

Abstract
We analyze the encounter of a peptide substrate with the native HIV-1 protease, the mechanism of substrate incorporation in the binding cleft, and the dissociation of products after substrate hydrolysis. To account for the substrate, we extend a coarse-grained model force field, which we previously developed to study the flap opening dynamics of HIV-1 protease on a microsecond timescale. Molecular and Langevin dynamics simulations show that the flaps need to open for the peptide to bind and that the protease interaction with the substrate influences the flap opening frequency and interval. On the other hand, release of the products does not require flap opening because they can slide out from the binding cleft to the sides of the enzyme. Our data show that in the protease-substrate complex the highest fluctuations correspond to the 17- and 39-turns and the substrate motion is anticorrelated with the 39-turn. Moreover, the active site residues and the flap tips move in phase with the peptide. We suggest some mechanistic principles for how the flexibility of the protein may be involved in ligand binding and release.
INTRODUCTION
Human immunodeficiency virus (HIV) type 1 protease (HIV-1 PR) cleaves the viral poly-proteins during the replication of the HIV virus. Because this process is indispensable for the life cycle of the virus, inhibitors of HIV-1 PR are widely used in the treatment of HIV/AIDS (1–3). However, due to the rapid development of drug resistance of the virus and side effects encountered upon treatment, new inhibitors of HIV-1 PR are constantly needed.
HIV-1 PR is a 198-residue homodimer whose active site is covered by two flexible β-hairpins, called flaps, controlling the entry of the polypeptide substrate (see Fig. 1). The flaps are believed to sterically restrict access of a polypeptide to the active site. The initial stage of the reaction involves the encounter of a substrate with the protease followed by its entry and proper incorporation in the binding site. After the substrate is cleaved, the products leave the binding site. Because a deeper understanding of the events associated with the ligand binding and dissociation from the active site of HIV-1 PR is important for the design of more potent and selective inhibitors, both experimental and theoretical studies aim at elucidating the internal dynamics of this enzyme.
FIGURE 1.

Heavy atom and one-bead coarse-grained representation of HIV-1 PR homodimer in the complex with a peptide substrate Lys-Ala-Arg-Val-Leu–Ala-Glu-Ala-Met (PDB entry code 1F7A (32)). The Cα nuclei of monomers and the substrate are denoted in blue, red, and green, respectively.
Crystal structures of ligand bound HIV-1 proteases show that the flaps are in their closed configuration, tightly cover the binding site, and make hydrogen bonds with the ligand. The flaps in the free HIV-1 PR are more loosely packed and less compact (Fig. 2, red ribbon) but still relatively closed over the active site. An x-ray structure of a mutant in which the flap tips separate as far as 12 Å was reported (4), but it was shown in recent molecular dynamics (MD) simulations that this widening of the flaps might be induced by crystal packing (5). Packing effects might, on the other hand, preclude crystallographic observation of the configuration of the protease with wider open flaps.
FIGURE 2.

(Left) Structure of free HIV-1 PR (1HHP) before docking (red), aligned to a representative structure of the complex (blue and green) extracted from an NVT simulation of HIV-1 PR with a docked substrate. (Right) Crystallographic structure of the complex (PDB entry 1F7A) for comparison. The root mean-squared deviation of the complex from the 1F7A configuration during the simulation ranges from 1.5 to 1.7 Å.
The dynamical picture from NMR experiments for the free HIV-1 PR reveals substantial conformational alterations in the flap region (6). The flap movements occur on a micro- to millisecond timescale with faster motions on a sub-nanosecond timescale. In the inhibitor-bound protease, flexibility of the flaps is suppressed (7,8).
The mobility of the flaps in the free protease has been studied by all-atom MD. With an external force, the flaps configuration changed from a closed to a semi-open form (9). Based on a 10-ns MD, a curling mechanism was proposed to be involved in flap opening but the short timescale of the simulations precluded reclosing (10). In a simulation of an unbound V82F/I84V HIV-1 PR mutant, only a slight flap separation was observed (11). More substantial flap opening was achieved in accelerated MD (12). Recently, a 42-ns unconstrained all-atom MD displayed both opening and reclosing of the flaps (13). MD with implicit solvation in internal torsion angle space at various temperatures was also performed and separation of the flaps was observed (14).
Analyses of the substrate fluctuations in the active site of the protease were also carried out with all-atom MD (15,16). These studies show that the substrate motion is correlated with the motion of residues 24–30 that form the cleavage site and residues 45–55, which form the flaps and especially with the flap tips (residues 48–51). The conformational flexibility of the substrate in the binding cleft was found to be crucial for the reaction. The connection between flap motion and the binding of a fullerene-based inhibitor was investigated with all-atom MD including the free energy profile obtained by thermodynamic integration (17). Other studies showed that the flaps close upon incorporation of the ligand in the binding cleft and provided a detailed all-atomic description of the closure (18,19). To the best of our knowledge, there are no studies that simulate the dynamics of product release after cleavage. An all-atomic quantum-mechanical molecular dynamics simulation of the peptide bond breakage was performed but, due to the picosecond timescale, the dissociation of the peptide bond only up to 3.6 Å was observed (20) and the cleaved peptide did not leave the binding pocket.
