved in regulation of RPTPs. Although many protein tyrosine phosphatases have two intracellular domains, only the plasma membrane-proximal domain (D1) has significant catalytic activity. Dimeric crystallographic structure of RPTPα-D1 revealed the presence of an NH2-terminal helix-turn-helix segment interacting with the opposing monomer, in such a manner that both catalytic sites in the dimer are blocked (24). On the other hand, it has been shown that dimerization of Sap-1/PTPRH, a single intracellular domain RPTP, does not occur through its catalytic domain (59). Given a fact that rPTPη also has a unique catalytic domain, we studied its behavior in solution using synchrotron small-angle x-ray scattering, homology modeling, sequence alignment, and fluorescence anisotropy. Here, we provide the first ab initio low-resolution structure of rPTPηCD in solution restored at 1.85-nm resolution from synchrotron small-angle x-ray scattering data. The results from SAXS reveal that rPTPηCD forms dimers in solution. The fluorescence anisotropy demonstrates that the dimeric form of rPTPηCD is the predominant species in solution with a monomer-dimer equilibrium dissociation constant of 21.6 ± 2.0 μM and a corresponding  equal to 6.2 kcal/mol. Ab initio SAXS model building, homology modeling, and sequence analysis data of the putative rPTPηCD dimeric interfaces taken together strongly indicate that rPTPη dimeric architecture might be closely related to that of RPTPα-D1. It is tempting to speculate that dimerization of rPTPηCD could be involved in the self-inhibition of its enzymatic activity. In this case the autoinhibitory mechanism would occur through the N-terminal wedge region-active site interactions as proposed for RPTPα-D1, a hypothesis amenable to further site-directed mutagenesis analysis, and in vivo and in vitro functional studies.
equal to 6.2 kcal/mol. Ab initio SAXS model building, homology modeling, and sequence analysis data of the putative rPTPηCD dimeric interfaces taken together strongly indicate that rPTPη dimeric architecture might be closely related to that of RPTPα-D1. It is tempting to speculate that dimerization of rPTPηCD could be involved in the self-inhibition of its enzymatic activity. In this case the autoinhibitory mechanism would occur through the N-terminal wedge region-active site interactions as proposed for RPTPα-D1, a hypothesis amenable to further site-directed mutagenesis analysis, and in vivo and in vitro functional studies.
Acknowledgments
This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (grants No. 99/03387-4 and No. 06/00182-8) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
References
- 1.Hunter, T. 1987. A 1001 protein-kinases. Cell. 50:823–829. [DOI] [PubMed] [Google Scholar]
- 2.Hunter, T. 1995. Protein-kinases and phosphatases: the Yin and Yang of protein-phosphorylation and signaling. Cell. 80:225–236. [DOI] [PubMed] [Google Scholar]
- 3.Tonks, N. K., and B. G. Neel. 1996. From form to function: signaling by protein tyrosine phosphatases. Cell. 87:365–368. [DOI] [PubMed] [Google Scholar]
- 4.Neel, B. G., and N. K. Tonks. 1997. Protein tyrosine phosphatases in signal transduction. Curr. Opin. Cell Biol. 9:193–204. [DOI] [PubMed] [Google Scholar]
- 5.Chagnon, M. J., N. Uetani, and M. L. Tremblay. 2004. Functional significance of the LAR receptor protein tyrosine phosphatase family in development and diseases. Biochem. Cell Biol. 82:664–675. [DOI] [PubMed] [Google Scholar]
