

Abstract
We extend our coarse-grained modeling strategy described in parts I and II of this investigation to account for nonuniform spatial distributions of hydrophobic residues on the solvent-exposed surfaces of native proteins. Within this framework, we explore how patchy surfaces can influence the solvent-mediated protein-protein interactions, and the unfolding and self-assembly behaviors of proteins in solution. In particular, we compare the equilibrium unfolding and self-assembly trends for three model proteins that share the same overall sequence hydrophobicity, but exhibit folded configurations with different solvent-exposed native-state surface morphologies. Our model provides new insights into how directional interactions can affect native-state protein stability in solution. We find that strongly-directional attractions between native molecules with patchy surfaces can help stabilize the folded conformation through the formation of self-assembled clusters. In contrast, native proteins with more uniform surfaces are destabilized by protein-protein attractions involving the denatured state. Finally, we discuss how the simulation results provide insights into the experimental solution behaviors of several proteins that display directional interactions in their native states.
INTRODUCTION
Maintaining the active, native protein conformation is an important part of the biotherapeutic drug development process because unfolded, denatured proteins often irreversibly aggregate (1) resulting in decreased drug efficacy and reduced shelf life (2–4). Environmental factors including temperature, protein concentration, pressure, and pH, as well as intrinsic protein characteristics such as sequence length, hydrophobicity, and the presence of disulfide bonds, determine whether a protein favors its biologically active, native-state or its inactive, denatured form (3,5). Most of what is known about how these factors affect protein stability comes from insightful analysis of experimental data (6–9). However, computer simulations and theory are playing an increasingly valuable complementary role (10–15) because they provide a logical framework that allows one to independently vary, and thus isolate for detailed study, the thermodynamic and molecular parameters thought to be most important for native-state stability. In particular, the development of new theoretical tools for modeling protein folding/unfolding equilibria in the complex mixtures relevant to biological and pharmaceutical systems remains a key challenge.
In Cheung and Truskett (16) and Shen et al. (17) (referred to here as parts I and II, respectively), we introduced a new coarse-grained modeling strategy for probing how various environmental conditions and molecular properties of proteins affect the equilibrium populations of their native and denatured states. This approach utilizes a heteropolymer collapse (HPC) theory to determine both the intrinsic (i.e., infinite dilution) folding thermodynamics and the coarse structural characteristics of globular proteins. This information is then used to estimate the effective protein-protein interactions in solution (16). Finally, the intrinsic free energy of folding and the effective protein interactions are incorporated into highly efficient transition-matrix Monte Carlo simulations (17) to study the equilibrium properties of protein solutions. Our preliminary investigations with this method focused on computing native-state stability (16,18) and fluid phase behavior (17) of model proteins of varying sequence hydrophobicity.
While the aforementioned modeling strategy is able to qualitatively capture some of the nontrivial experimental trends for the thermodynamics of globular protein solutions (16,17), to maintain reasonable simulation times it still invokes a highly simplified picture of protein structure. Perhaps most notably, the hydrophobic and polar residues are assumed to be uniformly distributed on the solvent-exposed surface of the proteins. This simplification prevents the approach from providing structural insights into a wide variety of assembly processes that are driven by directional protein-protein interactions, including the formation of ordered nonnative aggregates like amyloid fibrils (1,19–22) or native-state complexes/oligomers such as those observed in solutions of β-lactoglobulin (23), ribonuclease A (24–26), and the sickle variant of hemoglobin (27), to mention a few.
In this work, we extend the modeling approach detailed in parts I and II to account for nonuniform surface compositions of native-state proteins. Specifically, we investigate how changes to the surface distribution of nonpolar residues affect the native-state protein stability as a function of temperature and protein concentration. Importantly, our approach still maintains the practical requirement of computational efficiency allowing the simulation of hundreds of proteins, which is far greater than the number that can be studied using atomistic protein models.
By comparing model proteins with identical sequence length and fraction of hydrophobic residues but different surface residue morphologies, we find that the latter strongly affects both protein unfolding and the protein self-assembly process. Proteins with weakly-directional attractions behave like the nondirectional proteins studied in parts I and II, where attractions involving denatured proteins tend to destabilize native proteins. On the other hand, strongly-directional attractions between patchy native proteins can help to stabilize the folded proteins. The predicted stability behavior observed here qualitatively agrees with observations from experimental protein systems.
Our article is organized as follows. We first introduce a simple way to account for the possibility of surface patches on the native state that have either higher or lower hydrophobic residue composition than the rest of the solvent-exposed surface. The formation of these patches affects the two main inputs into our transition-matrix Monte Carlo simulations: the intrinsic free energy of folding and the protein-protein interactions. We discuss how HPC theory and our implementation of transition-matrix Monte Carlo simulations can be extended to account for these modified inputs. We then examine the simulated behavior of solutions of three model proteins that share the same sequence hydrophobicity, but fold into native states with different surface-residue segregation characteristics. Finally, we discuss how the results of our simulations may relate to the experimental folding behaviors of several proteins that exhibit directional native-native interactions.
Modifying HPC theory
In this section, we briefly review the basic physics, as well as the inputs and outputs, of Dill and co-workers' HPC theory (28,29), which was previously employed within the coarse-grained modeling strategy described in parts I and II. We then introduce a simple way to modify the theory to account for the formation of native protein states that display a nonuniform spatial distribution of solvent-exposed hydrophobic residues.
HPC theory begins from the reductionist perspective that proteins of Nr residues can be modeled as heteropolymers of Ns coarse-grained segments (Ns = Nr /1.4 (29)) with interactions that statistically reflect the aqueous-phase solubilities of the corresponding amino acids of the protein sequence (29). Similar to small globular proteins (30), heteropolymers can show equilibrium folding behavior that results from a competition between two driving forces: the tendency to adopt a compact native state to reduce the nonpolar surface area in contact with aqueous solution versus the drive to partially expand to a denatured form to realize more conformational degrees of freedom.
