

Abstract
We introduce an analytical method that combines in one pot the advantages of column chromatography separation and NMR structural analysis. The separation of the NMR spectra of the components of a mixture can be achieved according to their apparent diffusion rates [James, T. L. and McDonald, G. G. (1973) J. Magn. Reson. 58, 58–61]. We show that the separation of the spectral components, corresponding to single molecular species, can be enhanced by order of magnitudes upon addition of a typical stationary phase used in HPLC. The solid phase imbibed by the mixture for analysis is an heterogeneous ensemble, so that solid-state NMR methods (high-resolution magic angle spinning) are necessary to recover high-resolution spectra. We demonstrate applications of this combination of high-resolution magic angle spinning and NMR diffusometry on test mixtures for direct (silica gel) and inverse (C18) columns. However, many common chromatographic supports available for HPLC should be readily adaptable for use with this technique.
The challenge in the analysis of complex mixtures is 2-fold: to achieve an effective separation of the components and provide a proper structural characterization for each of them. The common solution is hyphenation, which is a sequential combination of chromatography and spectroscopic analysis such as mass or UV spectrometry. Because NMR is often the tool of choice for precise structural characterizations of organic molecules, an effort has been done to introduce NMR as a detector in hyphenated techniques (1). An alternative solution for the NMR characterization of mixture components exploits the behavior of the NMR signal of a molecule diffusing in an inhomogeneous magnetic field. In this case, the frequency of the observed target becomes time dependent, with consequent broadening of the resonance line (2). The apparent diffusion rate associated to a molecular resonance can be estimated by performing a series of experiments varying the amplitude of the inhomogeneous field and inverting the corresponding decay curve of the signal amplitude. Diffusion-ordered spectroscopy (DOSY) (3) is a particularly convenient means of displaying this information, organized in a bidimensional array with the NMR spectrum on one dimension and the apparent diffusion rate on the other one. As a collateral aspect, the DOSY display decomposes the overall NMR spectrum of a mixture into those of its components if these latter possess different molecular mobility, without previous collection of separate fractions (4–7). In optimal conditions, the diffusion experiment is capable of resolving contributions from molecules whose diffusion coefficients differ only by a few percent (8). In cases of serious signal overlapping, other methods for processing diffusion data achieve better spectral separation (9–11). We present here an analytical procedure that capitalizes on this idea, but in which the separation properties of the NMR method are enhanced by addition of a solid phase, a typical stationary phase material used for chromatographic columns. In liquid-phase chromatography, the translational velocities of the components of a mixture are selectively altered by interaction with the stationary phase, to collect them individually at the outlet of a separation column. The effective diffusion rate of each molecule in this condition may differ severely from its solution value. In fact, the mobility of molecules is altered if an equilibrium exists between a free and a complexed form. If the complex is in the fast exchange limit on the NMR time scale, an average diffusion rate is observed, weighed over the populations of the free and bound species (12). This property constitutes the basis of affinity NMR (13), which tests binding affinity of ligands to relevant receptor molecules (14), and has also been used to study peptide–micelle association (15) and screen new potential chromatographic materials (16). Indeed, enhanced resolution in DOSY spectra was achieved by addition of micelles, in the case of hydrophobic interactions (17).
We reproduced a model chromatographic column in a standard sample holder for solid-state NMR (a zirconia cylindrical rotor). The spatial variation of the magnetic susceptibility caused by the heterogeneous structure of the liquid/solid mix results in broad signals and a large loss of resolution, because of variation of the susceptibility and residual solid-state effects. High-resolution magic angle spinning (HRMAS) NMR has been designed to recover well-resolved spectra for similar cases (18–21), and very recently it was applied to the study of chromatographic phases (22). In this study, HRMAS coupled with diffusion-based NMR techniques (23–26) on a mixture in the presence of a silica gel was used to achieve well-separated and high-resolution spectra for the single components.
