

Abstract
To classify cancer specimens by their gene expression profiles, we created a statistical method based on Bayes' rule that estimates the probability of membership in one of two cancer subgroups. We used this method to classify diffuse large B cell lymphoma (DLBCL) biopsy samples into two gene expression subgroups based on data obtained from spotted cDNA microarrays. The germinal center B cell-like (GCB) DLBCL subgroup expressed genes characteristic of normal germinal center B cells whereas the activated B cell-like (ABC) DLBCL subgroup expressed a subset of the genes that are characteristic of plasma cells, particularly those encoding endoplasmic reticulum and golgi proteins involved in secretion. We next used this predictor to discover these subgroups within a second set of DLBCL biopsies that had been profiled by using oligonucleotide microarrays [Shipp, M. A., et al. (2002) Nat. Med. 8, 68–74]. The GCB and ABC DLBCL subgroups identified in this data set had significantly different 5-yr survival rates after multiagent chemotherapy (62% vs. 26%; P ≤ 0.0051), in accord with analyses of other DLBCL cohorts. These results demonstrate the ability of this gene expression-based predictor to classify DLBCLs into biologically and clinically distinct subgroups irrespective of the method used to measure gene expression.
Keywords: gene expression profile, Bayesian predictor, microarray
An initial gene expression profiling study of diffuse large B cell lymphoma (DLBCL) led to the proposal that this single diagnostic category consists of at least two molecularly distinct diseases (1). One DLBCL subgroup, termed germinal center B cell-like (GCB) DLBCL, expressed genes characteristic of normal germinal center (GC) B cells whereas the other subgroup, termed activated B cell-like (ABC) DLBCL, instead expressed genes characteristic of mitogenically activated blood B cells. Patients with GCB DLBCL were more often cured by combination chemotherapy than were patients with ABC DLBCL. Recently, in an expanded gene expression profiling study of 274 DLBCL patients, the two gene expression subgroups were again identified together with a new subgroup, termed type 3, which did not express the genes characteristic of either GCB or ABC DLBCL (2). As before, patients with GCB DLBCL had a more favorable clinical course, with a 5-yr survival rate of 60% compared with 5-yr survival rates of 35% and 38% for patients with ABC and type 3 DLBCL, respectively (2).
Another study used oligonucleotide microarrays to profile gene expression in 58 DLBCL biopsies (3) and attempted to identify the GCB and ABC DLBCL subgroups by using genes that were identified in the original profiling study as distinguishing these subgroups (1). Hierarchical clustering of the DLBCL cases based on expression of these genes resulted in two groups of patients that did not differ in clinical outcome (3), in apparent contrast with the two other studies (1, 2).
We were curious to see whether we could resolve the discrepancy between these gene expression profiling studies by using our current understanding of the gene expression differences between GCB and ABC DLBCL. As was pointed out (3), it is a challenging task to compare the results of these profiling studies because they used different microarray platforms that were only partially overlapping in gene composition [i.e., Lymphochip spotted cDNA microarrays (1) vs. Affymetrix (Santa Clara, CA) HU6800 oligonucleotide microarrays (3)]. Notably, the Affymetrix arrays lacked many of the genes on the Lymphochip microarrays that are selectively expressed in GCB DLBCLs and in normal GC B cells (1). As a consequence, the set of genes that was used to search for the DLBCL subgroups was missing some of the most discriminating genes and was correspondingly enriched for genes that are differentially expressed between the DLBCL subgroups with only modest statistical significance.
For this reason, we developed a classification method that focuses on those genes that discriminate the GCB and ABC DLBCL subgroups with highest significance. Our method does not merely assign a tumor to a DLBCL subgroup but also estimates the probability that the tumor belongs to the subgroup. We demonstrate that this method is capable of classifying a tumor irrespective of which experimental platform is used to measure gene expression. The GCB and ABC DLBCL subgroups defined by using this predictor have significantly different survival rates after chemotherapy.
