

Abstract
The current standard correlation coefficient used in the analysis of
microarray data was introduced by M. B. Eisen, P. T. Spellman, P. O. Brown,
and D. Botstein [(1998) Proc. Natl. Acad. Sci. USA 95,
14863–14868]. Its formulation is rather arbitrary. We give a
mathematically rigorous correlation coefficient of two data vectors based on
James–Stein shrinkage estimators. We use the assumptions described by
Eisen et al., also using the fact that the data can be treated as
transformed into normal distributions. While Eisen et al. use zero as
an estimator for the expression vector mean μ, we start with the assumption
that for each gene, μ is itself a zero-mean normal random variable [with
a priori distribution 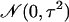 ], and
use Bayesian analysis to obtain a posteriori distribution of μ in
terms of the data. The shrunk estimator for μ differs from the mean of the
data vectors and ultimately leads to a statistically robust estimator for
correlation coefficients. To evaluate the effectiveness of shrinkage, we
conducted in silico experiments and also compared similarity metrics
on a biological example by using the data set from Eisen et al. For
the latter, we classified genes involved in the regulation of yeast cell-cycle
functions by computing clusters based on various definitions of correlation
coefficients and contrasting them against clusters based on the activators
known in the literature. The estimated false positives and false negatives
from this study indicate that using the shrinkage metric improves the accuracy
of the analysis.
], and
use Bayesian analysis to obtain a posteriori distribution of μ in
terms of the data. The shrunk estimator for μ differs from the mean of the
data vectors and ultimately leads to a statistically robust estimator for
correlation coefficients. To evaluate the effectiveness of shrinkage, we
conducted in silico experiments and also compared similarity metrics
on a biological example by using the data set from Eisen et al. For
the latter, we classified genes involved in the regulation of yeast cell-cycle
functions by computing clusters based on various definitions of correlation
coefficients and contrasting them against clusters based on the activators
known in the literature. The estimated false positives and false negatives
from this study indicate that using the shrinkage metric improves the accuracy
of the analysis.
Recent advances in technology, such as microarray-based gene expression analysis, have allowed us to “look inside the cells” by quantifying their transcriptional states. While the most interesting insight can be obtained from transcriptome abundance data within a single cell under different experimental conditions, in the absence of technology to provide one with such a detailed picture, we have to make do with mRNA collected from a small, frequently unsynchronized, population of cells. Furthermore, these mRNAs will give only a partial picture, supported only by those genes that we are already familiar with and possibly missing many crucial undiscovered genes.
Of course, without the proteomic data, transcriptomes tell less than half the story. Nonetheless, it goes without saying that microarrays have already revolutionized our understanding of biology even though they provide only occasional, noisy, unreliable, partial, and occluded snapshots of the transcriptional states of cells.
In an attempt to attain functional understanding of the cell, we try to understand the underlying structure of its transcriptional state-space. Partitioning genes into closely related groups has thus become the key mathematical first step in practically all statistical analyses of microarray data.
Traditionally, algorithms for cluster analysis of genomewide expression data from DNA microarray hybridization are based on statistical properties of gene expressions and result in organizing genes according to similarity in pattern of gene expression. If two genes belong to a cluster then one may infer a common regulatory mechanism for the two genes or interpret this information as an indication of the status of cellular processes. Furthermore, coexpression of genes of known function with novel genes may lead to a discovery process for characterizing unknown or poorly characterized genes. In general, since false negatives (FNs) (where two coexpressed genes are assigned to distinct clusters) may cause the discovery process to ignore useful information for certain novel genes, and false positives (FPs) (where two independent genes are assigned to the same cluster) may result in noise in the information provided to the subsequent algorithms used in analyzing regulatory patterns, it is important that the statistical algorithms for clustering be reasonably robust. Unfortunately, as the microarray experiments that can be carried out in an academic laboratory for a reasonable cost are small in number and suffer from experimental noise, often a statistician must resort to unconventional algorithms to deal with small-sample data.
A popular and one of the earliest clustering algorithms reported in the literature was introduced in ref. 1. In that paper, the gene-expression data are collected on spotted DNA microarrays (2) and based on gene expression in the budding yeast Saccharomyces cerevisiae during the diauxic shift (3), the mitotic cell division cycle (4), sporulation (5), and temperature and reducing shocks. Each entry in a gene expression vector represents a ratio of the amount of transcribed mRNA under a particular condition with respect to its value under normal conditions. All ratio values are log-transformed to treat inductions and repressions of identical magnitude as numerically equal but opposite in sign. It is assumed that the raw ratio values follow log-normal distributions, and hence, the log-transformed data follow normal distributions. Although our mathematical derivations will rely on this assumption for the sake of simplicity, we note that our approach can be generalized in a straightforward manner to deal with other situations where this assumption is violated.
