

Abstract
Structural changes in a 25-base DNA strand, induced by single 8-oxo-guanine or 8-oxo-adenine substitutions, were shown by using Fourier transform-infrared spectroscopy with multivariate statistics. Pronounced differences were demonstrated between the parent and derivatives with respect to base interactions and changes in the phospho-deoxyribose backbone. The greatest degree of change in the backbone likely occurred immediately adjacent to the 8-oxo group, potentially altering the stereochemistry at a distance. The 8-oxo lesions, formed from reactive oxygen species (e.g., hydroxyl radicals), may appreciably alter the conformational properties of strands at the replication fork, thus affecting the selectivity of polymerases, the proofreading capability of repair enzymes, and the fidelity of the transcriptional machinery.
Keywords: Fourier transform-infrared spectroscopy, DNA transcription, DNA replication, DNA repair, DNA polymerases
Mounting evidence suggests that free radicals [e.g., hydroxyl radicals (⋅OH)] give rise to mutagenic base lesions in DNA, such as 8-oxo-guanine (Go) (1–3) and 8-oxo-adenine (Ao) (4). The ⋅OH are believed to arise via the metal (e.g., Fe+2)-catalyzed decomposition of H2O2, such as in redox cycling of hormones (5) and xenobiotics (6). The 8-oxo lesions contribute to localized conformational changes in base pairing and vertical base stacking within the DNA helix, as suggested by x-ray diffraction studies (7–9).
An “uncompromised” DNA template is required for the accurate replication of the genome and the transcription of genes to produce functional RNA copies (10). Accordingly, it has been suggested that nucleotide-localized changes probably lead to alterations in the fidelity of DNA repair (8, 11) and create synthesis errors at the replication fork (2, 8, 10). Until recently, almost no information existed on whether the radical-induced changes in base structure also give rise to conformational alterations in the phospho-deoxyribose backbone. However, an x-ray diffraction study of a duplex crystal (7) containing Go [d(CCAGoCGCTGG)] showed that the conversion of guanine to Go does not significantly alter the glycosidic torsion preference of the molecule and that there is no steric interaction of the 8-oxygen with the phospho-deoxyribose backbone. Another study provided no direct evidence for backbone perturbations in relation to 8-oxo substituents in duplex crystals (8); however, additional studies suggested a possible minimal disturbance (9, 12). Nevertheless, given the limited number of studies undertaken, the question remains whether 8-oxo base lesions in other DNA forms would alter conformational structure, to include backbone components, thereby potentially affecting transcription and replication processes. In an attempt to answer this question, we used Fourier transform-infrared (FT-IR) spectroscopy with multivariate statistics to delineate structural differences between thin films of a 25-base DNA strand and comparable strands containing centrally located single Go and Ao groups. FT-IR/statistical analysis was previously used to study subtle changes in base and phospho-deoxyribose structures of DNA in relation to tumor progression (13–19). In the present study, a number of spectral differences attributable to base interactions and backbone conformations were identified between the 25-base parent strand and derivatives containing Go and Ao substituents.
Materials and Methods
Oligonucleotide Synthesis.
The oligonucleotides were synthesized on the ABI 3948 synthesis system (Applied Biosystems) from phosphoramidites of guanine, cytosine, adenine, thymine, Go, and Ao, and comprised the following sequences: 5′-GAAGACTAATTGAGAGGATTAACTG-3′; 5′-GAAGACTAATTGoAGAGGATTAACTG-3′; and 5′-GAAGACTAATTGAoGAGGATTAACTG-3′.