Methods
Gene Expression Data. DLBCL gene expression data generated by using Lymphochip microarrays were obtained from supporting information of ref. 2 at http://llmpp.nih.gov/DLBCL. DLBCL gene expression data generated by using Affymetrix HU6500 microarrays were obtained from supporting information of ref. 3 at www.genome.wi.mit.edu/MPR/lymphoma and were normalized as follows. We identified those genes that were listed as present on >50% of the samples and then multiplied the signal values on each array by a factor to make the median value of these genes equal to 1,000. After this normalization, we set all signal values that were <50 to a value of 50 and then applied a log2 transformation. All gene expression data used in the present analysis can be obtained from http://llmpp.nih.gov/DLBCLpredictor.
Formulation of the DLBCL Subgroup Predictor. We calculated a linear predictor score (LPS) for each sample X of the form
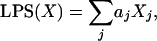 |
[1] |
where Xj represents the gene expression of gene j, and aj is a scaling factor whose value depends on the degree to which each gene discriminates the subgroups. The scaling factors were chosen to be the t statistics generated by a t test for the difference in expression between the two subgroups (4). Only the k genes with the most significant t statistics were used to form the LPS, with the optimal k determined empirically (see below). For genes represented by multiple features on the microarray, the feature with the most significant t statistic was used.
Because the LPS is a linear combination of gene expression values, its distribution within each subgroup should be approximately normal, provided it includes a sufficient number of genes and the correlation structure of those genes is not extreme. The mean and variance of these normal distributions can then be estimated from the LPSs calculated for the samples in each subgroup. Given the LPS distribution of each subgroup, it is possible to estimate the likelihood that a new sample is in each of the two subgroups by applying Bayes' rule, so that
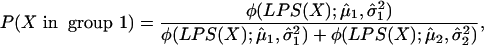 |
[2] |
where ϕ(x; μ, σ2) represents the normal
density function with mean μ, and variance σ2, and
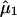 ,
,
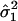 ,
,
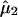 , and
, and
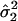 are the
observed mean and variance of the LPSs within subgroup 1 and subgroup 2,
respectively.
are the
observed mean and variance of the LPSs within subgroup 1 and subgroup 2,
respectively.
Because the samples that are used to estimate the distribution of the LPSs are also used to generate the model, there is a possibility of overfitting, resulting in a model that would indicate a larger separation between the subgroups' LPSs than would be found in independent data. Therefore, it is important to check the validity of the model on a separate validation data set. We constructed a training set consisting of 42 ABC DLBCL and 67 GCB DLBCL samples and a validation set consisting of 41 ABC DLBCL, 67 GCB DLBCL, and 57 type 3 DLBCL samples (2). To choose the optimal number of genes (k) to include in the model, multiple models with different numbers of DLBCL subgroup discrimination genes were evaluated on the training set by using a leave-one-out cross-validation procedure (5); a model including 27 genes had the lowest average error rate. We applied this model to the validation set and observed that the distribution of the LPSs within each subgroup matched the corresponding distributions in the training set, thus demonstrating that model overfitting was not an issue (data not shown). By using the probability estimates determined by Bayes' rule, we chose a cutoff of 90% certainty to decide final subgroup membership. Those samples for which there was <90% likelihood of being in either subgroup were termed “unclassified.”
To apply the DLBCL subgroup predictor to data obtained by using Affymetrix microarrays (3), we first excluded those Affymetrix microarray features with a median signal value of <200 across the samples and then averaged multiple microarray features representing the same gene, if present. Of the 27 genes in the DLBCL subgroup predictor described above, only 14 were represented on the Affymetrix microarrays and passed this filtering process. These 14 genes were used to create a new DLBCL subgroup predictor in which the LPS scaling coefficients were again calculated based on the DLBCL subgroup distinction in the Lymphochip data set (2). For each gene, we shifted and scaled the expression values in the Affymetrix data set to match the mean and variance of the corresponding expression values in the Lymphochip data set, to account for systematic measurement differences between the two microarray platforms. The adjusted expression values for the 14 genes in the predictor were used to calculate LPSs for each sample in the Affymetrix data set, and DLBCL subgroup membership was assigned as above based on a cutoff of 90% certainty.