The gene similarity metric used in ref. 1 was a form of correlation coefficient. Let Gi be the (log-transformed) primary data for gene G in condition i. For any two genes X and Y observed over a series of N conditions, the classical similarity score based on Pearson correlation coefficient is:
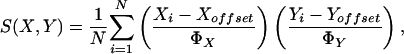 |
[1] |
where
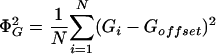 |
[2] |
and Goffset is the estimated mean of the observations, i.e.,
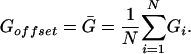 |
Note that ΦG is simply the (rescaled) estimated standard deviation of the observations. In the analysis presented in ref. 1, “values of Goffset which are not the average over observations on G were used when there was an assumed unchanged or reference state represented by the value of Goffset, against which changes were to be analyzed; in all of the examples presented there, Goffset was set to 0, corresponding to a fluorescence ratio of 1.0.” To distinguish this modified correlation coefficient from the classical Pearson correlation coefficient, we shall refer to it as Eisen correlation coefficient. Our main innovation is in suggesting a different value for Goffset, namely Goffset = γḠ, where γ is allowed to take a value between 0.0 and 1.0. Note that when γ = 1.0, we have the classical Pearson correlation coefficient and when γ = 0.0, we have replaced it by Eisen correlation coefficient. For a nonunit value of γ, the estimator for Goffset = γḠ can be thought of as the unbiased estimator Ḡ being shrunk toward the believed value for Goffset = 0.0. We address the following questions: What is the optimal value for the shrinkage parameter γ from a Bayesian point of view? (See ref. 6 for some alternate approaches.) How do the gene expression data cluster as the correlation coefficient is modified with this optimal shrinkage parameter?
To achieve a consistent comparison, we leave the rest of the algorithms undisturbed. Namely, once the similarity measure has been assumed, we cluster the genes by using the same hierarchical clustering algorithm as the one used by Eisen et al. (1). Their hierarchical clustering algorithm is based on the centroid-linkage method [referred to as the “average-linkage method” of Sokal and Michener (7) in ref. 1] and is discussed further below. The modified algorithm has been implemented by us within the New York University MicroArray Database system and can be freely downloaded from: bioinformatics.cat.nyu.edu/nyumad/clustering/. The clusters created in this manner were used to compare the effects of choosing differing similarity measures.
Model
We derive the proposed similarity metric. In our setup, the microarray data is given in the form of the levels of M genes expressed under N experimental conditions. The data can be viewed as
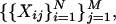 |
where M ≫ N and
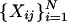 is the data vector for gene
j. We begin by rewriting S (see Eqs. 1 and 2)
in our notation:
is the data vector for gene
j. We begin by rewriting S (see Eqs. 1 and 2)
in our notation:
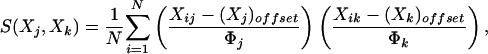 |
[3] |
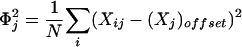 |
In the most general setting, we can make the following assumptions on the data distribution: let all values Xij for gene j have a normal distribution with mean θj and standard deviation βj (variance βj2); i.e.,
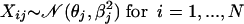 |
with j fixed (1 ≤ j ≤ M), where θj is an unknown parameter (taking different values for different j). To estimate θj, it is convenient to assume that θj is itself a random variable taking values close to zero:
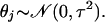 |
The assumed distribution aids us in obtaining the estimate of θj given in Eq. 6.
For convenience, let us also assume that the data are range-normalized, so that βj2 = β2 for every j. If this assumption does not hold on the given data set, it is easily corrected by scaling each gene vector appropriately. Following common practice, we adjusted the range to scale to an interval of unit length, i.e., its maximum and minimum values differ by 1. Thus,
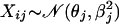 |
and
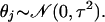 |
Replacing (Xj)offset in
Eq. 3 by the exact value of the mean θj
yields a clairvoyant correlation coefficient of
Xj and Xk. In
reality, since θj is itself a random variable, it
must be estimated from the data. Therefore, to get an explicit formula for
S(Xj, Xk)
we must derive estimators
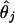 for all
j.
for all
j.
In Pearson correlation coefficient, θj is
estimated by the vector mean 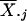 ; Eisen
correlation coefficient corresponds to replacing θj
by 0 for every j, which is equivalent to assuming
; Eisen
correlation coefficient corresponds to replacing θj
by 0 for every j, which is equivalent to assuming
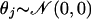 (i.e., τ2 = 0).
We propose to find an estimate of θj (call it
(i.e., τ2 = 0).