Four individual syntheses were carried out for each structure. Purity was established by matrix-assisted laser desorption time-of-flight mass spectrometry using PerSeptive Biosystems Voyager-DE (Applied Biosystems). Ions representing other structures were not found. Each sample was dissolved in distilled water (2.0 μg/μl), 2 M sodium acetate was added to a final concentration of 0.4 M, and the sodium salt of the oligonucleotide was precipitated with an equal volume of isopropanol. A DNA pellet was formed by centrifugation for 10 min at 1,700 rpm (4°C), and the supernatant was removed. Pellets were washed three times with 1.0 ml of 70% ethanol to remove residual sodium acetate, and each wash included centrifugation and removal of the supernatant. Residual ethanol was removed by using a SpeedVac sample concentrator (Savant), and the samples were lyophilized.
FT-IR Spectroscopy with Multivariate Statistics.
Using a FT-IR microscope spectrometer (System 2000, Perkin–Elmer) (17) analyses were performed on ≈1.0 μg films of the parent strand and strands containing Go and Ao. Baseline absorbance (the mean of 11 absorbances centered at the minimum of the flat region between 2,000 and 1,751 cm−1) was subtracted from each spectrum. After baseline subtraction, the absorbance at each wavenumber was divided by the mean absorbance between 1,750 and 770 cm−1 to yield a mean of 1.0 (19), thus removing the effects of differing film thickness.
Principal components (PCs) analysis (17, 20) was used to examine subtle spectral differences between structures (see Fig. 3). PC scores 2–10 were used. The first PC score, which is similar to the grand mean spectrum, was excluded. The other PC scores reflected aspects of spectral variation, such as changes in peak heights and peak locations or combinations of changes. The PC scores were calculated over the range 1,750 to 770 cm−1 by using standard methods (17, 20).
Figure 3.

Plot showing PC scores for 5′-GAAGACTAATTGAGAGGATTAACTG-3′ and derivatives containing a 5′ (12) Go and 5′ (13) Ao. See text for details.
A resampling test (the “bootstrap”) (21) was performed on the spectra to compare the parent structure to each derivative. The bootstrap was used to determine whether the differences between the mean spectra of the parent and the modified strands represented actual structural changes vs. those occurring by chance. Spectra of the strands were pooled (e.g., four parent strands and four Go derivatives). Spectra then were randomly selected from the pool (i.e., sampled with replacement from the pool) and placed into two groups of four spectra each, and the Euclidean distance between the spectral means of the two groups was calculated. The random selection and calculation of Euclidean distance was repeated 4 × 103 times. If a true difference did not exist between the parent and derivative (the null hypothesis), the Euclidean distance between the means for the two structures would fall in the middle of the distribution of distances based on random, repeated sampling. The fraction of resamplings with Euclidean distance greater than or equal to the distance between the two structures yields the P value for the comparison between the parent and derivative.
To determine which specific regions of the spectrum were most likely to differ nonrandomly for each comparison (parent to derivative), an unequal variance t test was performed on the mean absorbance at each integer wavenumber from 1,750 to 770 cm−1. Under the null hypothesis, P values ≤ 0.05 may arise by chance at an expected 5% of the wavenumbers. Because the mean of each spectrum was forced to equal 1.0, a substantial decrease in one region would increase the normalized absorbance in other regions to maintain the mean. The possibility for P values ≤ 0.05 caused by normalization was addressed by retaining the normalized mean of 1.0 for the parent and varying the mean of the derivative. The renormalized mean was varied from 0.8 to 1.2 in increments of 0.01, and the percentage of wavenumbers with P values ≤ 0.05 was calculated at each increment.
The differences between mean spectra are presented as Euclidean distance expressed as a percentage, which was calculated by using the following formula:
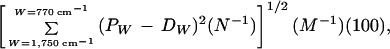 |
where PW is the absorbance at a given wavenumber for the parent structure; DW is the absorbance at a given wavenumber for the derivative; N is the number of wavenumbers, and M, used as the base for all percentages, is the mean normalized absorbance in the region 1,750–770 cm−1.