The inputs to HPC theory include temperature T (and, more generally, pressure (31), pH and ionic strength (32,33), the number of residues in the protein sequence Nr, the fraction of those residues that are hydrophobic Φ (e.g., based on an aqueous-phase solubility criterion (29,34)), and χ(T)—the free energy per unit kBT associated with hydrating a hydrophobic polymer segment. In parts I and II of our investigation, we invoked a simple parameterization for χ(T) that captures experimental trends for the partitioning of hydrophobic amino acids between an oily condensed phase and liquid water at ambient pressure (29).
The main thermodynamic output of HPC theory is the intrinsic free energy change 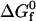 associated with the unimolecular folding process. It quantifies the difference in free energy between the native and denatured states in the absence of protein-protein interactions (i.e., in the limit of vanishing protein concentration). The main structural outputs of the theory include the ratio of the radii of gyration of the denatured and native states, RD/RN, and the fraction of solvent-exposed residues in the native state that are hydrophobic, Θ. It is assumed that the solvent-exposed residues in the denatured state have the same hydrophobic composition as the protein sequence. In the original formulation of this HPC theory, it is further assumed that there are no spatial correlations between solvent-exposed hydrophobic residues in either the denatured or the native state. Below, we discuss one way to relax this assumption.
associated with the unimolecular folding process. It quantifies the difference in free energy between the native and denatured states in the absence of protein-protein interactions (i.e., in the limit of vanishing protein concentration). The main structural outputs of the theory include the ratio of the radii of gyration of the denatured and native states, RD/RN, and the fraction of solvent-exposed residues in the native state that are hydrophobic, Θ. It is assumed that the solvent-exposed residues in the denatured state have the same hydrophobic composition as the protein sequence. In the original formulation of this HPC theory, it is further assumed that there are no spatial correlations between solvent-exposed hydrophobic residues in either the denatured or the native state. Below, we discuss one way to relax this assumption.
To facilitate the evaluation of 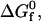 an imaginary two-step path for folding that connects the denatured state to the native state is constructed (29). In Step 1, the denatured heteropolymer collapses into a randomly-condensed configuration with the same radius of gyration as the native state, but with its hydrophobic residues uniformly distributed throughout the structure. In Step 2, the native state is formed from the randomly condensed state via internal residue rearrangement. The intrinsic free energy of folding is obtained by summing the contributions from these two steps,
an imaginary two-step path for folding that connects the denatured state to the native state is constructed (29). In Step 1, the denatured heteropolymer collapses into a randomly-condensed configuration with the same radius of gyration as the native state, but with its hydrophobic residues uniformly distributed throughout the structure. In Step 2, the native state is formed from the randomly condensed state via internal residue rearrangement. The intrinsic free energy of folding is obtained by summing the contributions from these two steps, 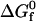 =
= 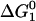 +
+ 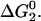 A complete description for Step 1 and the derivation for
A complete description for Step 1 and the derivation for 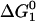 are presented in detail in Dill et al. (29).
are presented in detail in Dill et al. (29).
In Step 2, which describes the rearrangement of the randomly condensed state to form the native configuration, our approach departs from that of the original HPC theory (28,29). The original theory assumes that the solvent-exposed hydrophobic residues exhibit a uniform spatial distribution of composition Θ on the surface of the native state. Here, we assume that there are two patches on the surface of the native state that have a different composition of hydrophobic residues than the rest of the surface body. As is shown in Fig. 1, the size of the patch is defined by the polar angle α. The fractional patch hydrophobicity Θp and body hydrophobicity Θb are expressed as
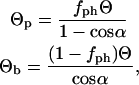 |
(1) |
where fph is the fraction of surface hydrophobic residues that are located on the patches. Increasing fph increases Θp, which results in higher surface anisotropy.
FIGURE 1.

Schematic of two native-state proteins with coarse-grained surface regions of different average hydrophobic residue composition: patch (shaded) versus body (open). The size of the patch is defined by the angle α. The hydrophobicities of the patch Θp and body Θb are determined by Eq. 1. Since the dashed line connecting the protein centers passes through a patch region on each molecule, these two proteins are currently in a patch-patch alignment.
In one sense, this approach is similar to a two-patch description that was recently introduced to model native-native protein interactions of sickle cell hemoglobin molecules (35) and also to other simple models for anisotropic native-state proteins that have been investigated recently (36–42). However, the coarse-grained strategy that we pursue here differs significantly from these earlier models in that it explicitly accounts for the possibility of protein unfolding, and it estimates the intrinsic properties of the native and denatured states using a statistical mechanical theory for heteropolymer collapse. This connection to the polymeric aspect of the protein allows our model to probe the global connections between native-state anisotropy, folding equilibria, and the thermodynamic behavior in protein solutions.
The aforementioned segregation of native-state surface hydrophobic residues into patches is favored by an increase in the number of hydrophobic segment contacts (estimated by the Bragg-Williams approximation (43)), but is opposed by the associated loss of entropy. Following similar logic to the development presented in Dill et al. (29), we arrive at the following free-energy change for Step 2:
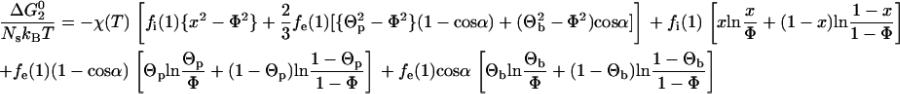 |
(2) |
Here, fi(1) = (1 − (4π /{3Ns})1/3)3 is the fraction of total residues buried in the interior of the native or the randomly condensed state (both have reduced segment density ρ = 1), fe(1) = 1 − fi(1) is the fraction of total residues that are solvent-exposed, and x is the hydrophobic composition of the native protein core. The numerical value that the average surface hydrophobicity Θ takes on is the one that minimizes 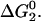 This value also minimizes the free energy of the native state since the properties of the randomly condensed state do not depend on Θ. The numerical value of x can be obtained by simultaneously applying a hydrophobic residue balance on the native state, Φ = fi(1)x + fe(1)Θp[1 − cos α] + fe(1)Θb cos α.