We propose to find an estimate of θj (call it
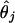 ) that takes into
account both the prior assumption and the data.
) that takes into
account both the prior assumption and the data.
First, let us obtain the posterior distribution of
θj from the prior
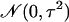 and the data. This derivation can be
done either from the Bayesian considerations or via the James-Stein shrinkage
estimators (see refs. 8 and
9). Here, we discuss the former
method.
and the data. This derivation can be
done either from the Bayesian considerations or via the James-Stein shrinkage
estimators (see refs. 8 and
9). Here, we discuss the former
method.
Assume initially that N = 1, i.e., we have one data point for each gene, and denote the variance by σ2 for the moment:
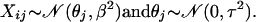 |
From these assumptions, we get (see ref. 10 for full details)
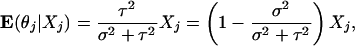 |
[4] |
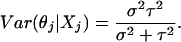 |
Now, if N > 1 is arbitrary, Xj
becomes a vector X.j. In ref.
10 we show (by using
likelihood functions) that the vector of values
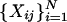 , with
, with
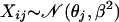 ,
can be treated as a single data point
,
can be treated as a single data point
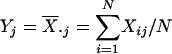 from the distribution
from the distribution
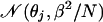 .
.
Thus, following the same derivation with σ2 = β2/N, we have a Bayesian estimator for θj given by E(θj|X.j):
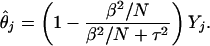 |
[5] |
Unfortunately, Eq. 5 cannot be used in Eq. 3 directly, because τ2 and β2 are unknown, so must be estimated from the data. The details of the estimation are presented in ref. 10.
The resulting explicit estimate for θj is
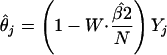 |
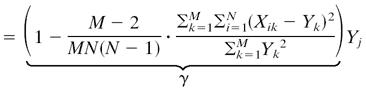 |
[6] |
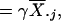 |
where
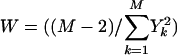 is an estimator for 1/(β2/N + τ2).
is an estimator for 1/(β2/N + τ2).
Finally, we substitute
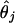 from Eq. 6
into the correlation coefficient in Eq. 3 wherever
(Xj)offset appears to obtain
an explicit formula for S(X.j,
X.k).
from Eq. 6
into the correlation coefficient in Eq. 3 wherever
(Xj)offset appears to obtain
an explicit formula for S(X.j,
X.k).
Algorithm and Implementation
The implementation of hierarchical clustering proceeds in a greedy manner, always choosing the most similar pair of elements (starting with genes at the bottom-most level) and combining them to create a new element. The “expression vector” for the new element is simply the weighted average of the expression vectors of the two elements that were combined. This structure of repeated pairwise combinations is conveniently represented in a binary tree, whose leaves are the set of genes and internal nodes are the elements constructed from the two children nodes. The algorithm is described below in pseudocode.
Hierarchical clustering pseudocode.
Given
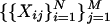
Switch:
Pearson: γ = 1;
Eisen: γ = 0;
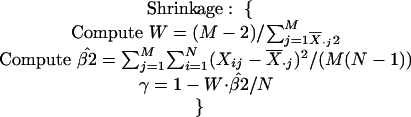 |
While (no. clusters > 1) do
- Compute similarity table:
- Find (j*, k*):
Create new cluster Nj*k* = weighted average of Gj* and Gk*.
Take out clusters j* and k*.
As each internal node can be labeled by a value representing the similarity between its two children nodes, one can create a set of clusters by simply breaking the tree into subtrees by eliminating all the internal nodes with labels below a certain predetermined threshold value.
The implementation of generalized hierarchical clustering with options to choose different similarity measures has been incorporated into the New York University MicroArray Database (NYUMAD), an integrated system to maintain and analyze biological abundance data along with associated experimental conditions and protocols. To enable widespread utility, NYUMAD supports the MAGE-ML standard (www.mged.org/Workgroups/MAGE/mage-ml.html) for the exchange of gene expression data, defined by the Microarray Gene Expression Data Group. More detailed information about NYUMAD can be found at http://bioinformatics.cat.nyu.edu/nyumad/.
Results
Mathematical Simulation. To compare the performance of these algorithms, we started with a relatively simple in silico experiment. In such an experiment, one can create two genes X and Y and simulate N (≈100) experiments as follows:
 |
where αi, chosen from a uniform distribution over a
range [L, H] ( ), is a “bias term”
introducing a correlation (or none if all αs are zero) between
X and Y.
), is a “bias term”
introducing a correlation (or none if all αs are zero) between
X and Y.
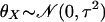 and
and
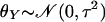 are the means
of X and Y, respectively. Similarly,
σX and σY are the standard
deviations for X and Y, respectively.
are the means
of X and Y, respectively. Similarly,
σX and σY are the standard
deviations for X and Y, respectively.