In this work, we aim to study the entire mechanism of the reaction dynamics, i.e., the diffusion of the substrate toward the enzyme, its proper entry, and accommodation in the binding site, which involves both flap opening and reclosing events, and the mobility of the substrate after the peptide bond cleavage. Because these events occur on a very long timescale, we performed molecular dynamics simulations with a reduced model, which was developed in our recent studies (21–23). Thanks to the extreme coarse-graining (i.e., one interacting center per amino acid), our model allows simulations on the timescale of tens of microseconds. Nevertheless, the model is sophisticated enough to reproduce high mobility of the flap region, i.e., the full flap opening dynamics. We previously reported (22) a large number of simulations in different statistical ensembles and an extensive analysis of the flap opening dynamics, thermodynamics, and kinetics of the unbound HIV-1 PR. Results are in agreement with the available experimental data and can be considered a validation of the model. Recently, we have also reported a combined coarse-grained (CG) and MD simulation where the approach of XK-263 inhibitor was studied with CG MD and the docking was refined with an all-atom MD simulation (24). Conversely, in this work the whole process is studied with CG MD. We parameterized a specific HIV-1 PR-substrate force field (FF) to obtain accurate structures of the complex. The results allow us to analyze not only the docking and accommodation of the substrate in the active site, but also the structural and dynamical changes induced by the substrate on HIV-1 PR upon binding. We subsequently simulate the release of the products after cleavage by ad hoc modification of the FF of the substrate to mimic the bond breakage. We study various binding paths through which the peptide approaches the enzyme.
METHODS
In our model, an amino acid is represented by a single bead placed on the Cα. HIV-1 PR in a coarse-grained representation is shown in Fig. 1. Such description is analogous to the elastic or Gaussian network models or Gō-type models (25–28). However, the latter approaches are not flexible enough to explore appropriately the nonnative configurations, which are far from equilibrium, due to an extremely simplified functional form choice and parameterization. Conversely, our force field resembles the all-atom molecular mechanics force fields and is based on a class of one-bead FF models (29–31). The effective potential energy U is the sum of three bonded terms (pseudo-bond, pseudo-bond angle, pseudo-dihedral) and three nonbonded terms:
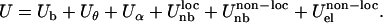 |
(1) |
The parameterization is based on the statistical analysis of a set of crystallographic structures (21). Particular care is devoted to the amino acid-dependent term Uθ, represented as a quartic double-well potential, which is responsible for the conformational changes involved in the flap opening (21). The correct hydrogen-bond topology is ensured by retaining a local bias in the term 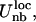 based on the native structure of the protease (PDB code 1HHP). This bias is applied, based on the spatial distance, to residues that are within 8 Å apart. The nonlocal nonbonded term,
based on the native structure of the protease (PDB code 1HHP). This bias is applied, based on the spatial distance, to residues that are within 8 Å apart. The nonlocal nonbonded term, 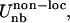 is a general one and is not dependent on initial structure. The local interactions contain electrostatics implicitly but for the nonlocal nonbonded interactions, we introduce the Coulombic term, which makes the interaction between charged amino acids stronger. The details of the force field and parameterization can be found in Tozzini et al. (22). For the electrostatic interactions,
is a general one and is not dependent on initial structure. The local interactions contain electrostatics implicitly but for the nonlocal nonbonded interactions, we introduce the Coulombic term, which makes the interaction between charged amino acids stronger. The details of the force field and parameterization can be found in Tozzini et al. (22). For the electrostatic interactions, 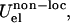 we used integer charges for Asp, Glu, Lys, and Arg according to their protonation in solution at pH 7 and a distance-dependent dielectric ε = 4r to account for the screening. We previously showed that this setup is an excellent compromise between simplicity-reduced computational cost and accuracy. The structures of intermediate steps of flap opening compare very well with the available x-ray structure (4). Additionally, the model allows for complete flap opening whose kinetics and thermodynamics are in agreement with experimental data (22).
we used integer charges for Asp, Glu, Lys, and Arg according to their protonation in solution at pH 7 and a distance-dependent dielectric ε = 4r to account for the screening. We previously showed that this setup is an excellent compromise between simplicity-reduced computational cost and accuracy. The structures of intermediate steps of flap opening compare very well with the available x-ray structure (4). Additionally, the model allows for complete flap opening whose kinetics and thermodynamics are in agreement with experimental data (22).
As a substrate, we used the nine-residue peptide Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met. The FF for the substrate and the substrate-HIV-1 PR interaction was extended following the same procedure as described in Tozzini et al. (22). 1F7A (32) PDB entry was chosen as the reference structure for the substrate-protease interaction part of the FF. To check the accuracy of the complex structure, a simulation at room temperature starting from the 1HHP (semi-open) structure with manually docked substrate was performed. Its analysis shows that the substrate correctly accommodates in the active site and the flaps close more tightly as an effect of the interaction with the substrate, acquiring a configuration that is very similar to the experimental substrate-protease complex (see Fig. 2). Consequently, in the present work we do not need to use an all-atom simulation for the last phases of binding and for the refinement of the complex structure as we did in our previous work (24), where a more generic CG FF was used for the ligand-protease interaction.