This value also minimizes the free energy of the native state since the properties of the randomly condensed state do not depend on Θ. The numerical value of x can be obtained by simultaneously applying a hydrophobic residue balance on the native state, Φ = fi(1)x + fe(1)Θp[1 − cos α] + fe(1)Θb cos α.
In this study, we focus in particular on aqueous solutions of three model proteins of identical molecular weight Ns = 110 (i.e., Nr = 154) and hydrophobic residue composition Φ = 0.4, parameters typical of medium-sized globular proteins (44). The only difference between the three model proteins is that their folded states have distinct surface residue distributions, which subsequently lead to different protein-protein interactions: nondirectional (i.e., no patches), weakly-directional (fph= 0.25, α = π/6), and strongly-directional (fph= 0.75, α = π/6). By examining the behavior of these three models, we can take a first step toward exploring the broader issue of how differences in surface characteristics of the native state can impact both the molecular stability and the global solution behaviors of proteins.
In Table 1, we present the surface hydrophobicity properties at T = 300 K (shown in parentheses), calculated by solving the modified HPC theory for the protein variants described in the previous paragraph. As a reference, we also show the hydrophobicity of the denatured protein, which we treat with an average surface hydrophobicity of Φ. As expected, by segregating the hydrophobic surface residues on the native protein, the Θp (Θb) for the weakly and strongly directional proteins are higher (lower) than for the nondirectional protein. This means that pairs of the strongly directional protein molecules can desolvate a higher number of hydrophobic residues by self-associating, but only if they do so with their hydrophobic patches mutually aligned. Given the patch geometry of the native state studied here, the possibility exists for equilibrium cluster formation of the strongly directional native proteins, a topic that we explore in greater detail in Results and Discussion.
TABLE 1.
The temperature and orientationally dependent attractive strengths relative to kBT for the nondirectional, weakly-directional, and strongly-directional proteins at T = 300 K
| Protein interaction | εpp/kBT (Θp) | εbb/kBT (Θb) | εpb/kBT |
|---|---|---|---|
| Nondirectional | 0.358 (0.153) | 0.358 (0.153) | 0.358 |
| Weakly directional | 1.25 (0.285) | 0.268 (0.132) | 0.578 |
| Strongly directional | 11.3 (0.859) | 0.003 (0.044) | 0.583 |
| Denatured | 1.74 (0.400) | 1.74 (0.400) | 1.74 |
Values inside the parentheses represent the surface hydrophobicity. The magnitude of the denatured-denatured protein attractions are given as reference with Φ shown in parentheses.
Coarse-grained protein-protein interactions
In our coarse-grained approach, we make the assumption that protein-protein attractions are primarily driven by the favorable differences in free energy associated with desolvating surface hydrophobic residues of the proteins that are otherwise solvated when they are in their isolated, infinite dilution state. While clearly an oversimplification, this assumption is supported in part by statistical analysis of protein-protein interfaces that reveals a higher fraction of hydrophobic residues in the vicinity of their binding sites (45). It is also supported by the strong role that hydrophobic interactions have been found to play in various protein aggregation processes (4).
The magnitudes of the solvent-mediated interprotein contact attractions in this model (εNN, εDD, and εND for native-native, denatured-denatured, and native-denatured protein pairs, respectively) depend on the strength of the intersegment hydrophobic attraction χ(T), but also on the segment densities and hydrophobic compositions of the solvent-exposed residues in the participating protein states. Mean-field expressions for these quantities (16) derived from the outputs of HPC theory are given by
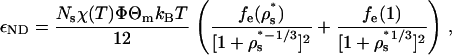 |
(3) |
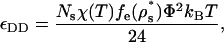 |
(4) |
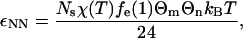 |
(5) |
where ρs* is the reduced segment density and fe(ρs*) = 1 − fi(ρs*) is the fraction of residues in the denatured state that are solvent-exposed, and fi(ρs*) = (1 − (4πρs*/{3Ns})1/3)3 is the fraction of residues that are on the interior of the protein. In the above, Θm and Θn denote the apparent surface hydrophobicities associated with different orientational states of participating native molecules m and n, respectively. For example, to determine the value of Θm for molecule m of a given pair interaction, one only needs to know the orientation of molecule m relative to that of the imaginary vector connecting its center of mass to that of the other participating protein. If this vector passes through a patch on molecule m's surface (see Fig. 1), then Θm = Θp; otherwise Θm = Θb, and so on. A discussion of similar relations for isotropic interprotein interactions can be found in part I of this study (16).
Table 1 lists the temperature-dependent native-native contact attractions for the patch-patch (pp), patch-body (pb), and body-body (bb) alignments for the three protein variants at T = 300 K. For comparison, we also present the magnitude of the denatured-denatured contact attraction. The main point is that the strongest effective interactions are the patch-patch hydrophobic interactions (≈11 kBT) of the strongly directional native proteins. The second strongest interactions are those between denatured proteins (≈1.7 kBT). For the weakly directional and nondirectional protein, the denatured-denatured attraction is energetically more favorable than the native-native interactions. As one might expect, the relative strengths of these various attractions can be qualitatively inferred from the solvent-exposed hydrophobic residue concentrations of the various proteins in their isolated, infinite dilution states (shown in parentheses in Table 1).