Note that, with this model
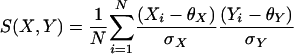 |
if the exact values of the mean and variance are used.
The model was implemented in mathematica
(11); the following parameters
were used in the simulation: N = 100, τ ∈ {0.1, 10.0}
(representing very low or high variability among the genes),
σX = σY = 10.0, and
α = 0 representing no correlation between the genes or
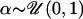 representing some correlation between the
genes. Once the parameters were fixed for a particular in silico
experiment, the gene-expression vectors for X and Y were
generated many thousand times, and for each pair of vectors
Sc(X, Y),
Sp(X, Y),
Se(X, Y), and
Ss(X, Y) were estimated by four
different algorithms and further examined to see how the estimators of
S varied over these trials. These four different algorithms estimated
S according to Eqs. 1 and 2 as follows: Clairvoyant
estimated Sc by using the true values of
θX, θY,
σX, and σY; Pearson
estimated Sp by using the unbiased estimators
X̄ and
Ȳ of θX and
θY (for Xoffset and
Yoffset), respectively; Eisen estimated
Se by using the value 0.0 as the estimator of
both θX and θY; and
Shrinkage estimated Ss by using the shrunk biased
estimators
representing some correlation between the
genes. Once the parameters were fixed for a particular in silico
experiment, the gene-expression vectors for X and Y were
generated many thousand times, and for each pair of vectors
Sc(X, Y),
Sp(X, Y),
Se(X, Y), and
Ss(X, Y) were estimated by four
different algorithms and further examined to see how the estimators of
S varied over these trials. These four different algorithms estimated
S according to Eqs. 1 and 2 as follows: Clairvoyant
estimated Sc by using the true values of
θX, θY,
σX, and σY; Pearson
estimated Sp by using the unbiased estimators
X̄ and
Ȳ of θX and
θY (for Xoffset and
Yoffset), respectively; Eisen estimated
Se by using the value 0.0 as the estimator of
both θX and θY; and
Shrinkage estimated Ss by using the shrunk biased
estimators 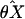 and
and
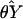 of
θX and θY, respectively.
In the latter three, the standard deviation was estimated as in Eq. 2.
The histograms corresponding to these in silico experiments can be
found in Fig. 1. Our
observations are summarized in Table
1.
of
θX and θY, respectively.
In the latter three, the standard deviation was estimated as in Eq. 2.
The histograms corresponding to these in silico experiments can be
found in Fig. 1. Our
observations are summarized in Table
1.
Fig. 1.

Histograms representing the performance of four different estimators of correlation between genes.
Table 1. Summary of observations from mathematical simulation of gene expression models of correlated and uncorrelated genes.
|
Parameters
|
|
Distributions
|
||||
|---|---|---|---|---|---|---|
| α | τ | Sc | Sp | Se | Ss | |
| 0 | 0.1 | μ | -0.000297 | -0.000269 | -0.000254 | -0.000254 |
| δ | 0.0996 | 0.0999 | 0.0994 | 0.0994 | ||
| 0 | 10 | μ | -0.000971 | -0.000939 | -0.00119 | -0.000939 |
| δ | 0.0994 | 0.100 | 0.354 | 0.100 | ||
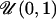 |
0.1 | μ | 0.331 | 0.0755 | 0.248 | 0.245 |
| δ | 0.132 | 0.0992 | 0.0915 | 0.0915 | ||
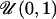 |
10 | μ | 0.333 | 0.0762 | 0.117 | 0.0762 |
| δ | 0.133 | 0.100 | 0.368 | 0.0999 | ||
The distributions of S as estimated by Sc
(Clairvoyant), Sp (Pearson), Se
(Eisen), and Ss (Shrinkage) are characterized by the means
μ and standard deviations δ. When there is no correlation (α =
0) and low noise (τ = 0.1), all methods do equally well. When there is no
correlation but the noise is high (τ = 10), all methods except Eisen do
equally well; Eisen has too many FPs. When the genes are correlated
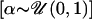 and the noise is low, all methods except
Pearson do equally well; Pearson has too many FNs. Finally, when the genes are
correlated and the noise is high, all methods do equally poorly, introducing
FNs; Eisen may also have FPs.
and the noise is low, all methods except
Pearson do equally well; Pearson has too many FNs. Finally, when the genes are
correlated and the noise is high, all methods do equally poorly, introducing
FNs; Eisen may also have FPs.
In summary, one can conclude that for the same clustering algorithm, Pearson tends to introduce more FNs and Eisen tends to introduce more FPs than Shrinkage. Shrinkage, on the other hand, reduces these errors by combining the good properties of both algorithms.