Differences between structures also might be reflected in peak shifts. Hence, the equal variance t test was used to compare peak locations. The plot of the grand mean of all 12 spectra was examined, and peaks were designated for each spectrum. A “window” was defined for each peak that did not overlap the window from an adjacent peak. The windows comprised 11–31 wavenumbers, depending on peak size. Peak location was determined within each window and for each individual spectrum. An equal variance t test was used to test for significant differences in peak locations between the parent and each derivative.
Results
In Figs. 1 and 2, A shows mean spectra for the parent and derivative structures, and B shows the P values (on a log10 scale) from a t test of absorbance at each integer wavenumber. The mean spectral differences for the region between 1,750 and 770 cm−1 were 5.1% (P = 0.01) for the parent/Go derivative comparison (Fig. 1 A and B) and 6.0% (P = 0.005) for the parent/Ao derivative comparison (Fig. 2 A and B). When the mean parent spectrum was compared with that of the Go and Ao derivatives with the unequal variance t test, P values were ≤0.05 over 32% and 35% of the spectral range, respectively. If the null hypothesis of no difference between pairs were true, then only 5% of the P values would be expected to be ≤0.05. Thus, the parent and each derivative differed significantly in specific regions over much of the spectral range.
Figure 1.

(A) Mean IR spectrum of 5′-GAAGACTAATTGAGAGGATTAACTG-3′ (black) is compared with the mean spectrum of the 5′ (12) Go derivative (red). (B) P values from unequal variance t tests between these spectra are depicted. Detail of the band assigned to antisymmetric stretching vibrations of the PO2- group (C) shows the + 3 wavenumber shift in the 1,233 cm−1 peak occurring in the Go derivative compared with the parent. Note the significant number of spectral areas with P < 0.01.
Figure 2.

(A) Mean IR spectrum of 5′-GAAGACTAATTGAGAGGATTAACTG-3′ (black) is compared with the mean spectrum of the 5′ (13) Ao derivative (red). (B) P values from an unequal variance t test between these spectra are depicted. Detail of the band assigned to antisymmetric stretching vibrations of the PO2- group shows the + 3 wavenumber shift in the 1,233 cm−1 peak occurring in the Ao derivative compared with the parent (C). Note the significant number of spectral areas with P < 0.01.
PC analysis resulted in nine PC scores (PC2–PC10), which accounted for >91% of the total experimental variance. Fig. 3 shows a plot of PC8 and PC10, the scores selected by a stepwise discriminant analysis to best differentiate the structures (20). No individual spectrum among the group of 12 differs from its most similar spectrum by more than 5%. The mean difference between the spectra and the centroid for their structure is 2–3%.
Absorbance differences between the parent and the Go derivative were apparent at 1,710 cm−1 and higher, as well as around 1,675 cm−1 (Fig. 1A). The corresponding P values ranged from 0.05 to ≈0.02 (Fig. 1B). Comparable differences for the parent/Ao derivative comparison (Fig. 2 A and B) were mostly in the range 1,710 to 1,625 cm−1 and reached minimum P values of ≈0.01 at 1,660 cm−1. These differences are in regions of the spectra assigned in each case to CO-stretching and NH2-bending vibrations (22–24). There were notable differences in band amplitudes (P = ≤0.05 to <0.001) for each parent/derivative comparison between ≈1,500 and 1,400 cm−1. This region has been attributed to weak NH vibrations, CH in-plane base deformations, and vertical base stacking interactions (22–24). The 1,233 cm−1 peak representing antisymmetric PO2− stretching vibrations shifts three wavenumbers higher in both the Go and Ao derivatives compared with the parent (P = 0.005 and 0.03, respectively) (Figs. 1C and 2C). In this peak region (1,270 to 1,180 cm−1) the absorbance of Go and Ao structures differs from the parent by 5% and 8%, respectively. The significant differences in absorbance in the vicinity of 1,230 cm−1 for both sets of parent/derivative comparisons are most probably related to these peak shifts (Figs. 1B and 2B). The absorbance differences for both comparisons between ≈1,100 and 1,150 cm−1 (the slope of the broad peak at ≈1,050 cm−1) are significant (P ≈ 0.005). The shoulder at ≈1,020 cm−1 is also significantly changed (P ≈ 0.001) in the parent/Go derivative comparison. A difference in absorbance also exists at ≈960 cm−1 in the parent/Ao comparison. Notably, these differences are generally in the region attributed to phospho-deoxyribose vibrations (22–24).