One can also use HPC theory to roughly estimate state-dependent, effective diameters of the various interactions (σNN, σDD, and σND). Here, as in parts I and II of this investigation, we take σDD/σNN ≈ RD/RN = 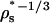 and σND/σNN ≈ (1 + RD/RN)/2 = (1 +
and σND/σNN ≈ (1 + RD/RN)/2 = (1 + 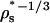 )/2. We then integrate these effective diameters and the contact energies of Eqs. 3–5 into a coarse-grained interprotein pair potential (46) that is known to qualitatively capture many aspects of protein solution thermodynamics and phase behavior (see, e.g., (46,47)):
)/2. We then integrate these effective diameters and the contact energies of Eqs. 3–5 into a coarse-grained interprotein pair potential (46) that is known to qualitatively capture many aspects of protein solution thermodynamics and phase behavior (see, e.g., (46,47)):
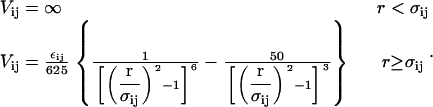 |
(6) |
In the above relation, r is the center-to-center distance separating interacting proteins of states i and j, and ij ∈ (NN, ND, DD).
To summarize, the coarse-grained model discussed here represents an effective binary mixture of orientation-dependent native and spherically symmetric denatured proteins (the aqueous solvent enters the picture through χ(T)) connected via the protein-protein interactions and the protein folding reaction. The links between the intrinsic native-state stability of the proteins 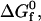 the physical parameters defining the protein-protein interactions (εij, σij), the protein sequence (Nr,Φ), and the interactions with the aqueous solvent χ(T) are established by the modified heteropolymer collapse model described in the last two sections. We approach this coarse-grained biomolecular system with the understanding that it parallels that of a classic reactive phase equilibria problem for a binary solution (see, e.g., (48)), which is readily amenable to the advanced Monte Carlo simulation techniques referred to in the next section.
the physical parameters defining the protein-protein interactions (εij, σij), the protein sequence (Nr,Φ), and the interactions with the aqueous solvent χ(T) are established by the modified heteropolymer collapse model described in the last two sections. We approach this coarse-grained biomolecular system with the understanding that it parallels that of a classic reactive phase equilibria problem for a binary solution (see, e.g., (48)), which is readily amenable to the advanced Monte Carlo simulation techniques referred to in the next section.
SIMULATION METHODS
To implement our coarse-grained strategy, we use highly efficient grand-canonical transition-matrix Monte Carlo (TMMC) simulations (49) adapted for the case of multicomponent mixtures (50,51) and simultaneous reaction (folding) equilibria (17). The details of the simulations are identical to those provided in part II of this investigation (17) (with one exception discussed below), and so we do not repeat them here.
The modification to the simulations in the present work is the use of aggregation-volume-bias (AVB) Monte Carlo moves. Such moves are needed because the native-state molecules of the strongly directional model protein can self-associate, forming long-lived chains of bonded configurations that prevent adequate sampling of phase space using standard Monte Carlo moves. AVB Monte Carlo moves circumvent this bottleneck by promoting the formation and destruction of bonded configurations by targeting trial displacements, insertions, and deletions to be attempted in the immediate vicinity of a randomly chosen molecule of interest. We performed AVB displacements using the so-called AVBMC2 implementation (52), and AVB insertions/deletions were implemented as described in Chen et al. (53). Sampling was further enhanced by combining this suite of moves with multiple first-bead trial insertions and configurational bias Monte Carlo (53–55). Grand-canonical TMMC simulations can be easily adapted to handle these specialized moves.
To examine the protein stability and self-assembly behavior, we tracked the temperature and protein-concentration dependencies of three different microscopic quantities for our model protein solutions: the total average folded fraction fN, the cluster fraction fclust, and the cluster average folded fraction fNc. The quantity fN is simply the average fraction of proteins in solution that are in their native state. The midpoint unfolding transition for the protein solution occurs when fN = 0.5. The quantity fclust is a measure of the fraction of proteins in a geometric cluster. A protein in state i is considered to be in the same cluster as a neighboring protein in state j if their center of masses are closer than 1.3 σij, where the magnitude of the effective pairwise attraction between the two proteins is >20% of its maximum value. The condition fclust = 0.5 can be viewed as a midpoint for a continuous self-assembly transition. Maxima in heat capacity have also been associated with self-assembly transitions (56,57), and indeed we have observed a close correspondence between these geometric and thermodynamic metrics in simulations of our system. Finally, the quantity fNc is the average fraction of native-state proteins within a geometric cluster. This metric can be used to help understand the origin of the clustering phenomenon in solution (e.g., packing versus protein-protein association effects).
RESULTS AND DISCUSSION
We now discuss the results of our grand-canonical TMMC simulations and modified coarse-grained modeling strategy, implemented here to probe how native-state surface anisotropy affects the unfolding behavior and equilibrium assembly behavior of proteins in solution. Specifically, we compare the simulated equilibrium unfolding curves (fN) and the self-association trends (fclust and fNc) for the nondirectional (i.e., no patches), the anisotropic weakly-directional (fph = 0.25, α = π/6), and the anisotropic strongly-directional (fph= 0.75, α = π/6) proteins described earlier.