Biological Example. We then proceeded to test the algorithms on a biological example. We chose a biologically well-characterized system and analyzed the clusters of genes involved in yeast cell cycle. These clusters were computed by using the hierarchical clustering algorithm with the underlying similarity measure chosen from the following three: Pearson, Eisen, or Shrinkage. As a reference, the computed clusters were compared to the ones implied by the common cell-cycle functions and regulatory systems inferred from the roles of various transcriptional activators (see Fig. 2).
Fig. 2.

Regulation of cell-cycle functions by the activators. [Reproduced with permission from ref. 12 (Copyright 2001, Elsevier)].
Note that our experimental analysis is based on the assumption that the groupings suggested by the chromatin immunoprecipitation analysis are, in fact, correct and thus, provide a direct approach to compare various correlation coefficients. It is quite likely that the chromatin immunoprecipitation-based groupings themselves contain many false relations (both positive and negative) and corrupt our inference in some unknown manner. Nonetheless, we observe that the trends of reduced false positives and negatives in shrinkage analysis with these biological data are consistent with the analysis based on mathematical simulation and hence, reassuring.
In the work of Simon et al. (12), genomewide location analysis was used to determine how the yeast cell-cycle gene expression program is regulated by each of the nine known cell-cycle transcriptional activators: Ace2, Fkh1, Fkh2, Mbp1, Mcm1, Ndd1, Swi4, Swi5, and Swi6. It was also found that cell-cycle transcriptional activators that function during one stage of the cell cycle regulate transcriptional activators that function during the next stage. This serial regulation of transcriptional activators together with various functional properties suggests a simple way of partitioning some selected cell-cycle genes into nine clusters, each one characterized by a group of transcriptional activators working together and their functions (see Table 2): for instance, group 1 is characterized by the activators Swi4 and Swi6 and the function of budding; group 2 is characterized by the activators Swi6 and Mbp1 and the function involving DNA replication and repair at the juncture of G1 and S phases, etc.
Table 2. Genes in our data set, grouped by transcriptional activators and cell cycle functions.
| Group | Activators | Genes | Functions |
|---|---|---|---|
| 1 | Swi4, Swi6 | Cln1, Cln2, Gic1, Gic2, Msb2, Rsr1, Bud9, Mnn1, Och1, Exg1, Kre6, Cwp1 | Budding |
| 2 | Swi6, Mbp1 | Clb5, Clb6, Rnr1, Rad27, Cdc21, Dun1, Rad51, Cdc45, Mcm2 | DNA replication and repair |
| 3 | Swi4, Swi6 | Htb1, Htb2, Hta1, Hta2, Hta3, Hho1 | Chromatin |
| 4 | Fkh1 | Hhf1, Hht1, Tel2, Arp7 | Chromatin |
| 5 | Fkh1 | Tem1 | Mitosis control |
| 6 | Ndd1, Fkh2, Mcm1 | Clb2, Ace2, Swi5, Cdc20 | Mitosis control |
| 7 | Ace2, Swi5 | Cts1, Egt2 | Cytokinesis |
| 8 | Mcm1 | Mcm3, Mcm6, Cdc6, Cdc46 | Prereplication complex formation |
| 9 | Mcm1 | Ste2, Far1 | Mating |
Upon closer examination of the data, we observed that the data in its raw “prenormalized” form is inconsistent with the assumptions used in deriving γ: (i) the gene vectors are not range-normalized, so βj2 ≠ β2 for every j, and (ii) the N experiments are not necessarily independent.
Range normalization and subsampling of experiments were used before clustering in an attempt to alleviate these shortcomings. The clusters on the processed data set, thresholded at the cut-off value of 0.60, are listed in Tables 3, 4, 5. The choice of the threshold parameter is discussed further in Discussion.
Table 3. Range-normalized subsampled data, γ = 0.0 (Eisen).
| Clusters | Activators | Genes |
|---|---|---|
| E58 | Swi4/Swi6 | Cln1, Och1 |
| E68 | Swi4/Swi6 | Cln2, Msb2, Rsr1, Bud9, Mnn1, Exg1 |
| Swi6/Mbp1 | Rnr1, Rad27, Cdc21, Dun1, Rad51, Cdc45, Mcm2 | |
| Swi4/Swi6 | Htb1, Htb2, Hta1, Hta2, Hho1 | |
| Fkh1 | Hhf1, Hht1, Arp7 | |
| Fkh1 | Tem1 | |
| Ndd1/Fkh2/Mcm1 | Clb2, Ace2, Swi5 | |
| Ace2/Swi5 | Egt2 | |
| Mcm1 | Mcm3, Mcm6, Cdc6 | |
| E29 | Swi4/Swi6 | Gic1 |
| E64 | Swi4/Swi6 | Gic2 |
| E33 | Swi4/Swi6 | Kre6, Cwp1 |
| Swi6/Mbp1 | Clb5, Clb6 | |
| Swi4/Swi6 | Hta3 | |
| Ndd1/Fkh2/Mcm1 | Cdc20 | |
| Mcm1 | Cdc46 | |
| E73 | Fkh1 | Tel2 |
| E23 | Ace2/Swi5 | Cts1 |
| E43 | Mcm1 | Ste2 |
| E66 | Mcm1 | Far1 |
Table 4. Range-normalized subsampled data, γ = 0.66 (Shrinkage).
