

Abstract
eIF3 in mammals is the largest translation initiation factor (~800 kDa) and is composed of 13 nonidentical subunits designated eIF3a–m. The role of mammalian eIF3 in assembly of the 48 S complex occurs through high affinity binding to eIF4G. Interactions of eIF4G with eIF4E, eIF4A, eIF3, poly(A)-binding protein, and Mnk1/2 have been mapped to discrete domains on eIF4G, and conversely, the eIF4G-binding sites on all but one of these ligands have been determined. The only eIF4G ligand for which this has not been determined is eIF3. In this study, we have sought to identify the mammalian eIF3 subunit(s) that directly interact(s) with eIF4G. Established procedures for detecting protein-protein interactions gave ambiguous results. However, binding of partially proteolyzed HeLa eIF3 to the eIF3-binding domain of human eIF4G-1, followed by high throughput analysis of mass spectrometric data with a novel peptide matching algorithm, identified a single subunit, eIF3e (p48/Int-6). In addition, recombinant FLAG-eIF3e specifically competed with HeLa eIF3 for binding to eIF4G in vitro. Adding FLAG-eIF3e to a cell-free translation system (i) inhibited protein synthesis, (ii) caused a shift of mRNA from heavy to light polysomes, (iii) inhibited cap-dependent translation more severely than translation dependent on the HCV or CSFV internal ribosome entry sites, which do not require eIF4G, and (iv) caused a dramatic loss of eIF4G and eIF2α from complexes sedimenting at ~40 S. These data suggest a specific, direct, and functional interaction of eIF3e with eIF4G during the process of cap-dependent translation initiation, although they do not rule out participation of other eIF3 subunits.
Eukaryotic translation initiation involves numerous initiation factors (eIFs)2 that participate in recruitment of initiator tRNA and mRNA to the 40 S ribosomal subunit, recognition of the initiator AUG codon, and joining of the 40 S and 60 S ribosomal subunits, culminating in formation of the first peptide bond (1). The factors required for recruitment of mRNA include eIF3, eIF4A, eIF4B, eIF4E, eIF4G, eIF4H, and PABP. eIF4E and PABP bind the 5′ cap and 3′ poly(A) tract of mRNA, respectively, whereas eIF4A unwinds 5′-terminal secondary structure in an ATP-dependent process that also involves the RNA-binding proteins eIF4B and eIF4H. eIF4G forms specific complexes with eIF4E, eIF4A, and PABP, thereby linking the processes of cap recognition, poly(A) binding, and secondary structure melting. eIF4G in turn is recruited to the 40 S ribosomal subunit via binding to the multisubunit complex eIF3.
eIF3 in mammals is the largest initiation factor (~800 kDa) and includes 13 nonidentical polypeptides designated eIF3a-m3 (1–4). In contrast, eIF3 in Saccharomyces cerevisiae contains only six subunits (eIF3a, -b, -c, -g, -i, and -j) (1, 5). Five of these (eIF3a, -b, -c, -g, and -i) are conserved in all eukaryotes and are considered to be the “conserved core” of eIF3 (2, 6). Most of the eight mammalian “non-core” subunits are found in other higher eukaryotes, Schizosaccharomyces pombe lacking only eIF3j, -k, and -l; Triticum aesitivum lacking eIF3j, -l, and -m; Arabidopsis thaliana lacking eIF3j and -m; and Drosophila melanogaster and Caenorhabditis elegans lacking eIF3m (4, 7–9). In addition to subunit composition, subunit-subunit interactions have been extensively characterized for both mammalian (10–12) and S. cerevisiae eIF3 (13).
Although the subunit compositions of S. cerevisiae and mammalian eIF3 are different, the functions of the core subunits appear to be the same, as suggested by the observation that the S. cerevisiae conserved core can substitute for mammalian eIF3 in an assay for Met-puromycin synthesis containing all other eIFs of mammalian origin (6). eIF3 is required for both 43 S and 48 S initiation complex formation and is also involved in dissociation of 80 S complexes and anti-association of 40 S and 60 S subunits (1). The 43 S complex consists of eIF3, eIF2·GTP·Met-tRNAi, eIF5, eIF1, eIF1A, and the 40 S ribosomal subunit (1). In S. cerevisiae, a multifactor complex has been identified consisting of eIF3, eIF2·GTP·Met-tRNAi, eIF5, and eIF1 (14). eIF3 stabilizes this complex through direct interactions with eIF1, eIF2, eIF5, and the 40 S ribosomal subunit (15). Most of these interactions have been mapped to individual eIF3 subunits. For example, eIF3a and eIF3c bind to eIF1 (14, 16), eIF3c binds to eIF5 (14), and eIF3a binds to eIF2β (16). eIF3 also binds eIF1A (17), although the subunit that mediates this interaction has not been reported. In mammals, eIF3 binds eIF1 (through eIF3c) (18) and eIF5 (subunit unknown) (19). eIF3 contacts the 40 S ribosomal subunit through eIF3b and -j in mammals (10) and S. cerevisiae (13). Finally, eIF3a in mammals (20) and eIF3g in plants (21) and S. cerevisiae (22) interacts with eIF4B.
Mammalian eIF3 participates in assembly of the 48 S complex through high affinity binding to eIF4G (23). This may not be the case for S. cerevisiae, where a requirement for eIF4G in mRNA recruitment to the 43 S complex has not been demonstrated (24), correlating with the lack of an observed eIF3-eIF4G interaction in this organism. Binding of eIF4G to eIF3 in mammals is regulated by insulin via association of mTOR with eIF3, specifically eIF3f (25, 26). Interactions with eIF3, eIF4E, eIF4A, PABP, and Mnk1/2 have been mapped to separate sites on eIF4G (27–32). The minimum portion of eIF4G that retains full affinity for eIF3 is located between amino acids 1015 and 1118 (23). The eIF4G-binding sites on all but one of these ligands have also been determined, viz. eIF4E (30, 33, 34), eIF4A (35), Mnk1 (32), Mnk2 (36), and PABP (29, 37, 38). The only eIF4G ligand for which this has not been determined is eIF3. However, the recently solved cryo-EM structure of an eIF4G·eIF3·40 S complex suggests a small surface contact between eIF4G and eIF3 (39), likely composed of one or a few eIF3 subunits.
The interaction between eIF3 and eIF4G is critical because it is essential for bringing mRNA and all of the proteins bound to eIF4G to the 43 S initiation complex. In this study, we have sought to identify the mammalian eIF3 subunit(s) that directly interact with eIF4G. Binding of peptides from partially proteolyzed eIF3 suggests one subunit, eIF3e, is more involved than the other 12. Adding FLAG-tagged eIF3e to a cell-free translation system decreases the overall rate of protein synthesis and polysome accumulation. FLAG-eIF3e inhibits eIF4G-dependent translation significantly more than eIF4G-independent (IRES-driven) translation. Finally, FLAG-eIF3e causes a dramatic reduction in the amount of eIF4G and eIF2 associated with complexes sedimenting at ~40 S. These functional activities would be expected for an eIF3 subunit involved in eIF4G binding.
EXPERIMENTAL PROCEDURES
Materials
An ECL+ Western blotting development kit and Low Range RainbowTM molecular weight markers were obtained from Amersham Biosciences. Sf9 cells and Sf-900 II SFM media were obtained from Invitrogen. [35S] Met was obtained from MP Biomedicals (Irvine, CA). S-protein-agarose and S-protein-coupled alkaline phosphatase was purchased from Novagen (Madison, WI). Beetle luciferin, mass spectrometry grade trypsin gold, RQ1 RNase-free DNase, and all DNA restriction enzymes were purchased from Promega. Ni2+-nitrilotriacetic acid-agarose was obtained from Qiagen (Chatsworth, CA). RNase A, EZView Red anti-FLAG M2 affinity gel, mouse monoclonal anti-FLAG M2 antibody, and FLAG peptide were from Sigma. pXL.HCV(40 –373).NS′ and pXL.CSFV(1– 442). NS′ were described previously (40, 41) and were kindly provided by Tatyana Pestova (State University of New York, Brooklyn).
Protein Expression and Purification
The plasmids encoding recombinant His6- and S-peptide-tagged eIF4G(1015–1118), 4 eIF4G(653–1118), and eIF4G(1118–1600) and purification of S-eIF4G(653–1118), and S-eIF4G(1118–1600) have been described previously (42). S-eIF4G(1015–1118) was expressed in Escherichia coli strain BL21(DE3) pLysS (Novagen) and purified by Ni2+-nitrilotriacetic acid-agarose chromatography. One-liter cultures were grown at 37 °C to A600 0.4–0.6, and expression was induced with 1 mM isopropyl β-D-thiogalactoside for 3.5 h. Cell pellets were harvested and stored at −80 °C. Thawed cell pellets were resuspended in lysis buffer containing Complete Mini EDTA-free Protease Inhibitor tablets (Roche Applied Science) and disrupted by sonication. Cleared extracts were incubated in batch with Ni2+-nitrilotriacetic acid-agarose (1 ml packed) with rotation for 2 h at 4 °C. Bound material was washed five times batchwise with 20 ml of Buffer A (20 mM Tris-HCl, 200 mM KCl, 10% glycerol, 20 mM imidazole, pH 7.6) and twice with 20 ml of Buffer B (same as Buffer A except 500 mM KCl), after which the resin was equilibrated in 10 ml of Buffer C (same as Buffer A except 100 mM KCl) and loaded into a gravity flow column for elution. S-eIF4G(1015–1118) was eluted with 5ml of Buffer D (same as Buffer C except 300 mM imidazole), and 1-ml fractions were collected. Fractions containing purified S-eIF4G(1015–1118) were passed over Econo-Pac© 10 DG columns (Bio-Rad) to exchange Buffer D with Buffer E (20 mM HEPES-KOH, 100 mM KCl, 10% glycerol, 1 mM EDTA, 2 mM DTT, pH 7.4).