We first discuss the physics, outlined in parts I and II, involved in the stability of the nondirectional protein as a function of protein concentration (Fig. 2 a, inset). The main trend for the folded fraction fN can be understood as a balance between two opposing factors: destabilizing interprotein attractions involving denatured molecules and stabilizing macromolecular crowding effects. At finite protein concentrations, a marginally stable protein can unfold in solution if:
It has enough local free volume to accommodate the transition from a compact state to the more expanded denatured configuration; and
It can simultaneously form enough interprotein hydrophobic contacts upon denaturing to overcome the intrinsic free energy penalty for unfolding.
FIGURE 2.

Protein concentration dependencies of the fraction of folded proteins fN (solid), the fraction of clustered proteins that are native fNc (dashed), and the fraction of proteins that are clustered fclust (dotted) as a function of protein concentration for the weakly-directional model protein (a), the nondirectional model protein (a, inset), and the strongly-directional model protein (b) at their respective infinite-dilution midpoint folding temperatures.
Assumption 2 is aided by the fact that attractions involving the denatured state are stronger than those involving only native molecules for the nondirectional protein (recall Table 1). Of course, increasing protein concentration decreases the probability of Assumption 1 but increases the likelihood of Assumption 2. The specific properties of the native and denatured states of the individual proteins will also generally affect both Assumption 1 and 2. We previously found that the attraction-induced destabilizing and crowding-induced stabilizing forces approximately balance each other at low to intermediate protein concentrations for nondirectional proteins of lower sequence hydrophobicity (e.g., Φ = 0.4), As discussed extensively in part I, these trends appear to be qualitatively reflected in the different experimental solution behaviors of real proteins with low Φ (such as ribonuclease A).
The self-assembly behavior of the nondirectional protein is shown in the inset of Fig. 2 a. The fraction of clustered proteins fclust increases with increasing protein concentration. Of course, this type of geometric clustering can occur for trivial packing reasons at high protein concentrations, where pairs of proteins are forced to adopt near-contact configurations that satisfy the above geometric criteria for fclust. Alternatively, protein clustering or self-assembly can also occur due to strong interprotein attractions, even at low concentrations. However, for the nondirectional protein, the interprotein attractions are relatively weak (see Table 1). Thus, increases in the cluster fraction are due to geometric packing effects.
Note that for the nondirectional protein, the native composition of the clusters fNc essentially tracks the fraction folded fN at higher protein concentrations, where most of the proteins are clustered. In contrast, at low protein concentrations where packing effects do not play a large role, the nondirectional proteins are less stable within the clusters (i.e., fNc < fN) because of the physical reasons listed previously:
There is enough free volume to accommodate the larger denatured state; and
Interactions involving denatured states are energetically more favorable than native-native interactions for this protein.
Given this delicate balance between attractions and crowding, and the fact that differences in protein surface properties significantly impact protein-protein interactions, one naturally expects that protein surface morphology may have a nontrivial effect on the concentration-dependent stability behavior of proteins in solution. In particular, as is shown in Table 1, anisotropic native-state proteins can have very strong interprotein attractions, even though their average surface hydrophobicity may be low. These favorable native-native interactions may significantly stabilize the patchy native-state against unfolding, a scenario that, as discussed above, does not occur for nondirectional proteins (16,17). If clustering is a result of highly favorable (i.e., native stabilizing) interprotein attractions, then one would expect to find fNc > fN, even at relatively low protein concentrations.
We now examine the trends for fN, fclust, and fNc for the weakly directional protein (Fig. 2 a). The qualitative trends for these stability and self-assembly metrics are similar to the nondirectional protein, and it appears that weak segregation of surface hydrophobic residues does not have a noticeable impact on either native-state stability or self-assembly behavior. Destabilizing attractions are approximately balanced by stabilizing crowding effects until very high protein concentrations where crowding physics dominate. Similar to the nondirectional protein, self-assembly is due mainly to packing constraints, since fclust = 0.5 occurs at relatively high protein concentrations.
Contrast this behavior to that of the strongly directional protein presented in Fig. 2 b. Interestingly, the relevant protein concentration range for the strongly directional protein is an order-of-magnitude less than that for the other protein variants. At these low concentrations, packing effects, which play a large role in the geometric clustering of the weakly-directional and nondirectional proteins, are negligible. Increases in fclust are therefore a result of different physics: specifically, the highly energetically favorable patch-patch alignment (see Table 1). The fact that the folded fraction within the clusters fNc rises above the average folded fraction fN indicates that the self-assembly behavior or clustering stabilizes the patchy native-state protein relative to the denatured state.
In Fig. 3, we plot the stability diagrams for the weakly directional and strongly directional proteins. The shaded regions indicate temperature and concentrations that favor the denatured state (fN < 0.5), while the white region indicates conditions that favor the native-state (fN > 0.5). The locus of the temperature-dependent concentrations, where fclust = 0.5 (squares), is also displayed. Proteins form geometric clusters for all states to the right of this curve. Two points are worth emphasizing here. First, as discussed above, the clusters that the weakly-directional proteins form in the native state are simply due to high concentration (i.e., packing effects) and are not due to directional native-native interactions. This observation is further supported by the very close agreement between the fclust = 0.5 loci of the weakly-directional protein solution (squares) and the nondirectional protein solution (triangles). The second point is that clustering occurs at lower protein concentrations for the denatured proteins, because of their larger radius of gyration and stronger interprotein attractions relative to the native protein attractions (see Table 1). For the strongly-directional proteins, the native-state global clustering trends are very different. Small increases in native protein concentration result in the self-assembly of clusters due to the fact that the patch-patch attractive interactions are much stronger than even the denatured-denatured interactions for this protein.
FIGURE 3.