| Clusters | Activators | Genes |
|---|---|---|
| S49 | Swi4/Swi6 | Cln1, Bud9, Och1 |
| Ace2/Swi5 | Egt2 | |
| Mcm1 | Cdc6 | |
| S6 | Swi4/Swi6 | Cln2, Gic2, Msb2, Rsr1, Mnn1, Exg1 |
| Swi6/Mbp1 | Rnr1, Rad27, Cdc21, Dun1, Rad51, Cdc45 | |
| S32 | Swi4/Swi6 | Gic1 |
| S65 | Swi4/Swi6 | Kre6, Cwp1 |
| Swi6/Mbp1 | Clb5, Clb6 | |
| Fkh1 | Tel2 | |
| Ndd1/Fkh2/Mcm1 | Cdc20 | |
| Mcm1 | Cdc46 | |
| S15 | Swi6/Mbp1 | Mcm2 |
| Mcm1 | Mcm3 | |
| S11 | Swi4/Swi6 | Htb1, Htb2, Hta1, Hta2, Hho1 |
| Fkh1 | Hhf1, Hht1 | |
| S60 | Swi4/Swi6 | Hta3 |
| S30 | Fkh1 | Arp7 |
| Ndd1/Fkh2/Mcm1 | Clb2, Ace2, Swi5 | |
| S62 | Fkh1 | Tem1 |
| S53 | Ace2/Swi5 | Cts1 |
| S14 | Mcm1 | Mcm6 |
| S35 | Mcm1 | Ste2 |
| S36 | Mcm1 | Far1 |
Table 5. Range-normalized subsampled data, γ = 1.0 (Pearson).
| Clusters | Activators | Genes |
|---|---|---|
| P1 | Swi4/Swi6 | Cln1, Och1 |
| P15 | Swi4/Swi6 | Cln2, Rsr1, Mnn1 |
| Swi6/Mbp1 | Cdc21, Dun1, Rad51, Cdc45, Mcm2 | |
| Mcm1 | Mcm3 | |
| P29 | Swi4/Swi6 | Gic1 |
| P2 | Swi4/Swi6 | Gic2 |
| P3 | Swi4/Swi6 | Msb2, Exg1 |
| Swi6/Mbp1 | Rnr1 | |
| P51 | Swi4/Swi6 | Bud9 |
| Ndd1/Fkh2/Mcm1 | Clb2, Ace2, Swi5 | |
| Ace2/Swi5 | Egt2 | |
| Mcm1 | Cdc6 | |
| P11 | Swi4/Swi6 | Kre6 |
| P62 | Swi4/Swi6 | Cwp1 |
| Swi6/Mbp1 | Clb5, Clb6 | |
| Swi4/Swi6 | Hta3 | |
| Ndd1/Fkh2/Mcm1 | Cdc20 | |
| Mcm1 | Cdc46 | |
| P49 | Swi6/Mbp1 | Rad27 |
| Swi4/Swi6 | Htb1, Htb2, Hta1, Hta2, Hho1 | |
| Fkh1 | Hhf1, Hht1 | |
| P10 | Fkh1 | Tel2 |
| Mcm1 | Mcm6 | |
| P23 | Fkh1 | Arp7 |
| P50 | Fkh1 | Tem1 |
| P69 | Ace2/Swi5 | Cts1 |
| P42 | Mcm1 | Ste2 |
| P13 | Mcm1 | Far1 |
Our initial hypothesis can be summarized as follows: Genes expressed during the same cell-cycle stage and regulated by the same transcriptional activators should be in the same cluster. We compared the performance of the similarity metrics based on the degree to which each of them deviated from this hypothesis. Below we list some of the observed deviations from the hypothesis.
Possible FPs.
Bud9 (group 1: budding), Egt2 (group 7: cytokinesis), and Cdc6 (group 8: prereplication complex formation) are placed in the same cluster by all three metrics: (E68, S49, and P51).