Recombinant baculoviruses for expression of FLAG-tagged eIF3 subunits, inserted into the FLAG-FastBac donor vector, were prepared as described previously (10, 11). Sf9 cells from frozen stocks were grown in 250-ml sterile flasks at 27 °C and 135 rpm in Sf-900 II SFM media in a New Brunswick Scientific Innova® 44 orbital shaker. For expression of recombinant proteins, cells were expanded to ~2 × 106 cells/ml in 50 ml of Sf-900 II SFM media prior to infection with recombinant baculoviruses expressing FLAG-eIF3e, FLAG-eIF3i, or FLAG-eIF3j. Cells were harvested after 72 h by centrifugation at 3000 rpm for 5 min followed by a single wash with 1× phosphate-buffered saline. A single cell pellet from each 50-ml culture was stored at −80 °C. Each thawed cell pellet was incubated on ice for 15 min, with occasional mixing, in 25 ml of Buffer F (20 mM Tris-HCl, 120 mM KCl, 10% glycerol, 5 mM β-mercaptoethanol, 1% Triton X-100, pH 7.5) containing one Complete Mini EDTA-free Protease Inhibitor tablet. Uncleared lysates for two pellets representing the same eIF3 subunit were then mixed and centrifuged at 10,000 × g for 20 min. Cleared lysates were incubated batch-wise with EZView Red Anti-FLAG M2 affinity gel (100 μl packed), equilibrated in Buffer F, for 2 h at 4 °C. The resin was washed three times in batch with 1 ml of Buffer F. FLAG-tagged proteins were eluted with three sequential 100-μl aliquots of Buffer G (20 mM Tris-HCl, 70 mM KCl, 10% glycerol, 1 mM DTT, 2 mM magnesium acetate, 200 μg/ml FLAG peptide, pH 7.5) at 4 °C for 45 min each. Recombinant protein purity was assessed by SDS-PAGE followed by Coomassie Blue staining and anti-FLAG Western blotting.
eIF3 from RRL (23) and HeLa cells (10) was purified as described previously. The concentrations of purified proteins were determined with the protein assay kit (Bio-Rad) with BSA as standard.
In Vitro Transcription
Unlabeled capped mRNAs were synthesized in vitro as described previously (43) except that the final volume of reactions was 100 μl; 1.5 μg of template DNA was used; and the final concentration of m7GpppG was 1.0 mM. The templates used were EcoRI-linearized pXL.HCV(40–373).NS′, EcoRI-linearized pXL.CSFV(1–442).NS′, HpaI-linearized pluc-A+ (43), and HpaI-linearized pluc-A60 (44). The mRNAs generated from these four templates are referred to as m7G-CycB2-HCV-NS′, m7G-CycB2-CSFV-NS′, m7G-Luc-A31, and m7G-Luc-A60, respectively. 32P-Labeled nucleotides were included when the pluc-A60 template was used to make 32P-labeled mRNA for measurement of polysomal distribution. Following incubation for 45 min at 37 °C, fresh T7 RNA polymerase was added, and the reaction was continued for an additional 45 min at 37 °C. DNA was digested by addition of 3.0 units of RQ1 RNase-Free DNase followed by incubation for 10 min at 65 °C. mRNA was then purified using an RNeasy mini kit (Qiagen). mRNA concentration and purity were assessed by absorbance at 260 and 280 nm.
In Vitro Translation
All in vitro translation reactions were performed at 30 °C with a micrococcal nuclease-treated RRL system (45). For measurement of translational inhibition utilizing the luciferase assay, m7G-Luc-A60 and nonradioactive amino acids were used. For measurement of translational inhibition utilizing m7G-CycB2-HCV-NS′ or m7G-CycB2-CSFV-NS′ bicistronic mRNAs, nonradioactive Met was omitted and [35S]Met was included at 0.5 μCi/μl. For time course measurements, reactions contained m7G-Luc-A31 and [35S]Met at 0.2 μCi/μl. When inhibition of protein synthesis was measured, FLAG-tagged eIF3 subunits and nonradiolabeled mRNA were added simultaneously. When polysomal distribution of mRNA was determined, reaction mixtures containing nonradiolabeled amino acids were preincubated with FLAG-tagged eIF3 subunits for 15 min at 30 °C prior to addition of 32P-labeled m7G-Luc-A60.
In all reaction mixtures, the volume of added Buffer G (except lacking FLAG peptide) was kept constant. For measurement of luciferase synthesis in the presence of FLAG-eIF3e, luciferase assays were performed using beetle luciferin following the manufacturer’s protocol (Promega). For inhibition of bicistronic mRNA translation, 2 μl of each reaction were analyzed by SDS-PAGE on 15% gels followed by Coomassie Blue staining. Newly synthesized protein was detected by exposure of each dried gel to a PhosphorScreen for 2 h and scanning with a STORM 860 PhosphorImager (Amersham Biosciences). Cyclin B2 (~50 kDa) and NS′ (~20 kDa) were quantitated with ImageQuant software, version 5.0 (Amersham Biosciences). Curves were fit, as if for competitive inhibition of an enzymatic reaction, with Kaleidagraph software (version 3.5; Synergy Software, Reading, PA) using the equation y = m1/(1 + M0/m2), where y = synthesis of cyclin B2, NS′, or luciferase in the presence of a FLAG-tagged eIF3 subunit; m1 = cyclin B2, NS′, or luciferase synthesized in the absence of the eIF3 subunit; M0 = concentration of the eIF3 subunit in μM, and m2 = the inhibition constant (KI). For time course measurements of inhibition of m7G-Luc-A31 mRNA translation, 10-μl aliquots of each reaction were removed at the indicated times and diluted 1:10 in 10 μg/ml BSA, 10 mM Met, and 10 μM cycloheximide on ice. Aliquots of 5 μl were spotted on Whatman 540 filter disks, and total trichloroacetic acid-precipitatable 35S radioactivity was measured by scintillation spectrometry.
Ultracentrifugal Analysis of Initiation Complexes and Polysomes
For polysomal distribution of radiolabeled mRNA, 100-μl translation reaction mixtures were layered immediately after incubation on a 10-ml 10–35% linear sucrose density gradient in Buffer K (50 mM Tris-HCl, 50 mM KCl, 10 mM MgCl2, 1 mM DTT, 50 μg/ml cycloheximide, pH 7.5) and centrifuged at 38,000 rpm in a Beckman SW41Ti rotor at 4 °C for 3 h. Fractions of 250 μl were collected using an ISCO gradient fractionator with continuous monitoring at 260 nm. The presence of 32P-labeled mRNA in each fraction was detected by Cerenkov radiation. For analysis of factors in the 40 S region, 100-μl translation reaction mixtures were incubated for 15 min in the absence or presence of FLAG-tagged eIF3e. Reaction mixtures were layered on a 10-ml 10–35% linear sucrose density gradient in Buffer L (20 mM HEPES-KOH, 100 mM KCl, 5 mM MgCl2, 2 mM DTT, pH 7.5) and centrifuged as above except for 5 h. Fractions of 500 μl were collected and immediately precipitated with 10% trichloroacetic acid overnight with 0.1 mg/ml linear polyacrylamide as carrier. Following centrifugation at 14,000 rpm for 20 min, pellets were washed twice with ice-cold 80% acetone, resuspended in 15 μl of 1× SDS-loading buffer, heated at 95 °C for 5 min, and proteins resolved by SDS-PAGE on 8% gels.
RNA Isolation and Real Time PCR
RNA was isolated from sucrose gradient fractions by treatment with 0.5% SDS and 40 μg/ml proteinase K, extraction with phenol/chloroform/ isoamyl alcohol (25:24:1) and chloroform/isoamyl alcohol (24: 1), and precipitation with 1.5 M LiCl and an equal volume of isopropyl alcohol overnight. Following centrifugation at 14,000 rpm for 20 min, pellets were washed once with 500 μl ice-cold 70% ethanol and resuspended in 20 μl of diethylpyrocarbonate-treated water. RNA was treated with RQ1 RNase-free DNase and then subjected to reverse transcription with random primers and reverse transcriptase from the TaqMan® reverse transcription reagents kit (Applied Biosystems), following the manufacturer’s protocol. Quantitative real time PCR was performed with primers specific for 18 S rRNA (forward, 5′-CGAGCCGC-CTGGATACC-3′; reverse, 5′-CAGTTCCGAAAACCAACA-AAATAG-3′). Products were amplified and detected with an iCycler IQ instrument and IQ SYBR Green Supermix (Bio-Rad). To calculate polysomal shifts, the threshold cycle (CT) of each reaction was subtracted from the CT of a control reaction lacking cDNA (ΔCT). Relative mRNA levels were calculated as 2ΔCT and graphically represented as the percentage of total 18 S rRNA present in each gradient.