For the intermolecular substrate-HIV-1 PR interactions, we used a phenomenological scaling factor R, with respect to the intramolecular interactions, that accounts for the possible range of the solvation effects. Different values ranging from 0.2 to 1 were considered. R ≃ 1 produces a faster binding kinetics, while R ≃ 0.2 reproduces the range of the binding enthalpy of the substrate which we estimated to be ∼5–6 kcal/mole from the values of equilibrium association constants of the order of 104 M−1 (33) and catalytic efficiency (kcat/Km) in the millimolar range for short peptides (34).
To limit the conformational phase space sampled in the simulation, the FF for the substrate was harmonically constrained to the extended conformation that it assumes in the active site. We used a single well potential for Uθ instead of the usual double well one, because the influence of the substrate conformational dynamics on the binding is beyond the scope of the present work.
The FF for the cleaved substrate is simply obtained by deleting the one Cα-Cα pseudo-bond, the two pseudo-bond angle, and three pseudo-dihedral terms involved in the cleaved peptide bond between Leu and Ala. Additionally, the local contacts between the Leu, Ala, and the substrate were eliminated, since the geometry-specific backbone hydrogen bonds that these residues make with the enzyme are no longer possible when the peptide bond is broken. However, these residues can still interact with HIV-1 PR with hydrophobic and electrostatic nonbonded interactions. This FF for the HIV-1 PR-substrate complex is a natural extension of the HIV-1 PR one, which we previously parameterized, and is fully consistent with it, thus the results can be directly compared with those in Tozzini et al. (22).
We performed multiple MD simulations in the NVT ensemble using the Berendsen thermostat (35), as well as Langevin Dynamics (36) (LD), which was implemented into DL_POLY package (37). Time step was set to 0.02 ps. Based on the results of our previous work (22), where we addressed the dependence of flap opening dynamics on the viscosity of the solvent either in Langevin or Brownian dynamics, the collision frequency parameter in LD was set to 2 ps−1. A value of 1 ps−1 leads to faster binding of the substrate and a value of 5 ps−1 results in higher damping and slows the binding event. One has to emphasize that the binding times are not necessarily realistic and what we aim to address in this work are the binding pathways not the timescales. For analysis of the internal fluctuations, we performed an independent fit of each trajectory frame on the same reference to eliminate the contribution from the translation and rotation of the protease. To analyze the principal directions of motions we applied Principal Components Analysis (38) of the GROMACS package (39) and visualized it with Essential Dynamics Software (40) under VMD (41). Table 1 reports some of the simulation parameters, others are as in Tozzini et al. (22). Results corresponding to these simulations are reported in the indicated figures in the next sections.
TABLE 1.
Details of simulations, which were used to prepare figures in Results
| No. | Thermostat | R | Simulation length | Figures |
|---|---|---|---|---|
| Native HIV-1 PR | ||||
| 1 | NVT | 1 | 10 μs | Figs. 7 and 8 (left) |
| HIV-1 PR:substrate complex | ||||
| 2 | NVT | 1 | 600 ns | Figs. 2, 7, 8 (right) and 9 |
| HIV-1 PR:substrate docking | ||||
| 3 | LD | 1 | 400 ns (side)* | Figs. 3, 4, and 5 B |
| 4 | LD | 1 | 400 ns (flaps)† | Fig. 5 A |
| 5 | LD | 1 | 1μs (termini)‡ | Fig. 5 C |
| 6 | LD | 0.2 | 1.2 μs | Fig. 6 A |
| 7 | LD | 0.2 | 400 ns | Fig. 6 B |
| HIV-1 PR:cleaved substrate | ||||
| 8 | LD | 0.2 | 300 ns | Figs. 3 and 4 (continuation of simulation No. 3) |
In some figures, for the sake of comparison, only parts of the trajectories were used. The temperature was set to 300 K in every simulation.
Starting position of the substrate as 80–100 Å from the side of the protease.
Starting position from the top above the flaps.
Starting position from the side of the termini.
RESULTS AND DISCUSSION
In this section, we report the analyses of the encounter of the substrate with the protease, the fluctuations of the complex, and the products' release after cleavage.
Substrate binding and product release pathways
We performed several simulations of the substrate encounter with the protease and its accommodation in the binding cleft. In the starting configuration the position of the substrate was chosen ∼80–100 Å away from the protease from its various sides.
A sample LD simulation (Table 1, No. 3) of the substrate encounter and accommodation in the active site is presented in Fig. 3. The corresponding center-of-mass distance between the protease and the substrate, and the flap tip distance are shown in Fig. 4. After initial diffusion (up to 20 ns, Fig. 3 A) toward the enzyme, the substrate waits in proximity to the binding site (Fig. 3 B). Later, between 60 and 100 ns (Fig. 3 C), the flaps open many times but the substrate is not correctly positioned to enter the binding site. Next, the substrate changes its position and moves to the other side of the binding cleft (Fig. 3 D). Flaps open again for a longer period of time and the substrate enters the binding cleft at ∼215 ns (Fig. 3 E), which results in closure of one flap. This is visible in smaller fluctuations of the flap tip distance after 215 ns (Fig. 4). When the substrate properly accommodates itself in the cleft the other flap closes (Fig. 3 F) at ∼250 ns. Subsequently, the fluctuations of the flap tips decrease in comparison with those in the beginning of the simulation when the substrate was outside the protease. Both the opening of the flaps and correct positioning of substrate must occur for the substrate incorporation in the cleft. The substrate may wander around the protease from one side of the active site to the other, exploring its surface and waiting for the flaps to open. In addition, the proximity of the substrate to the flaps influences their mobility, stabilizing slightly the open conformation with respect to the unbound protease case.