- 6.den Hertog, J. 1999. Protein-tyrosine phosphatases in development. Mech. Dev. 85:3–14. [DOI] [PubMed] [Google Scholar]
- 7.Mustelin, T., R. T. Abraham, C. E. Rudd, A. Alonso, and J. J. Merlo. 2002. Protein tyrosine phosphorylation in T cell signaling. Front. Biosci. 7:918–969. [DOI] [PubMed] [Google Scholar]
- 8.Fauman, E. B., and M. A. Saper. 1996. Structure and function of the protein tyrosine phosphatases. Trends Biochem. Sci. 21:414–417. [DOI] [PubMed] [Google Scholar]
- 9.Alonso, A., J. Sasin, N. Bottini, I. Friedberg, I. Friedberg, A. Osterman, A. Godzik, T. Hunter, J. Dixon, and T. Mustelin. 2004. Protein tyrosine phosphatases in the human genome. Cell. 117:699–711. [DOI] [PubMed] [Google Scholar]
- 10.Kolmodin, K., and J. Åqvist. 2001. The catalytic mechanism of protein tyrosine phosphatases revisited. FEBS Lett. 498:208–213. [DOI] [PubMed] [Google Scholar]
- 11.Jia, Z., D. Barford, A. J. Flint, and N. K. Tonks. 1995. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science. 268:1754–1758. [DOI] [PubMed] [Google Scholar]
- 12.Stuckey, J. A., H. L. Schubert, E. B. Fauman, Z. Y. Zhang, J. E. Dixon, and M. A. Saper. 1994. Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 A and the complex with tungstate. Nature. 370:571–575. [DOI] [PubMed] [Google Scholar]
- 13.Barford, D., A. K. Das, and M. P. Egloff. 1998. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 27:133–164. [DOI] [PubMed] [Google Scholar]
- 14.Zhang, L., M. L. Martelli, C. Battaglia, F. Trapasso, D. Tramontano, G. Viglietto, A. Porcellini, M. Santoro, and A. Fusco. 1997. Thyroid cell transformation inhibits the expression of a novel rat protein tyrosine phosphatase. Exp. Cell Res. 235:62–70. [DOI] [PubMed] [Google Scholar]
- 15.Martelli, M. L., F. Trapasso, P. Bruni, M. T. Berlingieri, C. Battaglia, M. T. Vento, B. Belletti, R. Iuliano, M. Santoro, G. Viglietto, and A. Fusco. 1998. Protein tyrosine phosphatase-eta expression is upregulated by the PKA-dependent and is downregulated by the PKC-dependent pathways in thyroid cells. Exp. Cell Res. 245:195–202. [DOI] [PubMed] [Google Scholar]
- 16.Trapasso, F., R. Iuliano, A. Boccia, A. Stella, R. Visconti, P. Bruni, G. Baldassarre, M. Santoro, G. Viglietto, and A. Fusco. 2000. Rat protein tyrosine phosphatase eta suppresses the neoplastic phenotype of retrovirally transformed thyroid cells through the stabilization of p27(Kip1). Mol. Cell. Biol. 20:9236–9246. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Florio, T., S. Arena, S. Thellung, R. Iuliano, A. Corsaro, A. Massa, A. Pattarozzi, A. Bajetto, F. Trapasso, A. Fusco, and G. Schettini. 2001. The activation of phosphotyrosine phosphatase eta (r-PTP eta) is responsible for the somatostatin inhibition of PC Cl3 thyroid cell proliferation. Mol. Endocrinol. 15:1838–1852. [DOI] [PubMed] [Google Scholar]
- 18.Iuliano, R., F. Trapasso, I. Le Pêra, F. Schepis, I. Samà, A. Clodomiro, K. R. Dumon, M. Santoro, L. Chiariotti, G. Viglietto, and A. Fusco. 2003. An adenovirus carrying the rat protein tyrosine phosphatase eta suppresses the growth of human thyroid carcinoma cell lines in vitro and in vivo. Cancer Res. 63:882–886. [PubMed] [Google Scholar]
- 19.Trapasso, F., S. Yendamuri, K. R. Dumon, R. Iuliano, R. Cesari, B. Feig, R. Seto, L. Infante, H. Ishii, A. Vecchione, M. J. During, C. M. Croce, and A. Fusco. 2004. Restoration of receptor-type protein tyrosine phosphatase eta function inhibits human pancreatic carcinoma cell growth in vitro and in vivo. Carcinogenesis. 25:2107–2114. [DOI] [PubMed] [Google Scholar]