The possibility existed that the significant differences in band amplitudes arose from artifacts of normalization. However, this possibility is not supported by evidence from incremental renormalization of spectra. When spectra of the derivative structures were renormalized to a mean absorbance of 0.8 to 1.2 in increments of 0.01, the percentage of P values ≤0.05 was still substantially above the 5% expected by chance. For the parent/Go derivative comparison, the number of P values ≤0.05 reached a minimum of 28.2% (at a normalized mean absorbance of 0.98). This percentage is not appreciably different from the 32.3% of P values ≤0.05 when the normalized mean of the Go derivative group was fixed at 1.0. For the parent/Ao derivative comparison, the number of P values ≤0.05 reached a minimum of 35.4% (also at 0.98) compared with 47.6% when the normalized mean of the Ao derivative was 1.0. In all comparisons, the percentage of P values ≤0.05 progressively escalated toward 100% when the mean normalized absorbance was varied more than ±0.03 from 1.0. The possibility that the P values ≤0.05 exceeding the 5% expected by chance (Figs. 1B and 2B) are an artifact of normalization is rejected; i.e., the data represent actual structural differences between the parent and derivatives.
Discussion
There is a keen interest in the acquisition of information on radical-induced conformational changes in DNA as a basis for understanding mechanisms relating to transcription and replication (2, 7, 8, 10). In this regard, FT-IR spectroscopy with multivariate statistics is known to be sensitive to subtle changes in the base and backbone structures of DNA in cancer-prone tissues and neoplasms [e.g., prostate (15) and breast (14, 18, 19)]. The FT-IR/statistical approach is thus an attractive means for acquiring information on conformational and other changes in a variety of synthetic and natural DNA structures (17).
In the present study, the tight clustering of points in each group of the PC plot shown in Fig. 3 [derived from PC analysis of nearly 1 × 106 correlations between absorbance/wavenumber values over the entire spectrum (18)] attests to the close structural similarity of the strands from each individual synthesis. Moreover, the complete separation between groups shows that each is structurally unique, thus demonstrating the ability of the FT-IR/statistical approach to discriminate between relatively high molecular weight DNA containing minor structural changes (e.g., those induced by ⋅OH) (17).
The parent and derivative spectra appear to be essentially indistinguishable visually (Figs. 1A and 2A), which might be expected considering that the only difference is a single 8-oxo group in the ≈8,000-Da strand. However, statistical analyses revealed striking differences in the spectra of the parent vs. the derivatives. Differences in vertical base stacking interactions are the likely triggers for the alterations in the phospho-deoxyribose structures, notably the pronounced change in the PO2− group of both derivatives (Figs. 1C and 2C). The spectral peak at ≈1,233 cm−1 is characteristic of the sodium salt of nucleic acids and does not represent vibrations associated with base or sugar structures (22–24). It is not known whether only backbone changes immediately adjacent to the 8-oxo group are detectable by the FT-IR/statistical approach. However, we suggest that the structural changes (e.g., in glycosidic bond angles) may well extend from the vicinity of the 8-oxo lesion to distant structures (“ripple effect”), although with diminishing magnitude.
Previously mentioned studies of double-stranded DNA crystals containing 8-oxo substituents suggested a possible minor perturbation (9, 12) or found virtually no effect on the phospho-deoxyribose structure (7, 8). It is uncertain whether the apparent differences between the present and previous findings are related to structure (e.g., single strand vs. duplex) or are attributable to the relatively high sensitivity of the FT-IR/statistical approach (17).