FIGURE 3.

Snapshots from a substrate docking, cleavage, and release simulation. Protease monomers are denoted as Cα trace in light and dark shading, and the substrate is represented as spheres. (A) Initial diffusion toward the protease (≈20 ns). (B) Substrate outside the protease waiting for the flaps to open. (C) Open flaps (≈80 ns) but the substrate is not correctly positioned to enter. (D) Substrate moves to the other side of the binding cleft (≈120 ns) and flaps open again (≈140 ns). (E) The substrate enters the binding site (≈215 ns) and one flap closes over the substrate. (F) Proper accommodation of substrate with both flaps closed (≈250 ns). (G) The breakage of the substrate after induced at 400 ns cleavage. (H) Release of the products from the binding cleft. (I) Diffusion of products away from the protease.
FIGURE 4.

Substrate docking and product release simulation with labels corresponding to the frames in Fig. 3. (Upper plot) Distance between the centers of mass of the protease and the substrate or two substrate parts after cleavage (products). Two distance scales are provided divided by black vertical line. (Lower plot) The corresponding flap tip (Ile50-Ile150) distance.
The asymmetric closure of flaps may arise from the fact that it was found in mutation experiments that substrate hydrogen bonding at the Gly49-Ile50 peptide bond in only one flap is sufficient for normal reaction to take place (42).
At 400 ns, we performed the Leu5-Ala6 bond breakage and continued the simulation for another 300 ns. The products left the active site on the nanosecond timescale and did not require flap opening for their release from the binding cleft (Fig. 3, G and H). Release was asymmetric, i.e., it took longer for one side of the substrate to leave the binding cleft than for the other. Similar to the substrate in some simulations, the products remain in the proximity of the HIV-1 PR surface (Fig. 3 H) at ∼20 Å away from its center of mass before definitely diffusing away at ∼550 ns (Fig. 3 I; see also Fig. 4). Immediately after the cleavage and release, the normal dynamics of HIV-1 PR is recovered, with average opening and closing intervals that are typical of the unbound protease (22). In addition, the fluctuations of the flap tip distances are higher when the substrate leaves its binding site (compare the inter-tip distance in the range 200–400 ns after the substrate binds and after 400 ns when the products had left the enzyme in Fig. 4). The graph also shows that the flaps can close more tightly when the substrate is bound (compare the shift in the minimum value of the inter-tip distance before and after the substrate release).
In the simulations after the cleavage, the new termini of the products were kept either charged or neutral because their protonation states during the release process are not known. We found that the product termini titration states do not affect their release from the enzyme. Their protonation state affects only the simulation time, which is required for their diffusion from the surface of the enzyme. If the termini are charged, the products more easily find a position with respect to the enzyme with favorable electrostatic interactions. Because we have two more charged residues in comparison with the neutral termini, it might take longer for these electrostatic interactions to allow the substrate to diffuse.
In the simulations of substrate association, the initial position of the substrate and type of the dynamics was subject to change. However, it did not affect the general binding pathway picture and incorporation scheme of the substrate. Fig. 5 shows the density of the substrate from simulations with different starting substrate positions with respect to HIV-1 PR, confirming that the substrate explores the surface of the enzyme.
FIGURE 5.

Density showing the areas explored by the substrate while encountering the protease from various starting positions between 80 and 100 Å from the surface of the protease. (A) From the flaps side (Table 1, No. 4). (B) From the side of the protease (Table 1, No. 3). (C) from the termini of the protease (Table 1, No. 5). HIV-1 PR is shown in its starting configuration with the flaps closed over the active site.
Even though the initial trajectory of the substrate depends on its starting configuration, exploration of the surface of the protease and waiting for the flaps to open is common for all the simulations. In Fig. 6 we show the behavior of the substrate and the flaps derived from other sample LD simulations (see Table 1, Nos. 6 and 7). Fig. 6 (top) shows that in the first 500 ns of the simulation, the flaps open a few times but the substrate is not in a good position to enter the binding site. It binds at ∼800 ns at a subsequent flap opening event, stabilizing the fluctuations of the flaps. A similar simulation but with a different initial position of the substrate leads to faster binding after 340 ns (Fig. 6, bottom). Multiple short opening events can be seen before a longer one, which leads to docking. Flap opening time is longer than in a simulation of the unbound protease due to the interaction of the enzyme with the substrate. The fluctuations of the flap tips are smaller after the binding.
FIGURE 6.

Two sample substrate docking simulations. Temperature (T), effective potential energy (E), distance of the center of mass of the protease and the substrate (dPr–subs), and flap tip distances (dinter–tip) are reported.