Purification and Gene Expression Analysis of B Cell Subpopulations. Bone marrow aspirates were separated into CD19+ and CD19– populations by magnetic sorting. The CD19+ cells were further fractionated by flow sorting into immature B cells (IgM+, IgD–, and CD10+) and mature B cells (IgM+, IgD+, and CD10–). The CD19– population was further purified by flow sorting to obtain plasma cells (CD138+, CD38high, and CD20–). CD19+ peripheral blood B cells were purified by magnetic sorting and fractionated further by flow sorting into naive B cells (IgD+ and CD27–), and two memory B cells types (IgM+, IgD+, and CD27+, see Fig. 2, rows 10 and 11; and IgM–, IgD–, and CD27+, see Fig. 2, rows 12 and 13). Total germinal center B cells (see Fig. 2, rows 9 and 10) and centrocytes (see Fig. 2, rows 7 and 8) were purified from tonsils as described (1). Total mRNA was linearly amplified two times (Ambion, Austin, TX), labeled with Cy5 dye, and hybridized to Lymphochip microarrays with a Cy3-labeled probe derived from pooled cell line mRNA as described (1).
Fig. 2.

Relationship of gene expression in normal B cell subpopulations to DLBCL subgroups. Relative gene expression in the indicated purified B cell subpopulations (see Methods) is depicted according to the color scale in Fig. 1. The P value of the difference in expression of these genes between the GCB and ABC DLBCL subgroups is shown, and the subgroup with the higher expression is indicated (blue, ABC DLBCL; orange, GCB DLBCL). (A) DLBCL subgroup distinction genes that are more highly expressed in germinal center B cells than at other B cell differentiation stages. (B) DLBCL subgroup distinction genes that are more highly expressed in plasma cells than at other B cell differentiation stages.
Results
The ABC and GCB subgroups of DLBCL were originally identified by applying a hierarchical clustering algorithm to gene expression data from DLBCL biopsies profiled by using Lymphochip microarrays (1, 2). We wished to create a statistical model of this distinction that could estimate the probability that a particular DLBCL case belongs to one or the other DLBCL subgroup. To ensure the reproducibility of our model, we divided the gene expression data from 274 DLBCL cases (2) into a training set that was used to create and optimize the model and a validation set that was used to evaluate its performance.
We selected genes to incorporate into the subgroup predictor based on several criteria. First, we identified those genes that were differentially expressed between the ABC and GCB DLBCL subgroups within the training set with high significance (P < 0.001). We narrowed this list further by considering only those genes that were most variably expressed within the training set (i.e., in the top third of genes with respect to variance). Finally, we eliminated genes that vary in expression due to differences in tumor cell proliferation rate or to differences in the host immune reaction in the lymph node, i.e., genes belonging to the previously described “proliferation” and “lymph node” gene expression signatures (2, 6). Because these two signatures can vary independently in expression within both DLBCL subgroups (2), we excluded them from the subgroup predictor so as not to obscure the distinction between the two subgroups.
For each DLBCL sample, the expression levels of these subgroup distinction genes were combined to create a “linear predictor score” (see Methods). The distributions of the linear predictor scores for GCB and ABC DLBCLs were used to estimate the probability that any particular DLBCL sample belonged to either subgroup by applying Bayes' rule (Fig. 1A). A DBLCL sample was classified into a subgroup if it had a ≥90% probability of belonging to that subgroup. Within the training set, we optimized the number of genes in the subgroup predictor based on the accuracy with which the predictor classified samples into the ABC and GCB subgroups defined by hierarchical clustering (2). The final subgroup predictor incorporated the 27 genes shown in Fig. 1 A and correctly classified 87% of the training set samples into the subgroup to which they had been assigned by hierarchical clustering (Fig. 1B). The reproducibility of the subgroup predictor was demonstrated by its ability to correctly classify 88% of the samples in the validation set (Fig. 1B). Interestingly, 56% of the DLBCLs that had been placed in the type 3 subgroup by hierarchical clustering were classified as either ABC or GCB DLBCL by the subgroup predictor. These results demonstrate that the subgroup predictor and hierarchical clustering produce similar but not identical classifications of the DLBCL samples.
Fig. 1.