Partial Trypsin Digestion of eIF3 and Binding of Fragments to S-eIF4G(1015–1118)
A reaction mixture (175 μl) containing 1.0 μM HeLa eIF3 and 43 μg/ml trypsin in Buffer H (20 mM Tris-HCl, 100 mM KCl, 10% glycerol, pH 7.6) was incubated at 25 °C. Aliquots containing ~32 μg of total HeLa eIF3 were removed at various times, trypsin inhibitor was added to each aliquot at a final concentration of 80 μg/ml, and samples were placed on ice for 10 min. S-eIF4G(1015–1118) was added to each sample to a final concentration of 3.0 μM in a final volume of 40 μl, and reactions were incubated with rotation at 4 °C for 45 min. Proteins were mixed with at least a 10-fold molar excess of S-protein agarose, previously equilibrated in Buffer I (20 mM Tris-HCl, 100 mM KCl, 10% glycerol, 1 mM EDTA, 2 mM DTT, 0.1% Tween 20, pH 7.6) and 0.3 mg/ml BSA. Following incubation for 1 h at 4 °C, the resin was washed three times with 100-μl aliquots of Buffer I. Samples bound to the resin were then eluted in 15 μl of 1×SDS-loading buffer and heated at 95 °C for 5 min. Acrylamidation of Cys residues was performed as described previously (46). Proteins were then resolved by SDS-PAGE on a pre-cast NOVEX 4–20% Tris-glycine polyacrylamide gel (Invitrogen) and visualized by Coomassie Blue staining. Prior to in-gel trypsin digestion of proteins and MALDI-TOF-MS analysis, each lane from the stained gel was cut into ~35 slices of equal width for a total of ~210 slices.
MALDI-TOF-MS Analysis of eIF3 Fragments
Mass spectrometric analysis was performed in the Louisiana State University Health Sciences Center-Shreveport Research Core Facility on a PerSeptive Biosystems Voyager-DE PRO Biospectrometry work station. Gel slices were minced and completely destained with three washes (400 μl each) of 50% acetonitrile, 25 mM NH4HCO3, pH 8.0. The polyacrylamide was dehydrated for 5 min with 100% acetonitrile followed by vacuum centrifugation in a Savant Speedvac for 30 min. In-gel digestion with trypsin was performed overnight at 37 °C by addition of 15 μl of enzyme (10 μg/ml) in 25 mM NH4HCO3, pH 8.0, to each gel slice. Peptides were extracted from gel slices twice with 30-μl portions of 50% acetonitrile and 5% trifluoroacetic acid. The extract was dried, dissolved in 10 μl of 0.1% trifluoroacetic acid, and purified on a ZipTip (C18; Millipore Corp., Bedford, MA). Peptides were eluted from the ZipTip with 3 μl of 10 mg/ml α-cyano-4-hydroxy-trans-cinnamic acid (Sigma) in 50% aceto-nitrile, 0.1% trifluoroacetic acid containing 0.8 ng/μl Calibration Mixture 1 (SequazymeTM peptide mass standards kit, Applied Biosystems) for internal calibration, and spotted on a MALDI plate. Data were summed over 100 acquisitions in delayed extraction mode. Data analysis was performed using Data Explorer software, version 4.0 (Applied Biosystems). Spectra were subjected to algorithms for base-line correction and noise removal at two S.D. values. Spectra were internally calibrated using one or more of the peptides present in Calibration Mixture 1 (des-Arg1-bradykinin, 904.4681 Da; angiotensin I, 1296.6853 Da; Glu1-fibrinopeptide B, 1570.6774 Da) and a trypsin autodigestion peak (842.5100 Da). Only masses ranging from 800 to 2800 Da in the generated spectra were utilized for peptide identification. Theoretical peptide masses arising from 0, 1, and 2 missed tryptic cleavages for all 13 human eIF3 subunits and S-eIF4G(1015–1118) were determined using the Peptide Mass tool at the ExPASy Proteomics site for a total of 3149 theoretical peptides. Theoretical masses for Cys-containing peptides were calculated with complete acrylamidation.
Peptide Mass Matching and Peptide Mapping Algorithm
To process the mass spectrometry data, we developed a suite of tools based on Perl scripting language (version 5.8.5) and the Gnuplot data and function plotting utility (version 4.0). Perl was used to write data processing scripts and to generate input files consisting of Gnuplot calls, which in turn produced visual representations of the results. The tab-delimited MALDI-TOF-MS outputs (peaks lists, which give masses of all peptides found after deisotoping and noise reduction) were imported into the software suite, one file for each of the ~210 gel slices. The files were imported in batch mode, as they were all located in one directory, and the suite automatically detected and processed all the files in a selected directory. The software proceeded by identifying the best match between an actual peak mass from the MALDI-TOF-MS output and an entry in the list of theoretical peptide masses. A variable window of permissible mass difference was used in the matching algorithm to accommodate experimental error in mass detection. If only one predicted peptide was identified within this window, that peptide was named as the best match. If several predicted peptides fit within this window, the one with smallest absolute difference was selected as the best match. Most of the data presented in this study were generated using a window of ±100 ppm. Another parameter that could be set was the number of missed tryptic cleavages. In this study, we found that the best signal-to-noise ratio was obtained when the number of missed tryptic cleavages was set to 0. A visual representation was then built in which each matched peptide was mapped to the corresponding protein from which it was derived. The data processing was performed on an SuSE 9.3 Linux-based server running on a dual Intel Xeon 3.06 GHz system equipped with 1 gigabyte of memory and 160-gigabyte local hard disk.
Competition of eIF3 Binding by and Direct Binding of FLAG-tagged eIF3 Subunits to eIF4G
For in vitro competition, 20-μl reaction mixtures contained equimolar concentrations of S-eIF4G(1015–1118) and HeLa eIF3, to which was added FLAG-tagged eIF3 subunits to achieve molar ratios of 1:1, 2:1, and 4:1 (subunit/eIF3). Each reaction mixture contained a final concentration of 0.3 mg/ml BSA and a constant amount of Buffer G (except lacking FLAG peptide). Reaction mixtures were kept at 4 °C for 2 h with rotation. A 10-fold molar excess of S-protein agarose, pre-equilibrated in the same buffer, was added, and rotation continued at 4 °C for 1 h. The resin was washed three times with 100 μl of Buffer G (except lacking FLAG peptide). Bound proteins were eluted in 20 μl of 1× SDS loading buffer, heated at 95 °C for 5 min, and resolved by SDS-PAGE on 8% gels. When testing the RNA dependence of direct interactions, reactions were set up as above but in the presence or absence of a final concentration of 20 μg/μl RNase A prior to pulldown with S-protein-agarose and SDS-PAGE analysis.
Western Blot Analysis
Proteins were transferred to an Immobilon-P membrane (Millipore) in a Mini Trans-Blot cell (Bio-Rad) at 100 V for 1 h (for in vitro competition experiments) or 30 V overnight (for sucrose density gradient analysis of initiation factors). Membranes were blocked in Buffer J (20 mM Tris-HCl, 150 mM NaCl, 0.05% Tween 20, pH 7.5) containing 5% milk proteins. Membranes were then washed four times briefly with Buffer J. Membranes were cut to expose different sections to various primary antibodies and incubated in Buffer J containing 1% milk proteins and a 1:1000 dilution of primary antibodies (rabbit polyclonal anti-eIF4G peptide 7 (47), goat polyclonal anti-eIF3 (48), rabbit polyclonal anti-eIF2α (Cell Signaling Technology), or mouse monoclonal anti-FLAG M2). Membranes were washed four times briefly with Buffer J and then incubated with Buffer J containing 1% milk proteins and a 1:1000 dilution of secondary antibody conjugated with either alkaline phosphatase or horseradish peroxidase, as indicated. Blots were developed with either the nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl-phosphate color development reagents (Promega) for alkaline phosphatase or ECL+ for horseradish peroxidase detection. Membranes were scanned with an HP Scanjet 3970 scanner (Hewlett Packard) for alkaline phosphatase or with a STORM 860 PhosphorImager for ECL+. Sometimes membranes were stripped by incubation in 62.5 mM Tris-HCl, 100 mM β-mercaptoethanol, 2% SDS, pH 6.7, for 30 min at 50 °C with agitation and then blocked and re-probed with antibodies as described above. Blocking and all incubations with primary and secondary antibodies were performed for 1 h at room temperature. Quantitation was performed with ImageQuant software as described above.
RESULTS
Initial Attempts to Identify a Subunit of eIF3 That Directly Interacts with eIF4G
We tried several approaches to identify an eIF4G-binding subunit of eIF3, but most of them produced ambiguous results. First, we used far Western analysis in which the subunits of rabbit eIF3 were resolved by SDS-PAGE and then exposed to S-eIF4G(653–1118),4 a recombinant fragment of human eIF4G that contains the eIF3-binding site as well as additional binding sites for eIF4A and RNA (42). Detection of bound S-eIF4G(653–1118) with S-protein-coupled alkaline-phosphatase revealed an interaction with eIF3b and/or eIF3c (which are not well resolved by SDS-PAGE), eIF3d, eIF3h, and eIF3i (data not shown). A similar experiment with 125I-labeled S-eIF4G(1015–1118), a smaller fragment of eIF4G that binds eIF3 with the same affinity but lacks the other ligand-binding sites (42), implicated the same subunits.