The gain in energy upon binding is between 20 and 60 kcal/mol, depending on the value of R. However, it is quite difficult to compare this value with experimental binding free energies (34), because the entropic part is not included in our calculation. This would be difficult to estimate because the phase space of the unbound substrate is not properly sampled, and it is likely to be large, considering possible conformations of the unbound substrate. However, rough estimate of the binding enthalpy can be made considering the number of hydrogen bond contacts between the substrate and the protease. Their number is either 16 or 24 if one also includes the water-mediated hydrogen bonds (32). Therefore, considering an average binding energy of 3–4 kcal/mole per each hydrogen bond, the binding energy we obtain from the simulation is in the correct range. Moreover, in our all-atom simulations of the docking of the XK-263 inhibitor, the difference in mean interaction energy inside and outside the protease is ∼30–40 kcal/mole (24).
Another conclusion that can be derived from this set of simulations is that the substrate needs to be on the proper side of the enzyme to enter the binding cleft. Also, the events of docking happen on a faster timescale in the NVT dynamics than in LD. LD has the effect of damping the dynamics, which was shown before in the flap opening frequencies and intervals (22).
INTERNAL FLUCTUATIONS OF THE COMPLEX
Residual fluctuations
To obtain information on the displacement of individual residues, we calculated the root mean-square fluctuations (RMSF) from the MD trajectories,
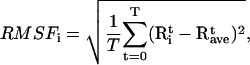 |
(2) |
where 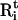 and
and 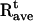 are the instantaneous and time-averaged coordinates of residue i (Cα bead in our case), respectively, and T is the number of trajectory frames. RMSF gives a measure of the fluctuations about the time-averaged structure. RMSF for the free and substrate bound protease are reported in Fig. 7 (Table 1, Nos. 1 and 2) and compared to the experimental temperature factors B of the 1HHP native structure according to the relation
are the instantaneous and time-averaged coordinates of residue i (Cα bead in our case), respectively, and T is the number of trajectory frames. RMSF gives a measure of the fluctuations about the time-averaged structure. RMSF for the free and substrate bound protease are reported in Fig. 7 (Table 1, Nos. 1 and 2) and compared to the experimental temperature factors B of the 1HHP native structure according to the relation 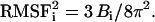 The parts that move the most in both structures are the flaps and the 17- and 39-turns or the so-called ear-cheek region (11). The fluctuations of the native protease compare well with the experiment except for the flap region because the flap opening is probably hindered in the crystal. A previous Gaussian network model study (43) showed smaller fluctuations of the flaps than we achieved, which is more in accord with that found by experiment in a crystal. However, our aim was to account for higher mobility of the flap region to simulate their full opening, allowing for substrate capture by the native enzyme. In the simulation with the docked substrate, the agreement is consistently very good, because in the complexed HIV-1 PR, the flaps do not open. A similar pattern of RMSF is obtained from the simulation of the free protease when the open conformations are excluded from the RMSF calculation (22). Stabilization of flaps upon formation of the HIV-1 PR:substrate complex was also determined in earlier all-atom MD simulation (10) and its principal components analysis (44).
The parts that move the most in both structures are the flaps and the 17- and 39-turns or the so-called ear-cheek region (11). The fluctuations of the native protease compare well with the experiment except for the flap region because the flap opening is probably hindered in the crystal. A previous Gaussian network model study (43) showed smaller fluctuations of the flaps than we achieved, which is more in accord with that found by experiment in a crystal. However, our aim was to account for higher mobility of the flap region to simulate their full opening, allowing for substrate capture by the native enzyme. In the simulation with the docked substrate, the agreement is consistently very good, because in the complexed HIV-1 PR, the flaps do not open. A similar pattern of RMSF is obtained from the simulation of the free protease when the open conformations are excluded from the RMSF calculation (22). Stabilization of flaps upon formation of the HIV-1 PR:substrate complex was also determined in earlier all-atom MD simulation (10) and its principal components analysis (44).
FIGURE 7.

Comparison of the experimental fluctuations of the native 1HHP protease with RMSF2 derived from simulations of the free HIV-1 PR and the complex with a peptide substrate. (Inset) Substrate density in the HIV-1 PR:substrate complex showing the asymmetric fluctuations of the termini.
The fluctuations of the substrate (green line) are asymmetric because the N-terminal lysine is more mobile than the C-terminal methionine. It has been shown by NMR experiments that an asymmetric ligand packs more tightly with one of the flaps (33). The density of the substrate within the active site is also shown as an inset in Fig. 7. The three-dimensional space occupied by the fluctuating substrate is indeed asymmetric at the extremities. The least fluctuating residue in the substrate is Glu at the P2′ position (with 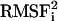 of 0.7 Å2). Other small fluctuations below 1 Å are for the residues of the cleaved bond (Leu and Ala). Even though the extremities of the peptide are mobile, the cleaved bond is stabilized within the active site cleft.
of 0.7 Å2). Other small fluctuations below 1 Å are for the residues of the cleaved bond (Leu and Ala). Even though the extremities of the peptide are mobile, the cleaved bond is stabilized within the active site cleft.