Performance of the DLBCL subgroup predictor using gene expression measurements from spotted cDNA microarrays. (A) The expression levels for the 27 genes in the subgroup predictor in 274 DLBCL samples (2) are depicted according to the color scale shown at the left. The 14 genes that were used to predict the DLBCL subgroups within the Affymetrix data set (Fig. 3) are indicated with asterisks. The probabilities that the DLBCL samples belong to the ABC or GCB subgroups are graphed at the top, and the DLBCL cases are arranged accordingly. The cases that belong to either the ABC or GCB DLBCL subgroups with ≥90% likelihood are indicated. (B) The assignments of the DLBCL cases to the ABC or GCB subgroups based on hierarchical clustering (2) vs. the subgroup predictor are compared within the training, validation, and total set of samples.
In previous work, the genes that were used to distinguish GCB and ABC DLBCLs were deliberately chosen to include those that were preferentially expressed in normal GC B cells (1, 2). In the present analysis, the subgroup predictor was not biased a priori to include such genes. We therefore investigated whether GC B cell-restricted genes were differentially expressed between the GCB and ABC DLBCL subgroups as defined by the subgroup predictor. Fig. 2A shows the expression of 38 genes that were more highly expressed in GC B cells than at other stages of B cell differentiation (P < 0.001) and that were differentially expressed between the DLBCL subgroups (P < 0.001). All but two of these GC B cell-restricted genes were more highly expressed in GCB than in ABC DLBCLs. This result demonstrates that the DLBCL subgroups defined by the subgroup predictor again seem to differ with respect to cell of origin, with GCB DLBCL retaining the gene expression program of normal GC B cells.
ABC DLBCLs, on the other hand, had higher expression of genes that are characteristic of plasma cells. Fig. 2B shows the expression of 24 genes that were more highly expressed in plasma cells than in B cells at earlier developmental stages (P < 0.001) and that were differentially expressed between the DLBCL subgroups (P < 0.001). The majority of these plasma cell-restricted genes were more highly expressed in ABC DLBCLs. Eight of these genes encode proteins that reside and function in the endoplasmic reticulum (ER) or golgi apparatus, suggesting that ABC DLBCLs have increased the intracellular machinery for protein secretion. Another gene in this list, XBP-1, encodes a protein that is required for plasma cell differentiation (7) and is involved in the response to unfolded proteins in the ER (8). ABC DLBCLs have not undergone full plasmacytic differentiation, however, because other key plasma cell genes such as Blimp-1 were not more highly expressed in ABC DLBCLs (data not shown).
We next applied the subgroup predictor to another published set of gene expression data from DLBCLs that was generated by using Affymetrix oligonucleotide microarrays (3). We first identified the 14 genes among the 27 genes in the subgroup predictor that were represented on the Affymetrix microarrays. With these 14 genes, we constructed and optimized a new subgroup predictor by using the DLBCL gene expression data from Lymphochip microarrays; the Affymetrix data were not used to adjust the model parameters. Given the inherent methodological differences between microarray platforms, it is expected that they will yield gene expression measurements that differ both in absolute level and variance across a group of samples. Therefore, in applying the subgroup predictor to the Affymetrix data set, we adjusted the Affymetrix data for each gene to match the mean and variance of the expression levels of that gene within the Lymphochip data.
In Fig. 3, the 58 DLBCL samples in the Affymetrix data set are arranged according to their probabilities of being ABC or GCB DLBCL based on the subgroup predictor. Several observations suggest that the subgroup predictor identified ABC and GCB DLBCL subgroups within the Affymetrix data set that are comparable to those found in the Lymphochip data set. First, the relative proportions of ABC DLBCLs (29%) and GCB DLBCLs (53%) are very similar to the corresponding proportions in the Lymphochip data set (34% and 49%, respectively). Second, 43 genes were found to be differentially expressed between the two DLBCL subgroups with high significance (P < 0.001) based on the Affymetrix data (Fig. 3); this number of genes is many more than would be expected by chance given that the Affymetrix arrays measure the expression of ≈5,720 genes (based on Unigene, www.ncbi.nlm.nih.gov/UniGene). Third, this list includes 22 genes that were not used in the subgroup predictor but were represented on both the Affymetrix and Lymphochip microarrays; a majority of these (i.e., 14) were also found to be differentially expressed between the two subgroups within the Lymphochip data set with high statistical significance (P < 0.001). Finally, the expression of the c-rel gene was previously found to correspond to amplification of the c-rel genomic locus in DLBCL tumor cells, an oncogenic event occurring in GCB DLBCLs but not in ABC DLBCLs (2). Within the Affymetrix data set, c-rel was differentially expressed between the two subgroups (P = 0.0025) and was highly expressed only in a subset of GCB DLBCLs (Fig. 3). Although genomic DNA is unavailable from these tumors, the subdivision of the DLBCLs in the Affymetrix data set based on gene expression seems to have correctly segregated those tumors with c-rel amplification into the GCB DLBCL subgroup.