- 20.Le Pêra, I., R. Iuliano, T. Florio, C. Susini, F. Trapasso, M. Santoro, L. Chiariotti, G. Schettini, G. Viglietto, and A. Fusco. 2005. The rat tyrosine phosphatase eta increases cell adhesion by activating c-Src through dephosphorylation of its inhibitory phosphotyrosine residue. Oncogene. 24:3187–3195. [DOI] [PubMed] [Google Scholar]
- 21.Iuliano, R., I. Le Pera, C. Cristofaro, F. Baudi, F. Arturi, P. Pallante, M. L. Martelli, F. Trapasso, L. Chiariotti, and A. Fusco. 2004. The tyrosine phosphatase PTPRJ/DEP-1 genotype affects thyroid carcinogenesis. Oncogene. 23:8432–8438. [DOI] [PubMed] [Google Scholar]
- 22.Ruivenkamp, C. A., T. van Wezel, C. Zanon, A. P. M. Stassen, C. Vlcek, T. Csikos, A. M. Klous, N. Tripodis, A. Perrakis, L. Boerrigter, P. C. Groot, J. Lindeman, et al. 2002. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat. Genet. 31:295–300. [DOI] [PubMed] [Google Scholar]
- 23.Ruivenkamp, C. A., M. Hermsen, C. Postma, A. Klous, J. Baak, G. Meijer, and P. Demant. 2003. LOH of PTPRJ occurs early in colorectal cancer and is associated chromosomal loss of 18q12–21. Oncogene. 22:3472–3474. [DOI] [PubMed] [Google Scholar]
- 24.Bilwes, A. M., J. den Hertog, T. Hunter, and J. P. Noel. 1996. Structural basis for inhibition of receptor protein-tyrosine phosphatase-alpha by dimerization. Nature. 382:555–559. [DOI] [PubMed] [Google Scholar]
- 25.Majeti, R., A. M. Bilwes, J. P. Noel, T. Hunter, and A. Weiss. 1998. Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science. 279:88–91. [DOI] [PubMed] [Google Scholar]
- 26.Jiang, G., J. den Hertog, J. Su, J. Noel, J. Sap, and T. Hunter. 1999. Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-alpha. Nature. 401:606–610. [DOI] [PubMed] [Google Scholar]
- 27.Jiang, G., J. den Hertog, and T. Hunter. 2000. Receptor-like protein tyrosine phosphatase alpha homodimerizes on the cell surface. Mol. Cell. Biol. 20:5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffmann, K. M. V., N. K. Tonks, and D. Barford. 1997. The crystal structure of domain 1 of receptor protein-tyrosine phosphatase mu. J. Biol. Chem. 272:27505–27508. [DOI] [PubMed] [Google Scholar]
- 29.Santos, M. A. M., S. M. Santos, H. C. Matozo, R. V. Portugal, R. Iuliano, A. Fusco, and I. Polikarpov. 2005. Expression, purification, and characterization of rat protein tyrosine phosphatase eta catalytic domain. Protein Exp. Purif. 41:113–120. [DOI] [PubMed] [Google Scholar]
- 30.Bradford, M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. [DOI] [PubMed] [Google Scholar]
- 31.Kellermann, G., F. Vicentin, E. Tamura, M. Rocha, H. Tolentino, A. Barbosa, A. Craievich, and I. Torriani. 1997. The small-angle x-ray scattering beamline of the Brazilian Synchrotron Light Laboratory. J. Appl. Crystallogr. 30:880–883. [Google Scholar]
- 32.Guinier, A., and G. Fournet. 1995. Small-Angle Scattering of X-rays. John Wiley & Sons, New York.
- 33.Svergun, D. I. 1992. Determination of the regularization parameter in indirect-transform methods using percentual criteria. J. Appl. Crystallogr. 25:495–503. [Google Scholar]
- 34.Svergun, D. I. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76:2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porod, G. 1982. General theory. In Small-Angle X-ray Scattering. O. Glatter and O. Kratky, editors. Academic Press, London, UK. 17–51.