Dynamical correlations
The extent of the correlation between the residues may be quantified by calculating a normalized covariance Cij between their fluctuations
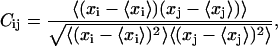 |
(3) |
where xi is a Cartesian coordinate of an atom and the brackets represent the time averages over all configurations obtained in the simulation. This so-called dynamical cross-correlation matrix provides useful information about the relation of motions of distant parts of the molecule. The value Cij varies from −1.0 to 1.0 for completely anticorrelated and correlated motions, respectively. To compare the fluctuations and their directionality between the free and bound protease, we calculated the correlation matrices for the unbound protease and the one complexed with a nine-amino-acid substrate. Fig. 8 reports the dynamical correlation matrices evaluated from MD trajectories (Table 1, Nos. 1 and 2) of the native protease (left) and complexed with a substrate (right). The secondary structure patterns are well conserved in both cases, as well as the interface contacts, which remain positively correlated. In the native protease matrix, spots of negative correlations are clearly visible in the intermonomer quadrant in the region of flap contacts (i.e., around residues 50 and 150). These reflect the counterphase movements of the flaps that move away from each other and from other regions of the opposite monomer as they open. In the simulation with the docked substrate the flaps do not open and, conversely, the substrate stabilizes the contact between the flap tips. Consequently, the negative correlation regions are generally decreased and in particular the interflap tip region is turned into a positive correlation region. The positive correlation between the substrate and the flap tips confirms that the substrate stabilizes the flaps. Furthermore, the substrate motion is positively correlated with the motion of the active site (residues 24–28), which reflects the fact that the substrate must stay in contact with the active site for the enzymatic reaction to take place. These features are in agreement with those observed in the all-atom MD simulations (15,16) and with an extended Gaussian network model (45). As we previously reported, the flexibility of the region enclosed by the 17- and 39-turns is essential for the flaps to open because a simulation where this site is artificially kept rigid does not show flap opening (21). This can explain the residual flexibility of these sites (small residual blue regions in the correlation matrix of the complex).
FIGURE 8.

Dynamical correlation matrices evaluated for the unbound protease (left) and for the complexed protease (right). Residues of the two monomers and the substrate are numbered subsequently and separated by black lines. Color levels: blue [−1,−0.2]; cyan [−0.2,0]; green [0,0.2]; yellow [0.2,0.7]; and red [0.7,1].
Principal components analysis
We performed principal components analysis based on the MD simulations of the complex (Table 1, No. 2) and projected the trajectory on the principal directions. The modes with the highest amplitudes include the side movement and contraction of the 17- and 39-turns. During the side movement we observe closing of residues 45–48 over the substrate, which is pushed toward the catalytic aspartates 25 and 125. In one of the modes we see the contraction of the 17- and 39-turns whose movements were found to be correlated with the flap opening (11,21,22,46) but the flaps do not open because the substrate is present. In the 10 modes with the highest eigenvalues, the substrate termini are mobile but the central part less so. The flap tip region is immobilized by the interaction with the substrate. Fig. 9 shows two of the principal modes from the simulation with a bound substrate. One of the modes also shows bending of the monomers around the C2V symmetry axis.
FIGURE 9.

Two principal directions of motions (denoted in black) derived from the simulations of the dynamics of the protease-substrate complex showing the side movement (A) and contraction of the turns (B). The 17- and 39-turns are shown in orange.
CONCLUSIONS
In this article, we extended our previously developed coarse-grained model for HIV-1 PR to include the protease-substrate interactions and applied it to the simulation of the substrate capture and product release processes. The extremely reduced cost of the model has allowed us to perform a large number of simulations of the substrate encounter and its final incorporation in the binding site and product release after cleavage. This enabled us to detect the mechanism of protease-substrate association. The substrate may approach the protease from various sides and wanders around the enzyme until it reaches the neighborhood of the binding site. Then a complex interplay between the flaps and the substrate occurs; the interaction with the substrate modifies the flap dynamics, making the opening more frequent and more stable. The capture occurs when the flaps are open and the substrate is properly oriented with respect to the active site. After the substrate entry, the flaps do not close symmetrically, but as one following the other. When the substrate is in the active site, the closed conformation is stabilized, as expected, at least until the substrate is cleaved. The structure of the complex accurately reproduces the crystallographic data and the interaction of the substrate with the flaps stabilizes the latter. Introduction of the appropriate bond cleavage results in the release of the products from the binding site without opening of the flaps. The products slide out on both sides and after some time diffuse away from the protease.
The presence of the substrate suppresses flap fluctuations but does not decrease the fluctuations of the 17- and 39-turns, which are believed to facilitate the flap opening. The motions of the substrate are positively correlated with the active site residues, as well as with the flap tips.
In conclusion, within a framework of the coarse-grained model, we have analyzed the main features of the substrate encounter and its dynamics in the active site of HIV-1 PR. Our results suggest that the presence of the substrate modifies the protease internal mobility in ways that favor the capture.
SUPPLEMENTARY MATERIAL
A sample movie of the substrate encounter and accommodation in the binding site of HIV-1 PR is available online as Supplemental Material, and can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank David Minh, Donald Hamelberg, and Krzysztof Nowinski for useful discussions. V.T. acknowledges the allocation of computer resources from INFM “Progetto Calcolo Parallelo 2005”.
J.T. was supported by the Warsaw University funds (grant No. 115/30/E-343/S/BST-1140/ICM/2006), Polish Ministry of Science and Higher Education (grant No. 3-T11F-005-30, 2006–2008), Fogarty International Center (National Institutes of Health research grant No. R03-TW07318) and Foundation for Polish Science. C.C. and J.A.M. acknowledge the National Institutes of Health, the National Science Foundation, Howard Hughes Medical Institute, the National Biomedical Computational Resource, the National Science Foundation Center for Theoretical Biological Physics, the W. M. Keck Foundation, and Accelrys, Inc.