Fig. 3.

Prediction of DLBCL subgroups using gene expression measurements from oligonucleotide microarrays. The DLBCL subgroup predictor was used to discover ABC and GCB subgroups within 58 DLBCL biopsies that were profiled on Affymetrix microarrays (3). The probabilities that each sample belongs to the ABC or GCB subgroups are indicated. The differential expression of genes between the two subgroups is depicted according to the color scale in Fig. 1. Also shown for each gene are the P values derived from Wilcoxon rank-sum tests of the difference in expression between the two subgroups by using Affymetrix microarray data or Lymphochip microarray data, if available. N.S., P > 0.05. Asterisks indicate genes that were included in the DLBCL subgroup predictor.
Two previous studies of DLBCL have demonstrated that the DLBCL subgroups have different survival rates after chemotherapy (1, 2). As expected, the GCB and ABC DLBCL subgroups defined within the Lymphochip data set by the subgroup predictor had distinct 5-yr survival rates of 59% and 31%, respectively (Fig. 4; P = 6.5 × 10–6). The cases that remained unclassified by using the subgroup predictor had an intermediate 5-yr survival rate of 47%. The distinction between the GCB and ABC DLBCL subgroups was associated with a 2.32 relative risk of dying. Previously, the DLBCL subgroup distinction was made by hierarchical clustering (2), and by this method the relative risk of dying associated with this distinction was 2.17. Among cases that were previously classified as type 3 by using hierarchical clustering (2), those that were now classified as GCB or ABC DLBCL had 5-yr survival rates of 50% and 22%, respectively. Taken together, these results suggest that the Bayesian method has somewhat improved the stratification of DLBCL patients into clinically distinct subgroups.
Fig. 4.

Differences in survival between DLBCL subgroups. The Kaplan–Meier plots display the survival of patients in the GCB and ABC DLBCL subgroups defined by using gene expression data from Lymphochip (2) (Left) or Affymetrix (3) (Right) microarrays. A log-rank test was used to calculate the P values.
Within the Affymetrix data set, the overall survival rates of GCB and ABC DLBCL patients were also different (P = 5.1 × 10–3), with GCB and ABC DLBCL patients having 5-yr survival rates of 62% and 26%, respectively (Fig. 4). In this independent set of patients, the relative risk of dying associated with the DLBCL subgroup distinction was 2.75.
Discussion
We have developed a statistical method that can define cancer subgroups based on gene expression differences irrespective of which DNA microarray platform is used. Using this method, we identified two subgroups of DLBCL within independent gene expression data sets generated by using spotted cDNA and oligonucleotide microarrays. This algorithm could form the basis of a robust diagnostic test that may prove useful in assessing the results of therapeutic trials in DLBCL, given that these two DLBCL subgroups are both biologically and clinically distinct.