Stability diagram for the weakly-directional model protein (a) and strongly-directional model protein (b) in the temperature versus protein concentration plane. The native state is thermodynamically favored (fN > 0.5) in the white region, while the denatured state is favored (fN < 0.5) in the shaded region. Also shown are the loci of conditions where fclust = 0.5 (squares). To the right of the fclust = 0.5 curve, more than half of the proteins are part of geometric clusters. In panel a, the nondirectional protein fclust = 0.5 locus (triangles) is shown as reference. For conditions where the denatured state is favored, note that the location of the fclust = 0.5 curve for both panels a and b are the same. This result is expected because the attractive strength and relative size of the denatured proteins are the same for all protein variants studied here.
The predicted behavior that strongly directional native-native attractions help stabilize the model protein against unfolding agrees with observations of some experimental protein systems. First consider the extreme example of the p53tet peptide fragment, which is not thermodynamically stable in its monomeric form due in part to its short chain length (64 residues), but instead is stable as a tetrameric protein. The surface buried at the monomer interfaces is mostly apolar, suggesting that the driving force for association and stabilization is hydrophobic (58). The stability results for this patchy protein indicate that the protein-protein associations are able to stabilize an inherently unstable protein (2,58).
In fact, this type of stability behavior is also observed for bovine β-lactoglobulin, which exists in a dimer form in solution but is also stable in its monomeric form. The monomer-monomer interface consists of a large hydrophobic patch (23). For β-lactoglobulin to denature under most conditions, the dimer must first dissociate (23,59). Here, the additional stabilization provided by the dimeric form has been found to be an important factor in preventing the nucleation and growth of β-lactoglobulin fibrils in solution (59).
Similarly, ribonuclease A is known to form dimers (as well as trimers and higher order oligomers) under a variety of experimental conditions (25,60,61). These oligomers can form two different conformers stabilized by specific interactions at the N- and C-termini of the molecules, which participate in the domain swapping mechanism (26). The formation of the N- and C-dimers may involve interactions of their exposed hydrophobic residues (61), similar to the patch-patch associations in the native aggregates formed in our simulations. The stability of these oligomers is temperature-dependent, and sufficiently high temperatures can result in an overall decrease in their presence (61), again in qualitative agreement with the cluster stability behavior observed for our strongly-directional model protein (Fig. 3).
Finally, the polymerization of the native form of sickle cell hemoglobin due to its strongly directional interactions in solution has been the focus of other recent computational studies (see, e.g., (35)), mostly due to its important biological implications. The directional interactions in the sickle variant are a consequence of a point mutation, and thus they are not present in the wild-type protein. Interestingly, self-association of native sickle hemoglobin can be extrapolated to occur for temperatures above the folding transition of the wild-type protein (see, e.g., (62)). This suggests that the strongly favorable native interactions of the sickle variant could play an active role in stabilizing the native (clustering) form over the denatured state.
CONCLUSIONS
In this article, we studied how anisotropic protein-protein attractions affect both the equilibrium fraction of native proteins and the protein self-assembly behavior in solution. We modified our original coarse-grained modeling strategy, described in Parts I and II, to account for nonuniform spatial distributions of the solvent-exposed hydrophobic residues on the native protein surface. Proteins with a high degree of hydrophobic residue segregation displayed anisotropic attractions that are very strong compared with those between nondirectional proteins with uniform surface hydrophobicity. These strongly-directional proteins were stabilized by native-native interprotein attractions, while proteins with weakly-directional behavior (i.e., a low degree of nonpolar residue segregation) were destabilized by interprotein attractions involving the denatured state.
We understand that we are still invoking a very simplified description of proteins in solution. Indeed, real proteins may contain asymmetric patches on their native states, which will lead to more complicated equilibrium unfolding and self-assembly behavior. However, the results of our coarse-grained approach still give meaningful physical insight into the observed experimental behavior for peptides and globular proteins that display directional behavior. Moreover, by taking this simplified approach, we can directly investigate the effects of individual interactions on the folded fraction of proteins in solution. Future directions for this work will be to include asymmetric surface patches, protein sequence effects, and the effects of other solution conditions (e.g., pH and denaturant concentration).
Acknowledgments
We thank Dr. Jack Douglas for useful suggestions pertaining to this work. This study utilized the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov) and the Texas Advanced Computing Center.
J.K.C. and T.M.T. gratefully acknowledge the financial support of the Merck Company Foundation. T.M.T. also acknowledges the financial support of the National Science Foundation grant No. CTS-0448721, the David and Lucile Packard Foundation, and the Alfred P. Sloan Foundation. J.R.E. acknowledges the support of the James D. Watson Investigator Program of the New York State Office of Science, Technology and Academic Research.
References
- 1.Fink, A. L. 1998. Protein aggregation: folding aggregates, inclusion bodies, and amyloids. Fold. Des. 3:R9–R23. [DOI] [PubMed] [Google Scholar]
- 2.Kendrick, B. S., T. Li, and B. S. Chang. 2002. Rational Design of Stable Protein Formulations, Chapt. 3. Kluwer Academic/Plenum, Dordrecht, The Netherlands.