Materials and Methods
All NMR experiments were performed on a Bruker (Wissembourg, France) Avance spectrometer operating at 400 MHz and equipped with an HRMAS probe head capable of producing z gradients with a strength of 53 G·cm–1. HRMAS NMR spectra were recorded at a spinning rate of 4 kHz at 300 K. Four-millimeter ZrO2 rotors with detection volumes of 12 and 50 μl were used for HRMAS 1H and HRMAS DOSY experiments, respectively. In HRMAS 1H experiments, the spectral width was typically 4,000 Hz, and the free induction decay contained 8–16 K data points. The 90° pulse duration was 5.2 μs.
Sample Preparation. Typically, 10 μl of a model mixture was added to a 4-mm ZrO2 rotor containing 50 mg of a specific chromatographic stationary phase. Roughly half of the stationary phase was placed in the rotor, and then the mixture was added with a syringe and the rotor was filled with the remaining part of the silica gel.
Sample I. The mixture for analysis was 5 μl of ethanol, 3 mg of naphthalene, and 10 μl of dec-1-ene dissolved in 100 μl of deuterated ethanol-d6. The stationary phase was a functionalized silica gel (C18 Chromabond, Macherey & Nagel).
Sample II. The mixture for analysis was 5 μl of heptane, 5 μl of ethanol, and 5 mg of 3,5-dichlorophenol dissolved in 100 μl of deuterated cyclohexane-d12. The stationary phase was a silica gel (230–400 mesh, Macherey & Nagel).
Diffusion Experiments. In a diffusion experiment, a series of pulsed-field gradient spin-echo spectra are recorded with incremented values of the gradient strength. In the simplest cases, the intensity of each resonance signal in the spectrum decays exponentially with the square of the gradient amplitude, g, and the diffusion time, Δ, according to the equation:
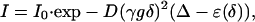 |
where I0 is the resonance intensity at zero gradient strength, D is the diffusion coefficient in the direction of the gradient, γ is the gyromagnetic ratio of the observed nucleus, δ is the duration of the gradient pulses, and ε(δ) is a correction factor that depends on both δ and the pulse sequence used.
The diffusion experiments were performed by using a stimulated-echo sequence incorporating bipolar gradient pulses and a longitudinal eddy current delay (27). The gradient strength was incremented in 16 steps from 2% to 95% of the maximum gradient strength in a customarily programmed scheme, which was optimized to achieve a 95% decrease in the resonance intensity at the largest gradient amplitudes for all of the mixture components. Diffusion times and gradient pulse duration were optimized for each experiment; typically, diffusion times between 100 and 700 ms and bipolar gradient pulses between 1.0 and 2.0 ms were used. For the particle size we used (37 μm, 400 mesh), this setup guarantees effective compensation of internal gradients by the bipolar pulses for diffusion constants in our range of measures (D is ≈10–9 m2/s or 1 μm/ms, associated to a rms displacement of 2 μm for a gradient pulse of 2 ms). The longitudinal eddy current delay was held constant to 5 ms in all experiments whereas the gradient pulse recovery time was set to 100 μs. For Fourier transformation and baseline correction, the diffusion dimension was processed by means of the DOSY options of the Bruker xwinnmr software package (version 3.0).
Results and Discussion
Fig. 1 shows a comparison of the NMR spectrum of sample II (described above), mixed with a silica gel. HRMAS is necessary to recover a resolution of sufficient quality for characterization of the molecular structure, although the signals remain broader than in the pure solution. Fig. 2 demonstrates the effect on molecular mobility upon introduction of a chromatographic column material, increasing the resolving power in the DOSY display. An inverse phase (C18) was tested in Fig. 2 A and B. The mixture for analysis consisted of naphthalene, ethanol, and heptane, dissolved in ethanol-d6. The effect of the addition of the solid phase on the variation of mobility is as expected, the molecules being “delayed” with respect to the solution-state DOSY in the same order as they would be in the corresponding chromatographic column. The fastest component is polar ethanol, whereas the slowest one is naphthalene, which is known to have high affinity for C18 columns. All molecular contributions appear to overlap in the solution spectra, while they assume up to almost 2 orders of magnitude difference in the apparent diffusion rate upon addition of the column material. In a second case (Fig. 2 C and D), we used a silica gel and a test mixture of molecules with different affinity for the silica: heptane, ethanol, and 3,5-dichlorophenol, dissolved in perdeuterated cyclohexane. In chromatographic conditions, the polar components are delayed. The most polar compound, 3,5-dichlorophenol, which would come out last in the corresponding HPLC experiment, is the most delayed. Ethanol assumes an intermediate value, whereas the heptane does not show any significant variation of its apparent diffusion rate with respect to the pure mixture. For both stationary phases, only one peak can be observed in the diffusion dimension for each molecule, which is the signature of fast exchange. No signals arising from column protons are observed, because of a combination of spectral editing and processing, which cuts off slow diffusing values.