To identify individual subunits that interact with eIF4G, and because some eIF3 subunits are not well resolved on SDS-PAGE, we also performed far Western experiments with lysates from E. coli cells that expressed eIF3 subunits individually (49). This indicated that 125I-labeled S-eIF4G(1015–1118) bound to eIF3e, eIF3f, eIF3g, and eIF3i (data not shown). Larger subunits (eIF3a, eIF3b, and eIF3c) may have been missed by this procedure because they are partially insoluble when produced in E. coli and are also partially degraded during expression or purification.
eIF3 subunits are both stable and soluble when expressed from recombinant baculoviruses in Sf9 insect cells (10). We therefore used purified FLAG-tagged eIF3 subunits expressed in Sf9 cells to test for direct interaction with S-eIF4G(1015–1118) in vitro. We detected binding to eIF3a, eIF3b, eIF3c, eIF3d, eIF3e, and eIF3i (data not shown). Because the eIF3-binding domain of eIF4G is no larger than 103 amino acids (23), it is unlikely that eIF4G simultaneously and specifically binds to all six of these eIF3 subunits. These results suggest that some eIF3 subunits may lose binding specificity when not present in the native eIF3 heteromultimer, e.g. because of loss of a specific conformation or exposure of buried surfaces. This is supported by previously observed differences in eIF3 subunit solubility and complex formation in the presence of other eIF3 subunits (10).
Because of these ambiguous results with individual eIF3 subunits, we attempted experiments utilizing native, intact eIF3. Initially we used the photoaffinity labeling reagent sulfosuccin-imidyl-2-[p-azidosalicylamido]ethyl-1,3′-dithiopropionate (SASD; Pierce), which is a cleavable, photoreactive, and hetero-bifunctional cross-linker capable of radioiodination that has been successfully used to detect numerous protein-protein interactions (50–52). We derivatized S-eIF4G(653–1118) with 125I-labeled SASD and reacted it with eIF3 from RRL. This resulted in the radiolabeling of eIF3a, eIF3b/c, eIF3d/l, and eIF3e (data not shown). (SDS-PAGE was not sufficient to resolve eIF3b from eIF3c or eIF3d from eIF3l.) As a control, we used S-eIF4G(1118–1600), which lacks the eIF3-binding site, and detected no labeling of eIF3 subunits. Although it is possible that all four (or six) eIF3 subunits directly interact with eIF4G under these conditions, it is more likely that all nearby eIF3 subunits are labeled, regardless of whether they make direct contact with S-eIF4G(653–1118), because of the relatively long spacer arm of SASD (18.9 Å) and the 49 primary amines in S-eIF4G(653–1118) that could be derivatized by SASD. As a result, this approach may identify an eIF4G-proximal surface of eIF3 rather than subunits that directly contact eIF4G.
Binding of Partially Proteolyzed eIF3 to S-eIF4G(1015–1118)
These ambiguous results prompted us to seek an approach that utilized native eIF3 but that could also rule out eIF3 subunits not directly involved in eIF4G binding. Recently, we identified domains of eIF4A that bind eIF4G by first subjecting eIF4A to partial trypsinolysis and then affinity purifying the fragments with the tagged eIF4A-binding domain of eIF4G (35). We adopted the same approach for eIF3, subjecting the complex to trypsinolysis for varying lengths of time followed by affinity purification with the minimal eIF3-binding domain of eIF4G, S-eIF4G(1015–1118). The rationale was that, after short times of trypsinolysis, most or all eIF3 subunits would be captured because of both primary and secondary interactions, whereas after long times, no subunits would be captured because the peptides would be too small for eIF4G recognition. However, at some intermediary point, a partially digested form of eIF3 would exist that retains binding specificity for eIF4G yet lacks most or all of the nonbinding subunits. The overall approach was to digest HeLa eIF3 for various lengths of time, select fragments that retain binding to S-eIF4G(1015–1118) with S-protein-agarose, and resolve the bound peptides and proteins by SDS-PAGE (Fig. 1A). A control reaction without trypsin showed that all eIF3 subunits were bound (Fig. 1A, lane 1), whereas a control without eIF3 showed that no proteins except S-eIF4G(1015–1118) were present (lane 7). The amount of peptide material bound to S-eIF4G(1015–1118) progressively decreased with longer times of trypsinolysis (Fig. 1A, lanes 2–6).
FIGURE 1. MALDI-TOF-MS analysis of S-eIF4G(1015–1118)-bound tryptic fragments of eIF3.

A, binding of partially trypsinized HeLa eIF3 to S-eIF4G(1015–1118). eIF3 was incubated with trypsin as described under ”Experimental Procedures.“ Untrypsinized eIF3 (lane 1), aliquots of the eIF3 tryptic digest from 2 min (lane 2), 5 min (lane 3), 15 min (lane 4), 30 min (lane 5), and 60 min (lane 6), as well as an equivalent reaction without eIF3 (60 min; lane 7) were incubated with S-eIF4G(1015–1118) as described under ”Experimental Procedures“ and analyzed by SDS-PAGE with Coomassie Blue staining. B–J, MALDI-TOF-MS analysis of lane 1 (C–J) and lane 6 (B). Each lane was cut into ~2-mm slices, digested to completion with trypsin, and peptides analyzed by MALDI-TOF-MS as described under ”Experimental Procedures.“ Matched peptides (black bars) are shown below the polypeptides (gray bars) from which they are derived. The x-axis identifies the amino acid location in the sequence. The peptide mapping outputs of selected gel slices (A) are shown in B–J. GenBankTM accession numbers for amino acid sequences corresponding to eIF3 subunits are as follows: eIF3a (Q14152), eIF3b (P55884), eIF3c (Q99613), eIF3d (O15371), eIF3e (P60228), eIF3f (O00303), eIF3g (O75821), eIF3h (O15372), eIF3i (Q13347), eIF3j (O75822), eIF3k (NP_037366), eIF3l (AF077207), and eIF3m (NP_006351). The amino acid sequence for recombinant S-eIF4G(1015–1118) used in this experiment in B–J (4G) was generated in Vector NTI, Suite 9.0.0 (Informax; North Bethesda, MD).
Because eIF3 subunits would be partially degraded during this procedure, the fragments bound to S-eIF4G(1015–1118) could no longer be identified by mobility on SDS-PAGE. We therefore used MALDI-TOF-MS to identify either intact or partially digested eIF3 subunits by excising gel slices, subjecting protein to complete in-gel digestion with trypsin, determining peptide masses, and matching them with the peptides predicted from complete trypsinolysis of eIF3. Because between 20 and 200 MALDI-TOF-MS peaks were typically obtained from a single gel slice, and because each of the six lanes corresponding to different times of trypsin digestion were cut into 30–40 slices, the number of peaks to be matched numbered in the thousands. Likewise, the peptides that could theoretically arise from either 0, 1, or 2 missed tryptic cleavages of the 13 eIF3 subunits and S-eIF4G(1015–1118) totaled 3149. It therefore became clear that we needed a high throughput approach for matching peaks generated by MALDI-TOF-MS to the composite list of theoretical peptides. To this end we developed a new algorithm for peptide matching and visualization (see “Experimental Procedures”).
The list of 3149 theoretical peptide masses was contained in one file, whereas the MALDI-TOF-MS peaks lists from each gel slice were contained in separate files. This allowed us to execute the peptide mass matching algorithm in parallel on a Linux server to match peaks to any of the 13 eIF3 subunits or to S-eIF4G(1015–1118). Mass changes due to oxidized Met and acrylamidated Cys residues were included in the theoretical peaks list. To reduce background because of spectrum noise and erroneous assignments, the algorithm assigned a match only if the peak was >5% of the base peak (highest peak on a given MALDI-TOF-MS spectrum) and had a mass within a specified window (usually ±100 ppm, which corresponds to ~0.1 Da for peptides in the range of 800–2800 Da). Once the peak matching algorithm was completed, matched peaks were automatically mapped to their corresponding amino acid sequence positions in the identified polypeptide. The visualization portion of the algorithm was important because MALDI-TOF-MS detects only about half of peptides present, and peak mass accuracy is generally ±50 ppm (46). This leads to a small but significant proportion of erroneously matched peaks. Visualization of peptide origin, and in particular the detection of contiguous peptides, improves discrimination between signal and noise and allows more certain assignment of the eIF3 subunit of origin. The output of the algorithm displayed peptide matches for all 13 eIF3 subunits as well as S-eIF4G(1015–1118) (Fig. 1, B–J).
We demonstrated that each eIF3 subunit, if present, could be detected by this approach by analysis of each slice of lane 1 in Fig. 1A, which corresponds to undigested eIF3 bound to S-eIF4G(1015–1118). When the window was set at ±100 ppm, all eIF3 subunits were identified except eIF3d and eIF3j (Fig. 1, C–J), the latter of which were detected at ±200 ppm. Slice 6 (Fig. 1C) yielded a large number of peaks, which resulted in more nonspecific matches than in other slices, but more matched eIF3a (170 kDa) than any other eIF3 subunit. eIF3a has 159 theoretical peptides arising from 0 missed tryptic cleavages, 82 of which are within the mass range of 800–2800 Da. The algorithm identified 21 peaks as being of eIF3a origin, or 26% of the theoretical total. Slice 7 (Fig. 1D) contained both eIF3b (116 kDa) and eIF3c (110 kDa), which were incompletely resolved on the gel. The N-terminal half of eIF3a was also found in this slice, likely due to partial degradation. In slice 11, the algorithm identified eIF3l (69 kDa) when the window was set at ±100 ppm (Fig. 1E) and eIF3d (66 kDa) when it was set at ±200 ppm (data not shown). eIF3e (52 kDa) was identified in slice 13 (Fig. 1F), whereas portions of eIF3e and eIF3f (47 kDa) were found in slice 14 (Fig. 1G). Portions of eIF3f, eIF3g (44 kDa), eIF3h (40 kDa), and eIF3m (42 kDa) were identified in slice 15 (Fig. 1H). Slice 16 (Fig. 1I) contained portions of eIF3h as well as intact eIF3i (36 kDa). eIF3j (35 kDa) was found in slice 17 but only when the window was set at ±200 ppm (data not shown). Finally, eIF3k (28 kDa) was detected in slice 24 (Fig. 1J). Thus, all 13 subunits could be identified in various portions of the gel by this approach. In addition, S-eIF4G(1015–1118) was detected near slice 20 in each lane (data not shown).