Hierarchical clustering was used in a previous attempt to uncover the ABC and GCB DLBCL distinction within the Affymetrix data set (3). The classification of the cases using this method differs substantially from the classification that we propose based on our subgroup predictor. By hierarchical clustering, 26 cases were classified as ABC DLBCL (3), but the subgroup predictor assigned 8 of these to the GCB DLBCL subgroup and found 6 to be unclassified. Conversely, among the 32 cases classified by hierarchical clustering as GCB DLBCL (3), 5 were assigned by the subgroup predictor to the ABC DLBCL subgroup and 4 were unclassified. The difference between these two classifications can be traced, in part, to differences in the genes used by each method. The hierarchical clustering classification used a set of genes that were differentially expressed between the two DLBCL subgroups in a pilot study of 42 cases (1). However, because many of the best DLBCL subgroup discrimination genes were absent from the Affymetrix arrays, the set of genes used for hierarchical clustering was enriched for those that distinguished the subgroups with lower statistical significance. By contrast, our subgroup predictor used only 14 of the genes that best discriminated the subgroups in a comprehensive study of 274 DLBCL cases (2). Further, our subgroup predictor allows for cases to be “unclassified” whereas the hierarchical clustering method assigned all cases to one of the two subgroups. DLBCL includes some cases that do not bear the hallmarks of either GCB or ABC DLBCL (2), and this possibility is better accommodated by our subgroup predictor.
The classification of the DLBCLs in the Affymetrix data set by our subgroup predictor recapitulates the previously published distinction between the DLBCL subgroups in several respects. First, many of the genes that relate the DLBCL subgroups to distinct stages of normal B cell differentiation (Fig. 2) are differentially expressed between the subgroups defined by our predictor. Second, in the Affymetrix data set, the c-rel gene was found to be highly expressed in a subset of GCB DLBCLs but not in ABC DLBCLs, consistent with the previous finding that the c-rel locus is amplified only in GCB DLBCLs (2). Finally, the GCB and ABC subgroups defined in the Affymetrix data set had significantly different survival rates after chemotherapy, as previously described for two other sets of DLBCL patients (1, 2). In this regard it is notable that protein kinase C β1 (PKCβ1) was previously shown to be more highly expressed in DLBCLs that were not cured by chemotherapy (3). In the present analysis, PKCβ1 was found to be more highly expressed in ABC DLBCLs than in GCB DLBCLs within the Lymphochip data set (P = 2.5 × 10–15) and within the Affymetrix data set (P = 2.1 × 10–4). Therefore, the association between PKCβ1 expression and poor prognosis is likely to reflect its preferential expression in ABC DLBCL, the subgroup with the inferior survival rate. Taken together, these data demonstrate that the GCB and ABC subgroups discovered in the Affymetrix data set share biological, pathogenetic, and clinical features with the corresponding subgroups defined in the Lymphochip data set.
As proposed previously and confirmed in the present study, the GCB DLBCL subgroup bears extensive gene expression similarity to normal germinal center B cells. This finding, together with the observation that GCB DLBCLs have ongoing somatic hypermutation of their Ig genes (9), suggests that these DLBCLs originate from germinal center B cells and retain many of their biological features. The cell of origin of ABC DLBCLs is more elusive. We found that many plasma cell-restricted genes were also preferentially expressed in ABC DLBCLs. Several of these genes encode resident ER and golgi proteins, suggesting that these DLBCLs have an enhanced secretory apparatus. The higher expression of XBP-1 in ABC DLBCLs is consistent with this hypothesis because it encodes a transcription factor that regulates the unfolded protein response in the ER. These considerations suggest that ABC DLBCLs may be derived from a B cell that is in the process of plasmacytic differentiation, such as the BCL-6–, IRF-4+ cells in the germinal center (10). Indeed, ABC DLBCLs have lower BCL-6 expression than GCB DLBCLs and express IRF-4 (Fig. 1), in keeping with this hypothesis. However, it is also possible that ABC DLBCLs may derive from another type of B cell that undergoes somatic hypermutation and plasmacytic differentiation outside of the germinal center (11, 12).
Many different methods have been formulated to predict cancer subgroups (4, 13–15). These methods assign tumors to one of two subgroups based on expression of a set of differentially expressed genes but do not provide a probability of membership in a subgroup. By contrast, our method uses Bayes' rule to estimate this probability, thus allowing one to vary the probability cutoff for assignment of a tumor to a subgroup. In tumor types in which unknown additional subgroups may exist, our method allows samples that do not meet the gene expression criteria of known subgroups to fall into an unclassified group with intermediate probability. A cancer subgroup predictor of the type we describe could potentially be used clinically to provide quantitative diagnostic information for an individual cancer patient.