Fig. 1.

1H NMR spectra at 400 MHz of sample II (see text) in the presence of a silica gel. (Upper) Solution-state NMR. (Lower) HRMAS NMR.
Fig. 2.

(A) DOSY spectrum of sample I (5 μl of ethanol, 3 mg of naphthalene, and 10 μl of dec-1-ene, dissolved in 100 μl of deuterated ethanol-d6). (B) HRMAS–DOSY spectrum of 20 μl of the same mixture as in A, but in the presence of 50 mg of a functionalized silica gel (C18 Chromabond, Macherey & Nagel). The peaks marked by * correspond to a water impurity. (C) DOSY spectrum of sample II (5 μl of heptane, 5 μl of ethanol, and 5 mg of 3,5-dichlorophenol dissolved in 100 μl of deuterated cyclohexane-d12). (D) HRMAS–DOSY spectrum of 12 μl of the same mixture as in C, but in the presence of 50 mg of a silica gel (230–400 mesh, Macherey & Nagel).
Conclusions
In conclusion, we have demonstrated a method for analysis of mixtures that incorporates the advantages of column liquid chromatography with NMR detection. The influence of stationary phases on mixture of organic compounds can be followed by NMR, provided high-resolution DOSY spectra can be achieved. Although its limitations in terms of resolution in both the NMR and diffusion dimension remains to be assessed, this HRMAS–DOSY method could profit from most of the know-how developed for HPLC. Moreover, the technique could be used to validate and investigate new chromatographic setups and, in general, to gain insight into the whole process of retention in HPLC.
Acknowledgments
A. Segre (Consiglio Nazionale delle Ricerche Montelibretti) provided the funding for a Ph.D. grant for S.V. and many useful comments and discussions. We are grateful to C. Delaurant and G. Herbette (Université d'Aix-Marseille III) for useful discussions about HPLC and providing the column material. S.V. performed most of the experiments. F.Z. optimized the sample conditioning and participated in the NMR experiments. S.C. designed the project and wrote this article.
Abbreviations: DOSY, diffusion-ordered spectroscopy; HRMAS, high-resolution magic angle spinning.
References
- 1.Albert, K., ed. (2002) On-Line LC-NMR and Related Techniques (Wiley, Chichester, U.K.).