References
- 1.de Clercq, E. 2002. New anti-HIV agents and targets. Med. Res. Rev. 22:531–565. [DOI] [PubMed] [Google Scholar]
- 2.Wlodawer, A. 2002. Rational approach to AIDS drug design through structural biology. Annu. Rev. Med. 53:595–614. [DOI] [PubMed] [Google Scholar]
- 3.Kurup, A., S. B. Mekapati, R. Garg, and C. Hansch. 2003. HIV-1 protease inhibitors: a comparative QSAR analysis. Curr. Med. Chem. 10:1679–1688. [DOI] [PubMed] [Google Scholar]
- 4.Martin, P., J. F. Vickrey, G. Proteasa, Y. L. Jimenez, Z. Wawrzak, M. A. Winters, T. C. Merigan, and L. C. Kovari. 2005. “Wide open” 1.3 Å structure of a multi-drug resistant HIV-1 protease represents a novel drug target. Structure. 13:1887–1895. [DOI] [PubMed] [Google Scholar]
- 5.Layten, M., V. Hornak, and C. Simmerling. 2006. The open structure of a multi-drug-resistant HIV-1 protease is stabilized by crystal packing contacts. J. Am. Chem. Soc. 128:13360–13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freedberg, D. I., R. Ishima, J. Jacob, Y. X. Wang, I. Kustanovich, J. M. Louis, and D. A. Torchia. 2002. Rapid structural fluctuations of the free HIV protease flaps in solution: relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Sci. 11:221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicholson, L. K., T. Yamazaki, D. A. Torchia, S. Grzesiek, A. Bax, S. J. Stahl, J. D. Kaufman, P. T. Wingfield, P. Y. S. Lam, P. K. Jadhav, C. N. Hodge, P. J. Domaille, and C. H. Chang. 1995. Flexibility and function in HIV-1 protease. Nat. Struct. Biol. 2:274–280. [DOI] [PubMed] [Google Scholar]
- 8.Ishima, R., D. I. Freedberg, Y. X. Wang, J. M. Louis, and D. A. Torchia. 1999. Flap opening and dimer-interface flexibility in the free and inhibitor-bound HIV protease, and their implications for function. Structure. 7:1047–1055. [DOI] [PubMed] [Google Scholar]
- 9.Collins, J. R., S. K. Burt, and J. W. Erickson. 1995. Flap opening in HIV-1 protease simulated by activated molecular dynamics. Nat. Struct. Biol. 2:334–338. [DOI] [PubMed] [Google Scholar]
- 10.Scott, W. R. P., and C. A. Schiffer. 2000. Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Structure. 8:1259–1265. [DOI] [PubMed] [Google Scholar]
- 11.Perryman, A. L., J.-H. Lin, and J. A. McCammon. 2004. HIV-1 protease molecular dynamics of a wild-type and of the V82F/I84V mutant: possible contributions to drug resistance and a potential new target site for drugs. Protein Sci. 13:1108–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamelberg, D., and J. A. McCammon. 2005. Fast peptidyl cis-trans isomerization within the flexible Gly-rich flaps of HIV-1 protease. J. Am. Chem. Soc. 127:13778–13779. [DOI] [PubMed] [Google Scholar]
- 13.Hornak, V., A. Okur, R. C. Rizzo, and C. Simmerling. 2006. HIV-1 protease flaps spontaneously open and re-close in molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 103:915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toth, G., and A. Borics. 2006. Flap opening mechanism of HIV-1 protease. J. Mol. Graph. Model. 24:465–474. [DOI] [PubMed] [Google Scholar]
- 15.Piana, S., P. Carloni, and U. Rothlisberger. 2002. Drug resistance in HIV-1 protease: flexibility-assisted mechanism of compensatory mutations. Protein Sci. 11:2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piana, S., P. Carloni, and M. Parrinello. 2002. Role of conformational fluctuations in the enzymatic reaction of HIV-1 protease. J. Mol. Biol. 319:567–583. [DOI] [PubMed] [Google Scholar]
- 17.Zhu, Z., D. I. Schuster, and M. E. Tuckerman. 2003. Molecular dynamics study of the connection between flap closing and binding of Fullerene-based inhibitors of HIV-1 protease. Biochemistry. 42:1326–1333. [DOI] [PubMed] [Google Scholar]
- 18.Hornak, V., A. Okur, R. C. Rizzo, and C. Simmerling. 2006. HIV-1 protease flaps spontaneously close to the correct structure in simulations following manual placement of an inhibitor into the open state. J. Am. Chem. Soc. 128:2812–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toth, G., and A. Borics. 2006. Closing of the flaps of HIV-1 protease induced by substrate binding: a model of a flap closing mechanism in retroviral aspartic proteases. Biochemistry. 45:6606–6614. [DOI] [PubMed] [Google Scholar]
- 20.Trylska, J., P. Grochowski, and J. A. McCammon. 2004. The role of hydrogen bonding in the enzymatic reaction catalyzed by HIV-1 protease. Protein Sci. 13:513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tozzini, V., and J. A. McCammon. 2005. A coarse grained model for the dynamics of flap opening in HIV-1 protease. Chem. Phys. Lett. 413:123–128. [Google Scholar]