Mcm2 (group 2: DNA replication and repair) and Mcm3 (group 8) are placed in the same cluster by all three metrics: (E68, S15, and P15).
For more examples, see ref. 10.
Possible FNs. Group 1: budding (Table 2) is split into five clusters by the Eisen metric: {Cln1, Och1} ∈ E58, {Cln2, Msb2, Rsr1, Bud9, Mnn1, Exg1} ∈ E68, Gic1 ∈ E29, Gic2 ∈ E64, and {Kre6, Cwp1} ∈ E33; into four clusters by the Shrinkage metric: {Cln1, Bud9, Och1} ∈ S49, {Cln2, Gic2, Msb2, Rsr1, Mnn1, Exg1} ∈ S6, Gic1 ∈ S32, and {Kre6, Cwp1} ∈ S65; and into eight clusters by the Pearson metric: {Cln1, Och1} ∈ P1, {Cln2, Rsr1, Mnn1} ∈ P15, Gic1 ∈ P29, Gic2 ∈ P2, {Msb2, Exg1} ∈ P3, Bud9 ∈ P51, Kre6 ∈ P11, and Cwp1 ∈ P62.
We introduced a new notation to represent the resulting cluster sets, and a scoring function to aid in their comparison.
Each cluster set can be written as follows:
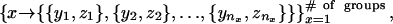 |
where x denotes the group number (as described in Table 2), nx is the number of clusters group x appears in, and for each cluster j ∈ {1,..., nx} there are yj genes from group x and zj genes from other groups in Table 2. A value of * for zj denotes that cluster j contains additional genes, although none of them are cell-cycle genes. The cluster set can then be scored according to the following measure:
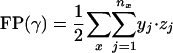 |
[7] |
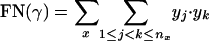 |
[8] |
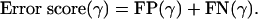 |
[9] |
Table 2 contains those genes from Fig. 2 that were present in our data set. Tables 3, 4, 5 contain these genes grouped into clusters by a hierarchical clustering algorithm using the three metrics (Eisen in Table 3, Shrinkage in Table 4, and Pearson in Table 5) thresholded at a correlation coefficient value of 0.60. Genes that have not been grouped with any others at a similarity of 0.60 or higher are absent from the tables; in the subsequent analysis they are treated as singleton clusters.
The subsampled data yielded the estimate γ ≃ 0.66. In our set notation, the resulting Shrinkage clusters with the corresponding error score computed as in Eq. 9 can be written as follows:
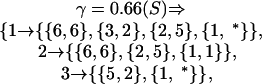 |
 |
The error scores for the Eisen (γ = 0.0) and Pearson (γ = 1.0) cluster sets, computed according to Eq. 9, are
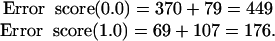 |
From the data shown in Tables 3, 4, 5, as well as by comparing the error scores, one can conclude that for the same clustering algorithm and threshold value, Pearson tends to introduce more FNs and Eisen tends to introduce more FPs than Shrinkage, as Shrinkage reduces these errors by combining the good properties of both algorithms. This observation is consistent with our mathematical analysis and the simulation presented above.
We have also conducted a more extensive computational analysis of Eisen's data, but omitted it from this article because of space limitations. This analysis appears in a full technical report available for download from www.cs.nyu.edu/cs/faculty/mishra/ (10).
Discussion
Microarray-based genomic analysis and other similar high-throughput methods have begun to occupy an increasingly important role in biology, as they have helped to create a visual image of the state-space trajectories at the core of cellular processes. This analysis will address directly to the observational nature of the new biology. As a result, we need to develop our ability to “see,” accurately and reproducibly, the information in the massive amount of quantitative measurements produced by these approaches or be able to ascertain when what we see is unreliable and forms a poor basis for proposing novel hypotheses. Our investigation demonstrates the fragility of many of these analysis algorithms when used in the context of a small number of experiments. In particular, we see that a small perturbation of, or a small error in the estimation of, a parameter (the shrinkage parameter) has a significant effect on the overall conclusion. The errors in the estimators manifest themselves by missing certain biological relations between two genes (FNs) or by proposing phantom relations between two otherwise unrelated genes (FPs).
A global picture of these interactions can be seen in Fig. 3, the receiver operator characteristic (ROC) figure, with each curve parametrized by the cut-off threshold in the range of [–1, 1]. An ROC curve (13) for a given metric plots sensitivity against (1–specificity), where
Fig. 3.

Receiver operator characteristic curves. Each curve is parametrized by the cut-off value θ ∈ {1.0, 0.95,..., –1.0}.