We analyzed each of the ~210 gel slices obtained from the six different time intervals of trypsinolysis (Fig. 1A, lanes 2–6). These samples contained portions of all eIF3 subunits (data not shown), indicating that at early time points all eIF3 subunits bind to S-eIF4G(1015–1118), either directly or indirectly, because subunit-subunit contacts had not yet been disrupted. Interestingly, no polypeptides larger than 60 kDa remained in Fig. 1A, lanes 2–4, which shows that intact eIF3a, eIF3b, eIF3c, eIF3d, and eIF3l are not necessary for binding of eIF3 to S-eIF4G(1015–1118). By contrast, in all 69 slices from Fig. 1A, lanes 5 and 6, only eIF3e was identified (Fig. 1B). Two tryptic fragments were observed by Coomassie Blue staining (Fig. 1A, lanes 6 and 7), both of which were identified as being derived from eIF3e. The smaller fragment (slice 192) was ~35 kDa as compared with 52 kDa for full-length eIF3e. Peptide mapping indicated that some of the C-terminal peptides observed for intact eIF3e (Fig. 1, F and G) were not found in slice 192 (Fig. 1B), suggesting that only the N-terminal two-thirds of eIF3e was bound to S-eIF4G(1015–1118) after extensive trypsinolysis.
The matches for MALDI-TOF-MS peaks found in slice 192 are given in Table 1. Out of 13 total peptides matched in slice 192, eight were from eIF3e. The actual mass of one peak (2239.1224 Da) matched to eIF3e (amino acids 173–191; theoretical mass 2239.0369 Da), but it could also have arisen from a trypsin autodigestion peak (theoretical mass 2239.1358 Da), because both are within ±100 ppm of the actual mass. Two of the peptides matched that were not from eIF3e are likely to be artifacts. A peak of 904.4695 Da was matched to two theoretical peptides, one from eIF3f and one from eIF3h, but it is also very close to the internal calibrant des-Arg1-bradykinin (904.4681 Da). Excluding these likely artifacts, 7 of 8 peptides mapped to eIF3e, whereas only one each mapped to eIF3a and eIF3g, making the correct assignment of eIF3e highly likely.
TABLE 1.
Assignment of MALDI-TOF-MS peaks found in gel slice 192
This list of matched peptides, using our novel peptide matching algorithm, corresponds to those peptides matched for slice 192 (Fig. 1B). Accuracy is comparable with all gel slices in this experiment.
| Theoretical peptidea | Proteinb | Positionsc | Modificationd | Theoretical masse | Actual massf | Differenceg | Abundanceh | Notesi |
|---|---|---|---|---|---|---|---|---|
| aa | Da | ppm | % BP | |||||
| ITKPGSIDSNNQLFAPGGR | eIF4G | 221–239 | 1972.0246 | 1972.0124 | 6 | 11.66 | ||
| IAHFLDR | eIF3e | 10–16 | 871.4784 | 871.4886 | −12 | 9.85 | ||
| NLYSDDIPHALR | eIF3e | 60–71 | 1413.7121 | 1413.7102 | 1 | 29.73 | ||
| QEYLDTLYR | eIF3e | 125–133 | 1200.5895 | 1200.5955 | −5 | 7.56 | ||
| FQYECGNYSGAAEYLYFFR | eIF3e | 137–155 | Acr: 141 | 2399.0437 | 2399.1377 | −39 | 9.13 | |
| VLVPATDR | eIF3e | 156–163 | 870.5043 | 870.4962 | 9 | 9.51 | ||
| LASEILMQNWDAAMEDLTR | eIF3e | 173–191 | MSO: 179, 186 | 2239.0369 | 2239.1224 | −38 | 67.7 | Trypsin |
| ETIDNNSVSSPLQSLQQR | eIF3e | 194–211 | 2015.9992 | 2015.9832 | 8 | 15 | ||
| LFIFETFCR | eIF3e | 338–346 | Acr: 345 | 1246.6288 | 1246.6375 | −7 | 6.73 | |
| AEEQMLK | eIF3a | 818–824 | MSO: 822 | 864.4131 | 864.4868 | −85 | 9.99 | |
| IGVDLIMK | eIF3f | 247–254 | MSO: 253 | 904.5172 | 904.4695 | 53 | 39.95 | Calibrant |
| DTLGPMQK | eIF3g | 173–180 | MSO: 178 | 905.4397 | 905.4725 | −36 | 6.01 | |
| TAQGSLSLK | eIF3h | 157–165 | 904.5098 | 904.4695 | 45 | 39.95 | Calibrant | |
Amino acid sequence of the matched peptide.
Protein from which the matched peptide is derived. eIF4G refers to recombinant S-eIF4G-(1015–1118) used in this study.
Amino acid position of the theoretical peptide in the corresponding protein. Amino acid positions for eIF4G correspond to the recombinant protein.
Oxidation of methionine (MSO) or acrylamidation of cysteine (Acr) residues are accounted for in theoretical mass. Amino acid position of modified residues in the matched peptide is indicated.
The mass of the theoretical peptide (in Da) was generated by the Peptide Mass tool at the ExPASy Proteomics site on line.
Mass of an actual peak generated by MALDI-TOF-MS (in Da) that is matched to a particular theoretical peptide.
Calculated difference in theoretical and actual masses expressed as parts/million.
Height of the peak as compared with the most abundant peak in a particular spectrum generated by MALDI-TOF-MS.
Matched peptides that are also within 100 ppm of either trypsin autodigestion or internal calibrant peaks are indicated.
Free FLAG-eIF3e Competes with Binding of Native eIF3 to, and Interacts Directly with, S-eIF4G(1015–1118)
We sought confirmation that eIF3e binds directly to S-eIF4G(1015–1118) by two methods, direct binding and specific competition of intact native eIF3 binding. To this end, FLAG-tagged eIF3e, eIF3i, and eIF3j were purified from extracts of Sf9 cells infected with recombinant baculoviruses as described under “Experimental Procedures” (Fig. 2A). Fig. 2B shows that FLAG-eIF3e was pulled down by S-protein-agarose in the presence (lane 1) but not the absence (lane 3) of S-eIF4G(653–1118), which contains all regions of eIF4G involved in interaction with RNA. This binding did not depend on the presence of RNA because it was not altered when RNase A was included (Fig. 2B, lane 2).
FIGURE 2. Free FLAG-eIF3e binds specifically to eIF4G in vitro in an RNA-independent manner and competes with binding of native HeLa eIF3.
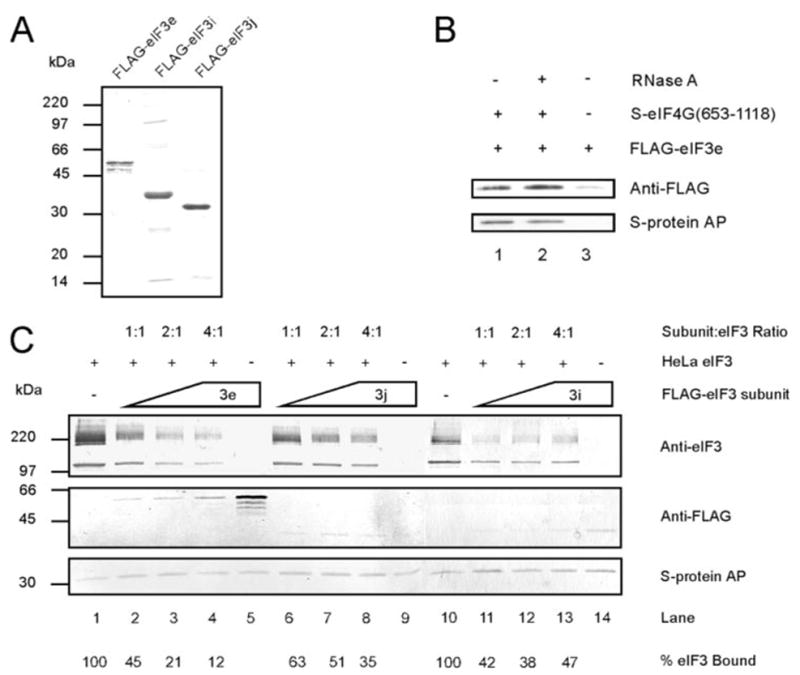
A, SDS-PAGE of purified FLAG-eIF3e, FLAG-eIF3i, and FLAG-eIF3j stained with Coomassie Blue. B, reactions contained 0.1 μM S-eIF4G(653–1118) and 0.4 μM FLAG-eIF3e in the absence (lane 1) or presence (lane 2) of 20 μg/μl RNase A. A control reaction contained 0.4 μM FLAG-eIF3e alone (lane 3). C, reactions contained equimolar concentrations of S-eIF4G(1015–1118) and HeLa eIF3 (0.2 μM) and increasing concentrations (0.2, 0.4, and 0.8 μM) of either FLAG-eIF3e (lanes 2– 4), FLAG-eIF3j (lanes 6 – 8), or FLAG-eIF3i (lanes 11–13). Separate reactions contained only 0.2 μM S-eIF4G(1015–1118) with either 0.8 μM FLAG-eIF3e (lane 5), FLAG-eIF3j (lane 9), or FLAG-eIF3i (lane 14). Lanes 1 and 10 contained 0.2 μM S-eIF4G(1015–1118) with 0.2 μM HeLa eIF3. The molar ratio of FLAG-eIF3 subunit to HeLa eIF3 is indicated above the lanes. The amount of eIF3 bound is displayed below the lanes as a percentage of the reaction lacking added FLAG-eIF3 subunits (lane 1 for FLAG-eIF3e and FLAG-eIF3j and lane 10 for FLAG-eIF3i). All binding reactions in B and C were performed and analyzed by Western blotting as described under ”Experimental Procedures.“ Native eIF3 subunits (eIF3a, eIF3b/c) and FLAG-tagged eIF3 subunits (FLAG-eIF3e, -eIF3i, and -eIF3j) were detected with anti-eIF3 and anti-FLAG M2 primary antibodies, respectively, and secondary antibodies coupled to alkaline phos-phatase. S-eIF4G(653–1118) and S-eIF4G(1015–1118) were detected using S-protein-coupled alkaline phosphatase.