- 3.Krishnamurthy, R., and M. C. Manning. 2002. The stability factor: importance in formulation development. Curr. Pharm. Biotechnol. 3:361–371. [DOI] [PubMed] [Google Scholar]
- 4.Shire, S. J., Z. Shahrokh, and J. Liu. 2004. Challenges in the development of high protein concentration formulations. J. Pharm. Sci. 93:1390–1402. [DOI] [PubMed] [Google Scholar]
- 5.Dill, K. A. 1990. Dominant forces in protein folding. Biochemistry. 29:7133–7155. [DOI] [PubMed] [Google Scholar]
- 6.Kauzmann, W. 1959. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 14:1–63. [DOI] [PubMed] [Google Scholar]
- 7.Privalov, P. L., and S. J. Gill. 1988. Stability of protein structure and hydrophobic interaction. Adv. Protein Chem. 39:191–234. [DOI] [PubMed] [Google Scholar]
- 8.Wang, W. 1999. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int. J. Pharm. 185:129–188. [DOI] [PubMed] [Google Scholar]
- 9.Chi, E. Y., S. Krishnan, T. Randolph, and J. Carpenter. 2003. Physical stability of proteins in aqueous solution: mechanism and driving forces in nonnative protein aggregation. Pharm. Res. 20:1325–1336. [DOI] [PubMed] [Google Scholar]
- 10.Dill, K. A., S. Bromberg, K. Z. Yue, K. M. Fiebig, D. Yee, P. D. Thomas, and H. Chan. 1995. Principles of protein folding—a perspective from simple exact models. Protein Sci. 4:561–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onuchic, J. N., Z. Luthey-Schulten, and P. G. Wolynes. 1997. Theory of protein folding: the energy landscape perspective. Annu. Rev. Phys. Chem. 48:545–600. [DOI] [PubMed] [Google Scholar]
- 12.Onuchic, J. N., H. Nymeyer, A. E. Garcia, J. Chahine, and N. D. Socci. 2000. The energy landscape theory of protein folding: insights into folding mechanisms and scenarios. Adv. Protein Chem. 53:87–130. [DOI] [PubMed] [Google Scholar]
- 13.Pande, V. S., A. Y. Grosberg, and T. Tanaka. 2000. Heteropolymer freezing and design: towards physical models of protein folding. Rev. Mod. Phys. 72:259–314. [Google Scholar]
- 14.Hall, D., and A. P. Minton. 2003. Macromolecular crowding: qualitative and semiquantitative successes, quantitative challenges. Biochim. Biophys. Acta. 1649:127–139. [DOI] [PubMed] [Google Scholar]
- 15.Daggett, V. 2006. Protein folding-simulation. Chem. Rev. 106:1898–1916. [DOI] [PubMed] [Google Scholar]
- 16.Cheung, J. K., and T. M. Truskett. 2005. Coarse-grained strategy for modeling protein stability in concentrated solutions. Biophys. J. 89:2372–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen, V. K., J. K. Cheung, J. R. Errington, and T. M. Truskett. 2006. Coarse-grained strategy for modeling concentrated protein solutions. II: Phase behavior. Biophys. J. 90:1949–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung, J. K., P. S. Raverkar, and T. M. Truskett. 2006. Analytical model for studying how environmental factors influence protein conformational stability in solution. J. Chem. Phys. 125:224903/1–224903/8. [DOI] [PubMed] [Google Scholar]
- 19.Harper, J., and P. T. Lansbury. 1997. Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66:385–407. [DOI] [PubMed] [Google Scholar]
- 20.Dobson, C. M. 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24:329–332. [DOI] [PubMed] [Google Scholar]
- 21.Lansbury, P. T. 1999. Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. USA. 96:3342–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly, J. W. 2002. Towards an understanding of amyloidogenesis. Nat. Struct. Biol. 9:323–325. [DOI] [PubMed] [Google Scholar]
- 23.Iametti, S., B. D. Gregori, G. Vecchio, and F. Bonomi. 1996. Modifications occur at different structural levels during the heat denaturation of β-lactoglobulin. Eur. J. Biochem. 237:106–112. [DOI] [PubMed] [Google Scholar]
- 24.Tsai, A. M., J. H. v. Zanten, and M. J. Betenbaugh. 1998. I. Study of protein aggregation due to heat denaturation: a structural approach using circular dichroism spectroscopy, nuclear magnetic resonance and static light scattering. Biotechnol. Bioeng. 59:273–280. [PubMed] [Google Scholar]
- 25.Libonati, M., and G. Gotte. 2004. Oligomerization of bovine ribonuclease A: structural and functional features of its multimers. Biochem. J. 380:311–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu, Y., and D. Eisenberg. 2002. 3D domain swapping: as domains continue to swap. Protein Sci. 11:1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrone, F. A. 2004. Polymerization and sickle cell disease: a molecular view. Microcirculation. 11:115–128. [DOI] [PubMed] [Google Scholar]
- 28.Dill, K. A. 1985. Theory for the folding and stability of globular proteins. Biochemistry. 24:1501–1509. [DOI] [PubMed] [Google Scholar]
- 29.Dill, K. A., D. O. V. Alonso, and K. Hutchinson. 1989. Thermal stability of globular proteins. Biochemistry. 28:5439–5449. [DOI] [PubMed] [Google Scholar]
- 30.Privalov, P. L. 1979. Stability of proteins: small globular proteins. Adv. Protein Chem. 33:167–241. [DOI] [PubMed] [Google Scholar]
- 31.Cheung, J. K., P. Shah, and T. M. Truskett. 2006. Heteropolymer collapse theory for protein folding in the pressure-temperature plane. Biophys. J. 91:2427–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stigter, D., D. O. V. Alonso, and K. A. Dill. 1991. Protein stability: electrostatic and compact denatured states. Proc. Natl. Acad. Sci. USA. 88:4176–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alonso, D. O. V., K. A. Dill, and D. Stigter. 1991. The three states of globular proteins: acid denaturation. Biopolymers. 31:1631–1649. [DOI] [PubMed] [Google Scholar]
- 34.Nozaki, Y., and C. Tanford. 1971. The solubility of amino acids and two glycine peptides in aqueous ethanol and dioxane solutions. Establishment of a hydrophobicity scale. J. Biol. Chem. 1971:2211–2217. [PubMed] [Google Scholar]
- 35.Shiryayev, A., X. Li, and J. D. Gunton. 2006. Simple model of sickle hemoglobin. J. Chem. Phys. 125:024902/1–024902/8. [DOI] [PubMed] [Google Scholar]
- 36.Sear, R. P. 1999. Phase behavior of a simple model of globular proteins. J. Chem. Phys. 111:4800–4806. [Google Scholar]
- 37.Lomakin, A., N. Asherie, and G. B. M. Benedek. 1999. Aeolotropic interactions of globular proteins. Proc. Natl. Acad. Sci. USA. 96:9465–9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curtis, R. A., H. W. Blanch, and J. M. Prausnitz. 2001. Calculation of phase diagrams for aqueous protein solutions. J. Phys. Chem. B. 105:2445–2452. [Google Scholar]
- 39.Hloucha, M., J. F. M. Lodge, A. M. Lenhoff, and S. I. Sandler. 2001. A patch-antipatch representation of specific protein interactions. J. Cryst. Growth. 232:195–203. [Google Scholar]
- 40.Song, X. 2002. Role of anisotropic interactions in protein crystallization. Phys. Rev. E. 66:011909/1–011909/4. [DOI] [PubMed] [Google Scholar]
- 41.Dixit, N. M., and C. F. Zukoski. 2002. Crystal nucleation rates for particles experiencing anisotropic interactions. J. Chem. Phys. 117:8540–8550. [Google Scholar]
- 42.Kern, N., and D. Frenkel. 2003. Fluid-fluid coexistence in colloidal systems with short-ranged strongly directional attraction. J. Chem. Phys. 118:9882–9889. [Google Scholar]
- 43.Hill, T. L. 1960. Introduction to Statistical Thermodynamics. Addison-Wesley, Reading, MA.