Sensitivity = fraction of positives detected by a metric
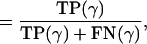 |
[10] |
Specificity = fraction of negatives detected by a metric
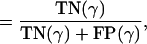 |
[11] |
and TP(γ), FN(γ), FP(γ), and TN(γ) denote the number of true positives, false negatives, false positives, and true negatives, respectively, arising from a metric associated with a given γ. (Recall that γ is 0.0 for Eisen, 1.0 for Pearson, and is computed according to Eq. 6 for Shrinkage, which yields 0.66 on this data set.) For each pair of genes, {j, k}, we define these events using our hypothesis (see above) as a measure of truth:
TP: {j, k} are in the same group (see Table 2) and {j, k} are placed in the same cluster;
FP: {j, k} are in different groups, but {j, k} are placed in the same cluster;
TN: {j, k} are in different groups and {j, k} are placed in different clusters; and
FN: {j, k} are in the same group, but {j, k} are placed in different clusters.
FP(γ) and FN(γ) were already defined in Eqs. 7 and 8, respectively, and we define
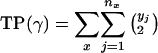 |
[12] |
and
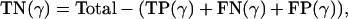 |
[13] |
where Total = 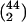 = 946 is the total no. of
gene pairs {j, k} in Table
2.
= 946 is the total no. of
gene pairs {j, k} in Table
2.
Fig. 3 suggests the best threshold to use for each metric and can also be used to select the best metric to use for a particular sensitivity.
The dependence of the error scores on the threshold can be more clearly seen from Fig. 4. It shows that the conclusions we draw above hold for a wide range of threshold values, and hence a threshold value of 0.60 is a reasonable representative value.
Fig. 4.

FN and FP curves, plotted as functions of θ.
As a result, to study the clustering algorithms and their effectiveness, one may ask the following questions. If one must err, is it better to err on the side of more FPs or more FNs? What are the relative costs of these two kinds of errors? Intelligent answers to our questions depend crucially on how the cluster information is used in the subsequent discovery processes.
Acknowledgments
This research was conducted under the support of the National Science Foundation's Qubic program, the Defense Advanced Research Projects Agency, a Howard Hughes Medical Institute biomedical support research grant, the U.S. Department of Energy, the U.S. Air Force, the National Institutes of Health, the National Cancer Institute, the New York State Office of Science, Technology, and Academic Research, a National Science Foundation Graduate Research Fellowship, and a New York University McCracken Fellowship.
Abbreviations: FN, false negative; FP, false positive; TP, true positive; TN, true negative.
References
- 1.Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. (1998) Proc. Natl. Acad. Sci. USA 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schena, M., Shalon, D., Heller, R., Chai, A., Brown, P. O. & Davis, R. W. (1996) Proc. Natl. Acad. Sci. USA 93, 10614–10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeRisi, J. L., Iyer, V. R. & Brown, P. O. (1997) Science 278, 680–686. [DOI] [PubMed] [Google Scholar]
- 4.Spellman, P. T., Sherlock, G., Zhang, M., Iyer, V. R., Anders, K., Eisen, M. B., Brown, P. O., Botstein, D. & Futcher, B. (1998) Mol. Biol. Cell 9, 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu, S., DeRisi, J. L., Eisen, M. B., Mulholland, J., Botstein, D., Brown, P. O. & Herskowitz, I. (1998) Science 282, 699–705. [DOI] [PubMed] [Google Scholar]
- 6.MacKay, D. J. C. (1992) Neural Comput. 4, 415–447. [Google Scholar]
- 7.Sokal, R. R. & Michener, C. D. (1958) Univ. Kans. Sci. Bull. 38, 1409–1438. [Google Scholar]
- 8.James, W. & Stein, C. (1961) in Proceedings of the Fourth Berkeley Symposium on Mathematical Statistics, ed. Neyman, J. (Univ. of California Press, Berkeley), Vol. 1, pp. 361–379. [Google Scholar]
- 9.Hoffman, K. (2000) Stat. Pap. 41, 127–158. [Google Scholar]
- 10.Cherepinsky, V., Feng, J., Rejali, M. & Mishra, B. (2003) NYU CS Technical Report 2003-845 (New York University, New York).
- 11.Wolfram, S. (1999) The Mathematica Book (Cambridge University Press, Cambridge, U.K.), 4th Ed.
- 12.Simon, I., Barnett, J., Hannett, N., Harbison, C. T., Rinaldi, N. J., Volkert, T. L., Wyrick, J. J., Zeitlinger, J., Gifford, D. K., Jaakkola, T. S. & Young, R. A. (2001) Cell 106, 697–708. [DOI] [PubMed] [Google Scholar]
- 13.Egan, J. P. (1975) Signal Detection Theory and ROC Analysis (Academic, New York).