Next we tested whether FLAG-eIF3e specifically competes with eIF3 for binding to S-eIF4G(1015–1118). If eIF3 binds to eIF4G through the eIF3e subunit, then simultaneous incubation with both eIF3 and FLAG-eIF3e should decrease the amount of eIF3 bound to S-eIF4G(1015–1118). HeLa eIF3 was incubated with S-eIF4G(1015–1118) alone or in the presence of FLAG-eIF3e at molar ratios of 1:1, 2:1, or 4:1 (subunit/eIF3) (Fig. 2C). Increasing concentrations of FLAG-eIF3e progressively decreased the amount of eIF3 bound to S-eIF4G(1015–1118) (Fig. 2C, lanes 2–4, top panel). Simultaneously, the amount of eIF3e bound to S-eIF4G(1015–1118) increased (Fig. 2C, lanes 2–4, middle panel). When FLAG-eIF3e was present at the highest of these concentrations (0.8 μM) but eIF3 was omitted, the amount of FLAG-eIF3e bound increased dramatically (Fig. 2C, cf. lane 5 and lane 4, middle panel). This is another indication that eIF3 and FLAG-eIF3e compete for binding to eIF4G.
Two recombinant subunits were used as negative controls, FLAG-eIF3j and FLAG-eIF3i. Neither of these had been detected in the S-eIF4G(1015–1118)-bound fraction after extensive trypsinolysis of eIF3 (Fig. 1). FLAG-eIF3j also caused a decrease in eIF3 binding to S-eIF4G(1015–1118), but it was not as marked as that of FLAG-eIF3e at any of the three concentrations (Fig. 2C, lanes 6–8, top panel). Furthermore, very little FLAG-eIF3j was bound to S-eIF4G(1015–1118) when present at the same molar concentrations as FLAG-eIF3e, and this did not increase with increasing FLAG-eIF3j concentrations (Fig. 2C, lanes 6–8, middle panel). Finally, no FLAG-eIF3j was bound to S-eIF4G(1015–1118) when eIF3 was omitted (Fig. 2C, lane 9, middle panel), suggesting that the observed binding of FLAG-eIF3j was due to its interaction with eIF3 and not S-eIF4G(1015–1118). FLAG-eIF3i also caused a decrease in binding of eIF3 to eIF4G, but as with FLAG-eIF3j, there was no further decrease at higher concentrations of FLAG-eIF3i (Fig. 2C, cf. lanes 11–13 and lane 10, top panel). Binding of eIF3i to S-eIF4G(1015–1118) was weak and similar to that of FLAG-eIF3j (Fig. 2C, lanes 11–13, middle panel) and increased only slightly in the absence of eIF3 (lane 14). Because binding of FLAG-eIF3i and FLAG-eIF3j to S-eIF4G(1015–1118) was much weaker than that of FLAG-eIF3e (Fig. 2C, cf. lanes 6–14 and lane 5, middle panel), it is likely that the observed decrease in eIF3 retention on S-protein-agarose (lanes 7–9 and 11–13, top panel) is because of an interaction of these subunits with eIF3 and not eIF4G. Finally, neither FLAG-eIF3i nor FLAG-eIF3j was able to bind S-protein-agarose alone (data not shown). These results support the view that eIF3e as an isolated polypeptide can bind to eIF4G and that it forms at least part of the eIF3-eIF4G interaction surface.
Free FLAG-eIF3e Decreases the Rate of Protein Synthesis
The preceding experiments were carried out with isolated proteins, but if free FLAG-eIF3e also prevents eIF4G binding to eIF3 in a complete protein synthesis system, it should cause an inhibition of translation, specifically translational initiation. To test this, we measured the effect of increasing concentrations of FLAG-eIF3e on luciferase synthesis from the capped monocistronic mRNA m7G-Luc-A60 (Fig. 3A). This led to a marked inhibition of luciferase synthesis with a KI of ~0.018 μM. For a simple competition model, 50% inhibition would be expected when the concentrations of FLAG-eIF3e and eIF4G are equal. The concentration of eIF4G in RRL has been reported to be ~0.04 μM (53, 54), but the concentrations of initiation factors in different batches of RRL can vary widely,5 so a KI of 0.018 μM is not unreasonable.
FIGURE 3. Free FLAG-eIF3e inhibits both the rate of protein synthesis and polysomal distribution of mRNA.
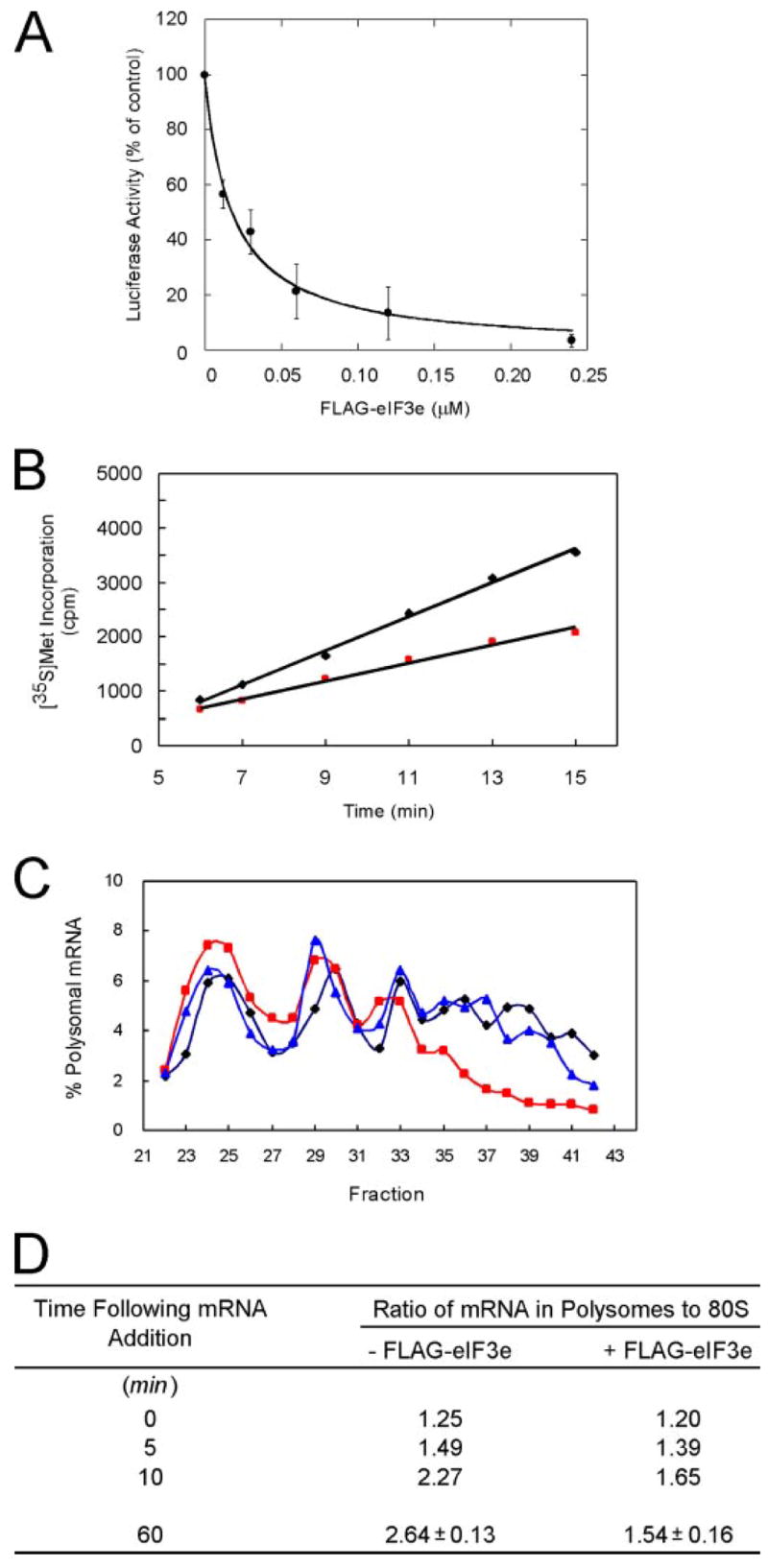
A, FLAG-eIF3e inhibits luciferase synthesis from m7G-Luc-A60. Translation reactions were programmed with m7G-Luc-A60 at a final concentration of 0.5 μg/ml in the presence of the indicated concentrations of FLAG-eIF3e. Luciferase synthesis was measured after 60 min as described under ”Experimental Procedures“ and is expressed as a percentage of a control reaction without FLAG-eIF3e. The average of three independent experiments is plotted and curve fitting performed as described under ”Experimental Procedures,“ yielding a KI value of 0.018 ± 0.002 μM with an R2 value of 0.996. B, FLAG-eIF3e decreases the rate of luciferase synthesis in an RRL translational system. Translation reactions (75 μl) were programmed with 15 μg/ml m7G-Luc-A31 in the absence (black diamonds) or presence (red squares) of 0.03 μM FLAG-eIF3e. Aliquots of 10 μl were removed at the indicated times. 35S radioactivity was analyzed as described under ”Experimental Procedures.“ C, FLAG-eIF3e decreases the amount of m7G-Luc-A60 mRNA present in heavy polysomes. Translation reactions were preincubated for 15 min with no added eIF3 subunit (black diamonds), 0.24 μM FLAG-eIF3e (red squares), or 0.24 μM FLAG-eIF3j (blue triangles). Then 32P-labeled m7G-Luc-A60 (specific activity ~2.75 μCi/μg) was added at a final concentration of 2.0 μg/ml and incubation continued for 60 min. Polysomes were fractionated by ultracentrifugation as described under ”Experimental Procedures.“ The presence of 32P-labeled mRNA in each fraction was detected as Cerenkov radiation and is displayed as % total mRNA. D, similar experiments were performed as in C with FLAG-eIF3e, except that incubation times were for either 0, 5, 10, or 60 min following mRNA addition. The ratio of mRNA present in polysomes (disomes, trisomes, and heavy polysomes) to that in monosomes (80 S) is shown. The results for 60 min represents the average of three independent experiments.