- 2.Stejskal, E. O. & Tanner, J. E. (1965) J. Chem. Phys. 42, 288–292. [Google Scholar]
- 3.Morris, K. F. & Johnson, C. S., Jr. (1992) J. Am. Chem. Soc. 114, 3139–3141. [Google Scholar]
- 4.Johnson, C. S., Jr. (1996) in Encyclopedia of Nuclear Magnetic Resonance, eds. Harris, R. K. & Grant D. M. (Wiley, Chichester, U.K.), Vol. 3, pp. 1626–1646. [Google Scholar]
- 5.Kärger, J. (1996) in Encyclopedia of Nuclear Magnetic Resonance, eds. Harris, R. K. & Grant, D. M. (Wiley, Chichester, U.K.), Vol. 3, pp. 1656–1663. [Google Scholar]
- 6.Morris, G. (2002) in Encyclopedia of Nuclear Magnetic Resonance, eds. Harris, R. K. & Grant, D. M. (Wiley, Chichester, U.K.), Vol. 9, pp. 35–44. [Google Scholar]
- 7.Johnson, C. S., Jr. (1999) Prog. Nucl. Magn. Reson. Spectrosc. 34, 203–256. [Google Scholar]
- 8.Barjat, H., Morris, G. A., Smart, S., Swanson, A. G. & Williams, S. C. R. (1995) J. Magn. Reson. B. 108, 170–172. [Google Scholar]
- 9.Stilbs, P., Paulsen, K. & Griffiths, P. C. (1996) J. Phys. Chem. 100, 8180–8189. [Google Scholar]
- 10.Antalek, B. & Winding, W. (1996) J. Am. Chem. Soc. 118, 10331–10332. [Google Scholar]
- 11.Antalek, B. (2002) Concepts Magn. Reson. 14, 225–258. [Google Scholar]
- 12.Johnson, C. S., Jr. (1993) J. Magn. Reson. A 102, 214–218. [Google Scholar]
- 13.Lin, M. F. & Shapiro, M. J. (1996) J. Org. Chem. 61, 7617–7619. [DOI] [PubMed] [Google Scholar]
- 14.John, S., Gounarides, J. S., Chen, A. & Shapiro, M. J. (1999) J. Chromatogr. B 725, 79–90. [DOI] [PubMed] [Google Scholar]
- 15.Deaton, K. R., Feyen, E. A., Nkulabi, H. J. & Morris, K. F. (2001) Magn. Reson. Chem. 39, 276–282. [Google Scholar]
- 16.Laverde, A., Da Conceição, G. J. A., Queiroz, S. C. N., Fujiwara, F. Y. & Marsaioli, A. J. (2002) Magn. Reson. Chem. 40, 433–442. [Google Scholar]
- 17.Morris, K. F., Stilbs, P. & Johnson, C. S., Jr. (1994) Anal. Chem. 66, 211–215. [Google Scholar]
- 18.Lippens, G., Bourdonneau, M., Dhalluin, C., Warras, R., Richert, T., Setharaman, C., Boutillon, C. & Piotto, M. (1999) Curr. Org. Chem. 3, 147–170. [Google Scholar]
- 19.Gross, J. D., Costa, P. R., Dubacq, J. P., Warschawski, D. E., Lirsac P. N., Devaux, P. F. & Griffin, R. G. (1995) J. Magn. Reson. B 106, 187–190. [DOI] [PubMed] [Google Scholar]
- 20.Gil, A. M., Duarte, I. F., Delgadillo, I., Colquhoun, I. J., Casuscelli, F., Humpfer, E. & Spraul, M. (2000) J. Agric. Food Chem. 48, 1524–1536. [DOI] [PubMed] [Google Scholar]
- 21.Chang, L. L., Lean, C. L., Bogdanova, A. & Wright, S. C. (1996) Magn. Reson. Med. 36, 653–658. [DOI] [PubMed] [Google Scholar]
- 22.Händel, H., Gesele, E., Gottschall, K. & Albert, K. (2003) Angew. Chem. Int. Ed. 42, 438–442. [DOI] [PubMed] [Google Scholar]
- 23.Maas, W. E., Laukien, F. H. & Cory, D. G. (1996) J. Am. Chem. Soc. 118, 13085–13086. [Google Scholar]
- 24.Warrass, R., Wieruszeski, J.-M. & Lippens, G. (1999) J. Am. Chem. Soc. 121, 3787–3788. [Google Scholar]
- 25.Pamapel, A., Michel, D. & Reszka, R. (2002) Chem. Phys. Lett. 357, 131–136. [Google Scholar]
- 26.Weybright, P., Millis, K., Campbell, N., Cory, D. G. & Singer, S. (1998) Magn. Reson. Med. 39, 337–345. [DOI] [PubMed] [Google Scholar]
- 27.Wu, D., Chen, A. & Johnson, C. S., Jr. (1995) J. Magn. Reson. A 115, 260–264. [Google Scholar]