- 44.Shen, M., F. Davis, and A. Sali. 2005. The optimal size of a globular protein domain: a simple sphere-packing model. Chem. Phys. Lett. 405:224–228. [Google Scholar]
- 45.Tsai, C., S. L. Lin, H. J. Wolfson, and R. Nussinov. 1997. Studies of protein-protein interfaces: a statistical analysis of the hydrophobic effect. Protein Sci. 6:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.ten Wolde, P. R., and D. Frenkel. 1997. Enhancement of protein crystal nucleation by critical density fluctuations. Science. 277:1975–1978. [DOI] [PubMed] [Google Scholar]
- 47.Petsev, D. N., X. Wu, O. Galkin, and P. G. Vekilov. 2003. Thermodynamic functions of concentrated protein solutions from phase equilibria. J. Phys. Chem. B. 107:3921–3926. [Google Scholar]
- 48.Sandler, S. I. 1999. Chemical and Engineering Thermodynamics, 3rd Ed. John Wiley and Sons.
- 49.Errington, J. R. 2003. Direct calculations of liquid-vapor phase equilibria from transition matrix Monte Carlo simulations. J. Chem. Phys. 118:9915–9925. [Google Scholar]
- 50.Errington, J. R., and V. K. Shen. 2005. Direct evaluation of multi-component phase equilibria using flat histogram methods. J. Chem. Phys. 122:064508. [DOI] [PubMed] [Google Scholar]
- 51.Shen, V. K., and J. R. Errington. 2005. Determination of fluid-phase behavior using transition-matrix Monte Carlo: binary Lennard-Jones mixtures. J. Chem. Phys. 122:064508/1–064508/17. [DOI] [PubMed] [Google Scholar]
- 52.Chen, B., and J. I. Siepmann. 2001. Improving the efficiency of the aggregation-volume-bias Monte Carlo. J. Phys. Chem. B. 105:11275–11282. [Google Scholar]
- 53.Chen, B., J. I. Siepmann, K. J. Oh, and M. L. Klein. 2001. Aggregation-volume-bias Monte Carlo simulations of vapor-liquid nucleation barriers for Lennard-Jonesium. J. Chem. Phys. 115:10903–10913. [Google Scholar]
- 54.Vlugt, T. J. H., M. G. Martin, B. Smit, J. I. Siepmann, and R. Krishna. 1998. Improving the efficiency of the configurational-bias Monte Carlo algorithm. Mol. Phys. 94:727–733. [Google Scholar]
- 55.Frenkel, D., and B. Smit. 2002. Understanding Molecular Simulations: from Algorithms to Applications, 2nd Ed. Academic Press, London.
- 56.Starr, F. W., J. F. Douglas, and S. C. Glotzer. 2003. Origin of particle clustering in a simulated polymer nanocomposite and its impact on rheology. J. Chem. Phys. 119:1777–1788. [Google Scholar]
- 57.Van Workum, K., and J. F. Douglas. 2005. Equilibrium polymerization in the Stockmayer fluid as a model of supermolecular self-organization. Phys. Rev. E. 71:031502/1–031502/15. [DOI] [PubMed] [Google Scholar]
- 58.Johnson, C. R., and E. Freire. 1996. Structural stability of small oligomeric proteins. Tech. Protein Chem. VII:459–467.
- 59.Hamada, D., and C. M. Dobson. 2002. A kinetic study of β-lactoglobulin amyloid fibril formation promoted by urea. Protein Sci. 11:2417–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park, C., and R. T. Raines. 200. Dimer formation by a “monomeric” protein. Protein Sci. 9:2026–2033. [DOI] [PMC free article] [PubMed]
- 61.Gotte, G., F. Vottariello, and M. Libonati. 2003. Thermal aggregation of ribonuclease A. J. Biol. Chem. 278:10763–10769. [DOI] [PubMed] [Google Scholar]
- 62.Vaiana, S. M., M. A. Rotter, A. Emanuele, F. A. Ferrone, and M. B. Palma-Vittorelli. 2005. Effect of T-R conformational change on sickle-cell hemoglobin interactions and aggregation. Proteins. 58:426–438. [DOI] [PubMed] [Google Scholar]