Inhibition of translation by eIF2α kinases leads to biphasic kinetics (55), whereas interference with mRNA recruitment to the 48 S initiation complex should produce diminished but nonetheless linear rates of protein synthesis. To distinguish between these mechanisms, we measured the rate of [35S]Met incorporation in RRL programmed with m7G-Luc-A31 mRNA in the presence or absence of 0.03 μM FLAG-eIF3e (Fig. 3B). Protein synthesis was linear with time in both cases, but FLAG-eIF3e resulted in a 47% decrease in the rate. This is consistent with slower mRNA recruitment to the 43 S initiation complex, i.e. a defect in 48 S complex formation. As a control, we also added 0.03 μM FLAG-eIF3j in a separate experiment and observed no effect on the rate of protein synthesis (data not shown).
The inhibition of protein synthesis by free FLAG-eIF3e could be due to an effect on either initiation or elongation/termination. These can be distinguished by examining changes in mRNA distribution on polysomes. Inhibitors of initiation cause a shift of mRNA distribution from higher to lower polysomes, whereas inhibitors of elongation/termination have the opposite effect (56). We therefore determined the effect of FLAG-eIF3e on the polysomal distribution of mRNA. The RRL system was preincubated with a concentration of FLAG-eIF3e sufficient to inhibit protein synthesis ~80% prior to addition of 32P-labeled m7G-Luc-A60 mRNA, after which incubation was continued for 1 h. Reaction mixtures were then fractionated by ultracentrifugation on sucrose gradients containing cycloheximide, to prevent any further elongation, analyzed for A260 (not shown), and 32P radioactivity plotted as % of total (Fig. 3C). mRNA was found in the messenger ribonucleoprotein complex region of the gradient (not shown), 80 S initiation complexes, and monosomes (which co-sediment), disomes (two ribosomes per mRNA), trisomes, and larger polysomes that were at least partially resolved up to 7-mers. FLAG-eIF3e caused a 20% increase of mRNA in the 80 S peak and a progressive decrease as polysome size increased, with almost none in 7-mer (Fig. 3C, red squares). FLAG-eIF3j, by contrast, caused no significant shift in polysomal distribution (Fig. 3C, blue triangles). The total radioactivity in monosomes plus polysomes was the same ±5% for reactions containing no exogenous eIF3 subunit, FLAG-eIF3e, or FLAG-eIF3j, ruling out the trivial explanation that the eIF3 subunit preparations contained RNase. The shift of mRNA from higher to lower polysomes caused by addition of FLAG-eIF3e indicates an effect on translation initiation rather than elongation/termination.
To assess more quantitatively the polysomal distribution of mRNA, we calculated the ratio of mRNA present in polysomes (disomes and higher) to that in monosomes (Fig. 3D). In the absence of FLAG-eIF3e, mRNA in polysomes increased with time as expected. In the presence of FLAG-eIF3e, the increase was less pronounced. The average values from three independent experiments are shown at the 60-min point. These results indicate that the rate of mRNA recruitment to polysomes is diminished by FLAG-eIF3e.
Free FLAG-eIF3e Inhibits Cap-dependent Translation More Severely than Translation Driven by eIF4G-independent IRESes
A prediction of the model that free FLAG-eIF3e prevents association of eIF4G with eIF3 is that translational mechanisms not requiring eIF4G would be insensitive to inhibition by FLAG-eIF3e. Initiation of translation from most eukaryotic cellular mRNAs occurs by a cap-dependent mechanism, but some mRNAs, both viral and cellular, are internally initiated via an IRES (57). IRESes differ in their requirements for initiation factors (58). Initiation from the HCV and CSFV IRESes does not require any part of eIF4G but does require eIF2·GTP·Met-tRNAi and eIF3 (40, 41). We reasoned that if eIF3e plays an important role in the eIF3-eIF4G interaction, then cap-dependent translation should be more sensitive to inhibition by FLAG-eIF3e than translation driven by the HCV or CSFV IRESes.
To test this, we carried out in vitro translation of bicistronic mRNAs in which expression from the first cistron (cyclin B2) was via the cap-dependent pathway but expression from the second cistron (NS′) required an IRES. Two bicistronic mRNAs were used as follows: m7G-CycB2-HCV-NS′ (Fig. 4A) and m7G-CycB2-CSFV-NS′ (Fig. 5A). Translation reactions contained either no FLAG-tagged eIF3 subunit (Figs. 4B and 5B, lane 2) or increasing amounts of either FLAG-eIF3e (lanes 3–7) or FLAG-eIF3j (lanes 8–12). A control reaction lacking mRNA (Figs. 4B and 5B, lane 1) aided in identification of mRNA-specific translation products. FLAG-eIF3e inhibited cap-dependent translation (CycB2) more than it inhibited IRES-driven translation (NS′) for both the HCV (Fig. 4B) and CSFV (Fig. 5B) IRESes. Addition of FLAG-eIF3j resulted in a less pronounced decrease in cap-dependent translation. At the highest concentration used (0.24 μM), FLAG-eIF3j caused a 14–24% decrease in cap-dependent translation as compared with a 91–94% decrease in the presence of FLAG-eIF3e, depending on which bicistronic mRNA was used. This is similar to the effect of FLAG-eIF3j on competition of native eIF3 for binding to S-eIF4G(1015–1118) (Fig. 2C).
FIGURE 4. Free FLAG-eIF3e inhibits cap-dependent translation more severely than HCV IRES-mediated translation.
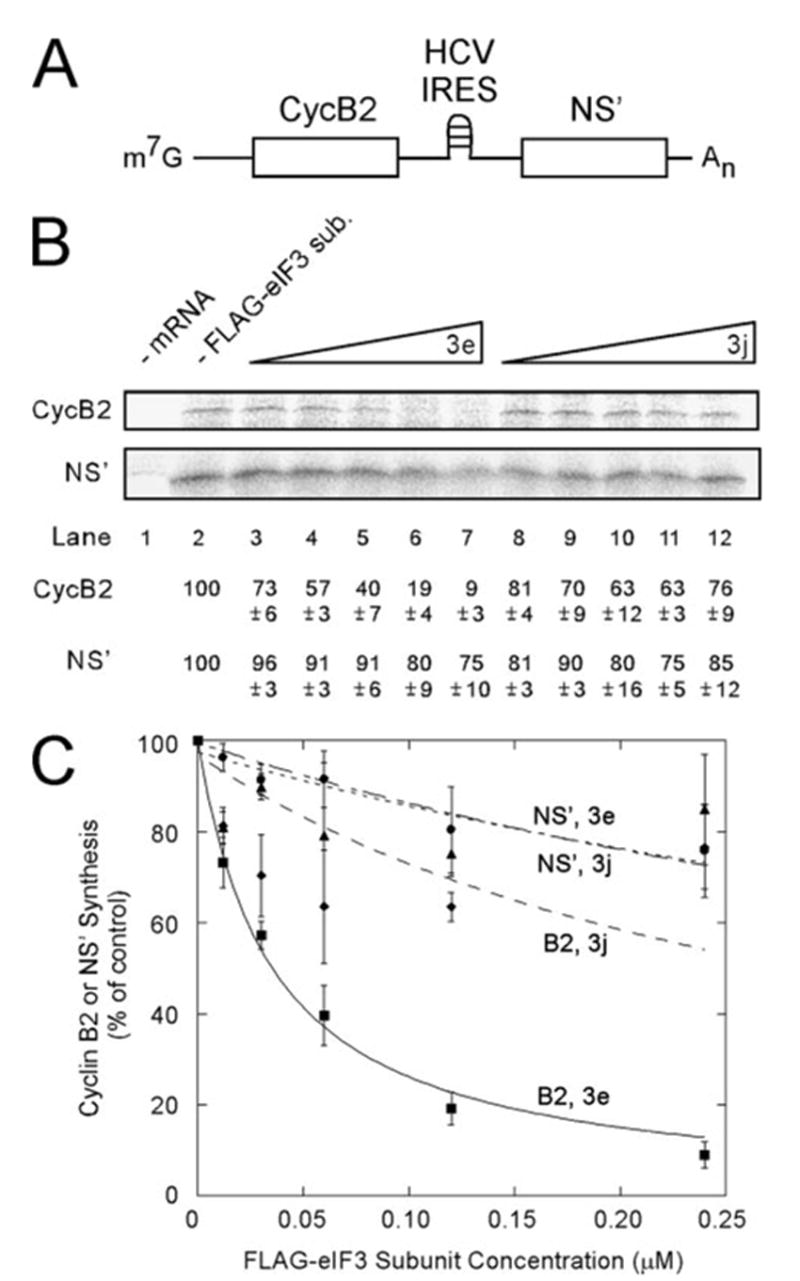
A, schematic representation of the bicistronic mRNA used in this experiment. B, in vitro translation from m7G-CycB2-HCV-NS′ in the presence of FLAG-eIF3e or FLAG-eIF3j. In vitro translation reactions (25 μl) contained the indicated concentrations of FLAG-eIF3e (lanes 3–7) or FLAG-eIF3j (lanes 8 –12) and m7G-CycB2-HCV-NS′ at 15 μg/ml. Control reactions contained no added mRNA (lane 1) and no added FLAG-tagged eIF3 subunits (lane 2). After a 90-min incubation, radiolabeled translation products were resolved by SDS-PAGE and detected by Phospho-rImager. The percent of product synthesized in each reaction is displayed below each lane as a percentage of the reaction without added FLAG-eIF3 subunits (lane 2). C, the average values from three independent experiments similar to B are plotted as described under ”Experimental Procedures.“ A KI of 0.035 ± 0.002 μM was determined for inhibition of cap-dependent translation by FLAG-eIF3e with an R2 value of 0.998. Squares, CycB2 with FLAG-eIF3e; circles, NS′ with FLAG-eIF3e; diamonds, CycB2 with FLAG-eIF3j; and triangles, NS′ with FLAG-eIF3j.