- 36.Kozin, M. B., and D. I. Svergun. 2001. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34:33–41. [Google Scholar]
- 37.Svergun, D. I., and M. H. Koch. 2002. Advances in structural analysis using small-angle scattering in solution. Curr. Opin. Struct. Biol. 12:654–660. [DOI] [PubMed] [Google Scholar]
- 38.Svergun, D. I., C. Barberato, and M. H. Koch. 1995. CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28:768–773. [Google Scholar]
- 39.Sali, A., and T. L. Blundell. 1993. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815. [DOI] [PubMed] [Google Scholar]
- 40.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. [Google Scholar]
- 41.Morris, A. L., M. W. MacArthur, E. G. Hutchinson, and J. M. Thornton. 1992. Stereochemical quality of protein structure coordinates. Proteins. 12:345–364. [DOI] [PubMed] [Google Scholar]
- 42.Hooft, R. W. W., G. Vriend, C. Sander, and E. E. Abola. 1996. Errors in protein structures. Nature. 381:272. [DOI] [PubMed] [Google Scholar]
- 43.Bowie, J. U., R. Luthy, and D. Eisenberg. 1991. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 253:164–170. [DOI] [PubMed] [Google Scholar]
- 44.Luthy, R., J. U. Bowie, and D. Eisenberg. 1992. Assessment of protein models with three-dimensional profiles. Nature. 356:83–85. [DOI] [PubMed] [Google Scholar]
- 45.www.piercenet.com/files/TR0031dh5-Calc-FP-ratios.pdf. 2007. [Online].
- 46.www.markergene.com/product_sheets/pis0955.pdf. 2007. [Online].
- 47.Pace, C. N., F. Vajdos, L. Fee, G. Grimsley, and T. Gray. 1996. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4:2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lima, L. M. T. R., and J. L. Silva. 2004. Positive contribution of hydration on DNA binding by E2c protein from papillomavirus. J. Biol. Chem. 279:47968–47974. [DOI] [PubMed] [Google Scholar]
- 49.Malencik, D. A., and S. R. Anderson. 1984. Peptide binding by calmodulin and its proteolytic fragments and by troponin C. Biochemistry. 23:2420–2428. [DOI] [PubMed] [Google Scholar]
- 50.McKinney, R., L. Thacker, and G. A. Hebert. 1976. Conjugation methods in immunofluorescence. J. Dent. Res. 55:A38–A44. [DOI] [PubMed] [Google Scholar]
- 51.Xu, G.-J., and G. Weber. 1982. Dynamics and time-averaged chemical potential of proteins: importance in oligomer association. Proc. Natl. Acad. Sci. USA. 79:5268–5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weber, G. 1992. Protein Interactions. Chapter XIV. Chapman and Hall, New York.
- 53.Shore, J. D., and S. K. Chakrabarti. 1976. Subunit dissociation of mitochondrial malate dehydrogenase. Biochemistry. 15:875–879. [DOI] [PubMed] [Google Scholar]
- 54.Silva, J. L., C. F. Silveira, A. Correia Jr., and L. Pontes. 1992. Dissociation of a native dimer to a molten globule monomer: effects of pressure and dilution on the association equilibrium of arc repressor. J. Mol. Biol. 223:545–555. [DOI] [PubMed] [Google Scholar]
- 55.Moreau, V. H., A. C. da Silva, R. M. Siloto, A. P. Valente, A. Leite, and F. C. Almeida. 2004. The bZIP region of the plant transcription factor opaque-2 forms stable homodimers in solution and retains its helical structure upon subunit dissociation. Biochemistry. 43:4862–4868. [DOI] [PubMed] [Google Scholar]
- 56.Stoker, A. W. 2005. Protein tyrosine phosphatases and signaling. J. Endo. 185:19–33. [DOI] [PubMed] [Google Scholar]
- 57.Grasberger, B., A. P. Minton, C. DeLisi, and H. Metzger. 1986. Interaction between proteins localized in membranes. Proc. Natl. Acad. Sci. USA. 83:6258–6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fan, Q. R., and W. A. Hendrickson. 2005. Structure of human follicle-stimulating hormone in complex with its receptor. Nature. 433:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walchli, S., X. Espanel, and R. H. van Huijsduijnen. 2005. Sap-1/PTPRH activity is regulated by reversible dimerization. Biochem. Biophys. Res. Commun. 331:497–502. [DOI] [PubMed] [Google Scholar]