Acknowledgments
We thank Sandra Weller and Jean-Claude Weill for help in purification of B cell populations.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GC, germinal center; GCB, GC B cell-like; DLBCL, diffuse large B cell lymphoma; ABC, activated B cell-like; LPS, linear predictor score; ER, endoplasmic reticulum.
References
- 1.Alizadeh, A. A., Eisen, M. B., Davis, R. E., Ma, C., Lossos, I. S., Rosenwald, A., Boldrick, J. C., Sabet, H., Tran, T., Yu, X., et al. (2000) Nature 403, 503–511. [DOI] [PubMed] [Google Scholar]
- 2.Rosenwald, A., Wright, G., Chan, W. C., Connors, J. M., Campo, E., Fisher, R. I., Gascoyne, R. D., Muller-Hermelink, H. K., Smeland, E. B., Giltnane, J. M., et al. (2002) N. Engl. J. Med. 346, 1937–1947. [DOI] [PubMed] [Google Scholar]
- 3.Shipp, M. A., Ross, K. N., Tamayo, P., Weng, A. P., Kutok, J. L., Aguiar, R. C. T., Gaasenbeek, M., Angelo, M., Reich, M., Pinkus, G. S., et al. (2002) Nat. Med. 8, 68–74. [DOI] [PubMed] [Google Scholar]
- 4.Radmacher, M. D., McShane, L. M. & Simon, R. (2002) J. Comput. Biol. 9, 505–511. [DOI] [PubMed] [Google Scholar]
- 5.Hills, M. (1966) J. R. Stat. Soc. B 28, 1–31. [Google Scholar]
- 6.Shaffer, A. L., Rosenwald, A., Hurt, E. M., Giltnane, J. M., Lam, L. T., Pickeral, O. K. & Staudt, L. M. (2001) Immunity 15, 375–385. [DOI] [PubMed] [Google Scholar]
- 7.Reimold, A. M., Iwakoshi, N. N., Manis, J., Vallabhajosyula, P., Szomolanyi-Tsuda, E., Gravallese, E. M., Friend, D., Grusby, M. J., Alt, F. & Glimcher, L. H. (2001) Nature 412, 300–307. [DOI] [PubMed] [Google Scholar]
- 8.Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., Clark, S. G. & Ron, D. (2002) Nature 415, 92–96. [DOI] [PubMed] [Google Scholar]
- 9.Lossos, I. S., Alizadeh, A. A., Eisen, M. B., Chan, W. C., Brown, P. O., Botstein, D., Staudt, L. M. & Levy, R. (2000) Proc. Natl. Acad. Sci. USA 97, 10209–10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falini, B., Fizzotti, M., Pucciarini, A., Bigerna, B., Marafioti, T., Gambacorta, M., Pacini, R., Alunni, C., Natali-Tanci, L., Ugolini, B., et al. (2000) Blood 95, 2084–2092. [PubMed] [Google Scholar]
- 11.Weller, S., Faili, A., Garcia, C., Braun, M. C., Le Deist, F. F., de Saint Basile, G. G., Hermine, O., Fischer, A., Reynaud, C. & Weill, J. (2001) Proc. Natl. Acad. Sci. USA 98, 1166–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.William, J., Euler, C., Christensen, S. & Shlomchik, M. J. (2002) Science 297, 2066–2070. [DOI] [PubMed] [Google Scholar]
- 13.Golub, T. R., Slonim, D. K., Tamayo, P., Huard, C., Gaasenbeek, M., Mesirov, J. P., Coller, H., Loh, M. L., Downing, J. R., Caligiuri, M. A., et al. (1999) Science 286, 531–537. [DOI] [PubMed] [Google Scholar]
- 14.Ramaswamy, S., Tamayo, P., Rifkin, R., Mukherjee, S., Yeang, C. H., Angelo, M., Ladd, C., Reich, M., Latulippe, E., Mesirov, J. P., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 15149–15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudoit, S., Fridlyand, J. & Speed, T. P. (2002) J. Am. Stat. Assoc. 97, 77–87. [Google Scholar]