The sensitivity of x-ray crystallography is generally about 2 Å (7), certainly sufficient to detect localized structural differences between native and modified substrates. The FT-IR/statistical approach detects stretching and bending vibrations of chemical bonds and has similar resolution potential to that of x-ray crystallography. Crystallography, although superior for space resolution, samples the time-averaged properties of these bonds over hours, or at least minutes. However, FT-IR spectroscopy samples over a much shorter time scale and thus has the potential for detecting dynamic changes in structure (although not in individual bonds). Moreover, the hydration state of the sample is different for each type of analysis. Consequently, the degrees of freedom of the atom ensemble in the crystal state are lower than those of the more randomly trapped sample (i.e., thin film) used in the present work.
An additional difference between the two analytical procedures relates to the nature of the experimental material. Clearly, the single polynucleotide strand has significantly greater conformational flexibility than an equivalent length of double-stranded helix; the bending and stretching of interatomic bonds would be severely dampened in the double strand. In fact, high-resolution x-ray crystallography (25) has shown that a localized modification of the base-pairing pattern within a double helix causes a small “aneurism” that does not rearrange the stereochemistry far beyond the site of occurrence. However, there exist changes in the base stacking in close proximity to the mismatched bases; that is, the stereochemical change is dampened and remains relatively confined by buttressing of the complementary strand in the double helix.
An interesting question arises: which of the two DNA states depicts the “true” situation? We suggest that both are equally relevant. The findings reported here with the single-stranded DNA document a more general potential for conformational flexibility. The previously reported crystallographic evidence from the double-stranded DNA (7–9) attests to the stabilizing effects (stiffening) of the double helix. During biological function (transcription as well as replication), the enzymatic machinery “reads” the coded information at the level of hydrogen bonds and it does this within areas of locally “melted” (single-stranded) DNA. Thus, the dynamics and conformation of a short (e.g., 3–7 bases long) stretch of polynucleotide could determine the fidelity of the overall process. In conclusion, we believe the results of our work offer a dynamic approach for examining how oxidative lesions affect DNA structure in relation to the fidelity of transcription and replication.
Acknowledgments
Helpful comments were made by Drs. Karl Erik Hellström, Michael Kahn, and Evangelos N. Moudrianakis. Technical assistance was provided by Dr. Yingzhong Su and Paul M. Johnson, statistical assistance was provided by Dr. Steven E. McKinney, and editorial assistance was provided by Dr. Virginia M. Green. Oligonucleotides were provided by Macromolecular Resources, Colorado State University, Fort Collins. This study was supported by grants from the National Cancer Institute (CA79690 and CA79479) and a grant from the National Institutes of Environmental Health Sciences (P42 ES0469) as a subcontract from the University of Washington Superfund Basic Research Program.
Abbreviations
- FT-IR
Fourier transform-infrared
- Go
8-oxo-guanine
- Ao
8-oxo-adenine
- PC
principal component
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.230438797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.230438797
References