- 22.Tozzini, V., J. Trylska, C. E. Chang, and J. A. McCammon. 2007. Flap opening dynamics in HIV-1 protease explored with a coarse-grained model. J. Struct. Biol. 157:606–615. [DOI] [PubMed] [Google Scholar]
- 23.Chang, C., T. Shen, J. Trylska, V. Tozzini, and J. A. McCammon. 2006. Gated binding of ligands to HIV-1 protease: Brownian dynamics simulations in a coarse-grained model. Biophys. J. 90:3880–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang, C., J. Trylska, V. Tozzini, and J. A. McCammon. 2007. Binding pathways of ligands to HIV-1 protease: coarse-grained and atomistic simulations. Chem. Biol. Drug Design. 69:5–13. [DOI] [PubMed] [Google Scholar]
- 25.Go, N., and H. Abe. 1981. Noninteracting local-structure model of folding and unfolding transition in globular proteins. I. Formulation. Biopolymers. 20:991–1011. [DOI] [PubMed] [Google Scholar]
- 26.Haliloglu, T., I. Bahar, and B. Erman. 1997. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 79:3090–3093. [Google Scholar]
- 27.Tirion, M. 1996. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 77:1905–1908. [DOI] [PubMed] [Google Scholar]
- 28.Hoang, T. X., and M. Cieplak. 2000. Sequencing of folding events in Gō-type proteins. J. Chem. Phys. 113:8319–8328. [Google Scholar]
- 29.Levitt, M., and A. Warshel. 1975. Computer simulation of protein folding. Nature. 253:694–698. [DOI] [PubMed] [Google Scholar]
- 30.McCammon, J. A., S. H. Northrup, M. Karplus, and R. M. Levy. 1980. Helix-coil transitions in a simple polypeptide model. Biopolymers. 19:2033–2045. [Google Scholar]
- 31.Wade, R. C., M. E. Davis, B. A. Luty, J. D. Madura, and J. A. McCammon. 1993. Gating of the active-site of triose phosphate isomerase—Brownian dynamics simulations of flexible loops in the enzyme. Biophys. J. 64:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prabu-Jeyabalan, M., E. Nalivaika, and C. A. Schiffer. 2000. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J. Mol. Biol. 301:1207–1220. [DOI] [PubMed] [Google Scholar]
- 33.Katoh, E., J. M. Louis, T. Yamazaki, A. M. Gronenborn, D. A. Torchia, and R. Ishima. 2003. A solution NMR study of the binding kinetics and the internal dynamics of an HIV-1 protease-substrate complex. Protein Sci. 12:1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boross, P., P. Bagossi, T. D. Copeland, S. Oroszlan, J. M. Louis, and J. Tozser. 1999. Effect of substrate residues on the p2′ preference of retroviral proteinases. Eur. J. Biochem. 264:921–929. [DOI] [PubMed] [Google Scholar]
- 35.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. Dinola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 36.Loncharich, R. J., B. R. Brooks, and R. W. Pastor. 1992. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N-methylamide. Biopolymers. 32:523–535. [DOI] [PubMed] [Google Scholar]
- 37.Smith, W., and T. R. Forester. 1996. DL_POLY_2.0: a general-purpose parallel molecular dynamics simulation package. J. Mol. Graph. 14:136–141. [DOI] [PubMed] [Google Scholar]
- 38.Amadei, A., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Proteins Struct. Funct. Genet. 17:412–425. [DOI] [PubMed] [Google Scholar]
- 39.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory. J. Mol. Model. (Online). 7:306–317. [Google Scholar]
- 40.Mongan, J. 2004. Interactive essential dynamics. J. Comput. Aided Mol. Design. 18:433–436. [DOI] [PubMed] [Google Scholar]
- 41.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 42.Baca, M., and B. H. Kent. 2000. Protein backbone engineering through total chemical synthesis: new insight into the mechanism of HIV-1 protease catalysis. Tetrahedron. 56:9503–9513. [Google Scholar]
- 43.Bahar, I., A. R. Atilgan, M. C. Demirel, and B. Erman. 1998. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 80:2733–2736. [Google Scholar]
- 44.Kurt, N., W. R. P. Scott, C. A. Schiffer, and T. Haliloglu. 2003. Cooperative fluctuations of unliganded and substrate-bound HIV-1 protease: a structure-based analysis on a variety of conformations from crystallography and molecular dynamics simulations. Proteins Struct. Funct. Genet. 51:409–422. [DOI] [PubMed] [Google Scholar]
- 45.Micheletti, C., P. Carloni, and A. Maritan. 2004. Accurate and efficient description of protein vibrational dynamics: comparing molecular dynamics and Gaussian models. Proteins Struct. Funct. Bioinf. 55:635–645. [DOI] [PubMed] [Google Scholar]
- 46.Perryman, A. L., J.-H. Lin, and J. A. McCammon. 2006. Restrained molecular dynamics simulations of HIV-1 protease: the first step in validating a new target for drug design. Biopolymers. 82:272–284. [DOI] [PubMed] [Google Scholar]