- 1.Cheng K C, Cahill D S, Kasai H, Nishimura S, Loeb L A. J Biol Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- 2.Feig D I, Reid T M, Loeb L A. Cancer Res. 1994;54:1890s–1894s. [PubMed] [Google Scholar]
- 3.Feig D I, Loeb L A. Biochemistry. 1993;32:4466–4473. doi: 10.1021/bi00067a040. [DOI] [PubMed] [Google Scholar]
- 4.Kamiya H, Miura H, Murata-Kamiya N, Ishikawa H, Sakaguchi T, Inoue H, Sasaki T, Masutani C, Hanaoka F, Nishimura S, et al. Nucleic Acids Res. 1995;23:2893–2899. doi: 10.1093/nar/23.15.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liehr J G. Eur J Cancer Prev. 1997;6:3–10. doi: 10.1097/00008469-199702000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Frenkel K, Wei L, Wei H. Free Radical Biol Med. 1995;19:373–380. doi: 10.1016/0891-5849(95)00046-z. [DOI] [PubMed] [Google Scholar]
- 7.Lipscomb L A, Peek M E, Morningstar M L, Verghis S M, Miller E M, Rich A, Essigmann J M, Williams L D. Proc Natl Acad Sci USA. 1995;92:719–723. doi: 10.1073/pnas.92.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McAuley-Hecht K E, Leonard G A, Gibson N J, Thomson J B, Watson W P, Hunter W N, Brown T. Biochemistry. 1994;33:10266–10270. doi: 10.1021/bi00200a006. [DOI] [PubMed] [Google Scholar]
- 9.Leonard G A, Guy A, Brown T, Téoule R, Hunter W N. Biochemistry. 1992;31:8415–8420. doi: 10.1021/bi00151a004. [DOI] [PubMed] [Google Scholar]
- 10.SenGupta D J, Blackwell L J, Gillette T, Borowiec J A. Chromosoma. 1992;102:S46–S51. doi: 10.1007/BF02451785. [DOI] [PubMed] [Google Scholar]
- 11.Berwick M, Vineis P. J Natl Cancer Inst. 2000;92:874–897. doi: 10.1093/jnci/92.11.874. [DOI] [PubMed] [Google Scholar]
- 12.Oda Y, Uesugi S, Ikehara M, Nishimura S, Kawase Y, Ishikawa H, Inoue H, Ohtsuka E. Nucleic Acids Res. 1991;19:1407–1412. doi: 10.1093/nar/19.7.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Closas M, Hankinson S E, Ho S, Malins D C, Polissar N L, Schaefer S N, Su Y, Vinson M A. J Natl Cancer Inst Monogr. 2000;27:147–156. doi: 10.1093/oxfordjournals.jncimonographs.a024238. [DOI] [PubMed] [Google Scholar]
- 14.Malins D C, Polissar N L, Schaefer S, Su Y, Vinson M. Proc Natl Acad Sci USA. 1998;95:7637–7642. doi: 10.1073/pnas.95.13.7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malins D C, Polissar N L, Gunselman S J. Proc Natl Acad Sci USA. 1997;94:259–264. doi: 10.1073/pnas.94.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malins D C, Polissar N L, Gunselman S J. Proc Natl Acad Sci USA. 1997;94:3611–3615. doi: 10.1073/pnas.94.8.3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malins D C, Polissar N L, Su Y, Gardner H S, Gunselman S J. Nat Med. 1997;3:927–930. doi: 10.1038/nm0897-927. [DOI] [PubMed] [Google Scholar]
- 18.Malins D C, Polissar N L, Gunselman S J. Proc Natl Acad Sci USA. 1996;93:14047–14052. doi: 10.1073/pnas.93.24.14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malins D C, Polissar N L, Nishikida K, Holmes E H, Gardner H S, Gunselman S J. Cancer. 1995;75:503–517. doi: 10.1002/1097-0142(19950115)75:2<503::aid-cncr2820750213>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 20.Fisher L D, Van Belle G. Biostatistics: A Methodology for the Health Sciences. New York: Wiley; 1993. [Google Scholar]
- 21.Efron B, Tibshirani R J. An Introduction to the Bootstrap. New York: Chapman & Hall; 1993. [Google Scholar]
- 22.Tsuboi M. In: Basic Principles in Nucleic Acid Chemistry. Ts'o P O P, editor. New York: Academic; 1974. pp. 399–452. [Google Scholar]
- 23.Tsuboi M. Appl Spectrosc Rev. 1969;3:45–90. [Google Scholar]
- 24.Taillandier E, Liquier J. Methods Enzymol. 1992;211:307–335. doi: 10.1016/0076-6879(92)11018-e. [DOI] [PubMed] [Google Scholar]
- 25.Kennard O. In: Biomolecular Stereodynamics IV. Sarma R H, Sarma M H, editors. New York: Adenine; 1986. pp. 117–138. [Google Scholar]