FIGURE 5. Free FLAG-eIF3e specifically inhibits cap-dependent translation more severely than CSFV IRES-mediated translation.
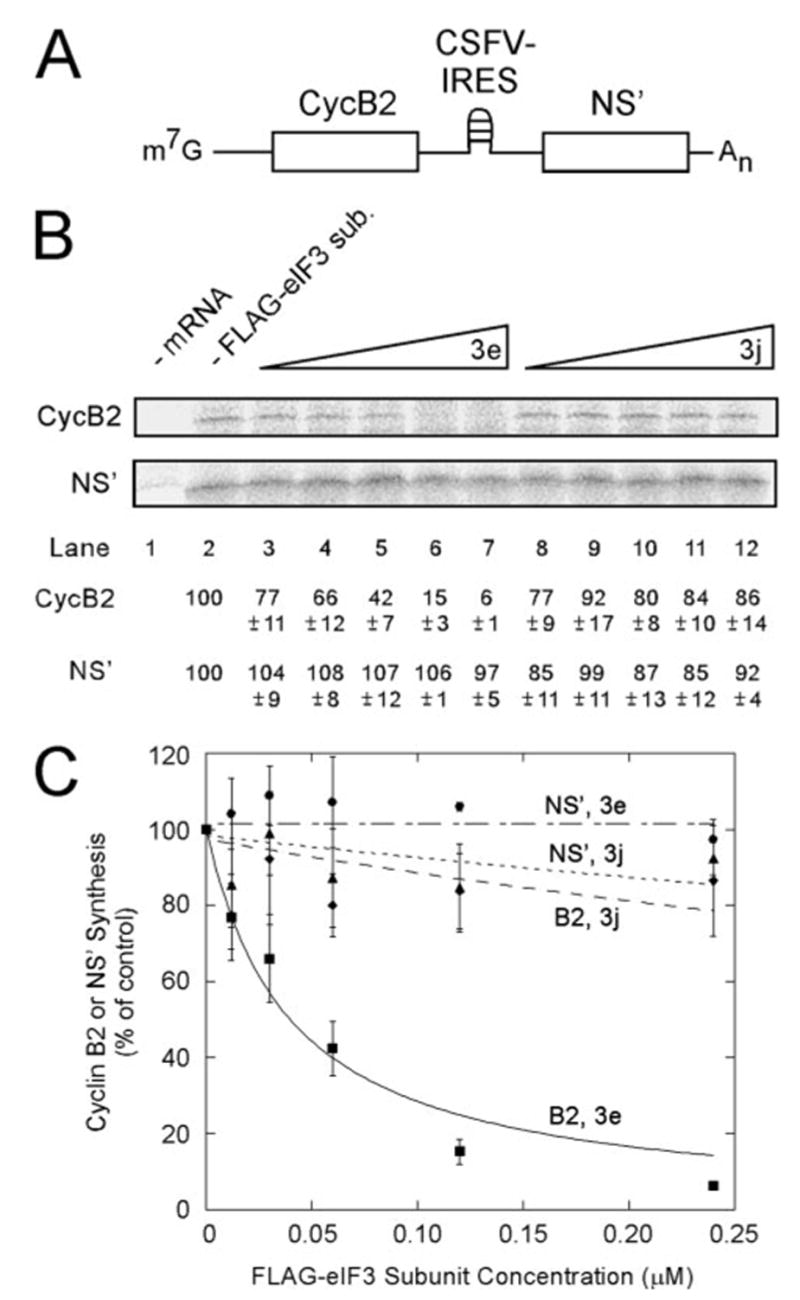
The experiment was performed as in Fig. 4 except m7G-CycB2-CSFV-NS′ was used. An estimated KI of 0.0396 ± 0.0039 μM was determined for inhibition of cap-dependent translation by FLAG-eIF3e with an R2 value of 0.992.
Experiments were performed in triplicate and the results quantitated. KI values for inhibition of cap-dependent translation of m7G-CycB2-HCV-NS′ and m7G-CycB2-CSFV-NS′ were determined with a curve-fitting program for competitive inhibition and yielded values of 0.035 and 0.040 μM, respectively (Figs. 4C and 5C, squares). These are similar to the KD values of the S-eIF4G(1015–1118)-eIF3 interaction (0.059 μM) measured by surface plasmon resonance (23). By contrast, the data for inhibition of cap-dependent translation by FLAG-eIF3j were fit poorly by the competitive inhibition equation (Figs. 4C and 5C, diamonds) suggesting some other mechanism for inhibition. Translation from the CSFV IRES (Fig. 5C) was more refractory to inhibition by either FLAG-eIF3e (circles) or FLAG-eIF3j (triangles) than that of the HCV IRES (Fig. 4C), which is consistent with the observation that the CSFV IRES is more potent translationally than the HCV IRES (57). The absence of significant inhibition of translation driven by the HCV or CSFV IRESes by FLAG-eIF3e (circles) also indicates that FLAG-eIF3e does not inhibit polypeptide elongation or termination, confirming the conclusions drawn from the poly-some shift experiment (Fig. 3, C and D). The results with both bicistronic vectors indicate that free FLAG-eIF3e inhibits cap-dependent translation more severely than HCV or CSFV IRES-dependent translation, which is consistent with a mechanism by which FLAG-eIF3e competes with eIF3 for binding to eIF4G during initiation of cap-dependent translation.
Free FLAG-eIF3e Blocks Association of eIF4G and eIF2α with the 40 S Ribosomal Subunit
The foregoing experiments are consistent with inhibition of 48 S initiation complex formation by FLAG-eIF3e but do not test it directly. We therefore carried out translation reactions with or without 0.050 μM FLAG-eIF3e, which is expected to inhibit cap-dependent translation ~80%. These reaction mixtures were subjected to ultracentrifugal analysis, and the individual fractions analyzed for initiation factors by Western blotting and 18 S rRNA by real time PCR. A representative absorbance profile is presented in Fig. 6A; no differences were detected in reactions containing FLAG-eIF3e (data not shown). The location of 18 S rRNA indicates that “40 S” initiation complexes (43 S and 48 S) are in fractions 9–11 (Fig. 6B). FLAG-eIF3e caused a slight shift of 18 S rRNA to slower sedimentation within the 40 S region, which may indicate loss of 48 S complexes and gain of 43 S complexes. Western blots (Fig. 6C) were performed in triplicate and quantitated, and the results are presented in Fig. 6D for reactions with (filled bars) or without (open bars) FLAG-eIF3e. eIF3 (represented by eIF3b) was present in the 40 S region and also in fractions 5–7, which would correspond to the ~20 S multifactor complex observed in S. cerevisiae (14). The distribution of eIF3 was not altered by FLAG-eIF3e. By contrast, eIF4G was dramatically shifted out of the 40 S region and into the 20 S region by FLAG-eIF3e. Similar behavior was seen for eIF2α. FLAG-eIF3e itself had a bimodal distribution. Because this protein is only 52 kDa, its presence at S values of 20–40 indicates that it is incorporated into various complexes. If it were entirely in the eIF3 complex, its distribution would have been similar to that of eIF3b. The peak in fractions 3–6 coincides with the fractions that increased in eIF4G when reactions contained FLAG-eIF3e (Fig. 6D, panel eIF4G, filled bars). Thus, the faster sedimenting peak of FLAG-eIF3e may represent its binding to eIF3, whereas the slower sedimenting peak may represent an eIF4G·FLAG-eIF3e complex.
FIGURE 6. Free FLAG-eIF3e blocks association of eIF4G and eIF2α with the 40 S ribosomal subunit.
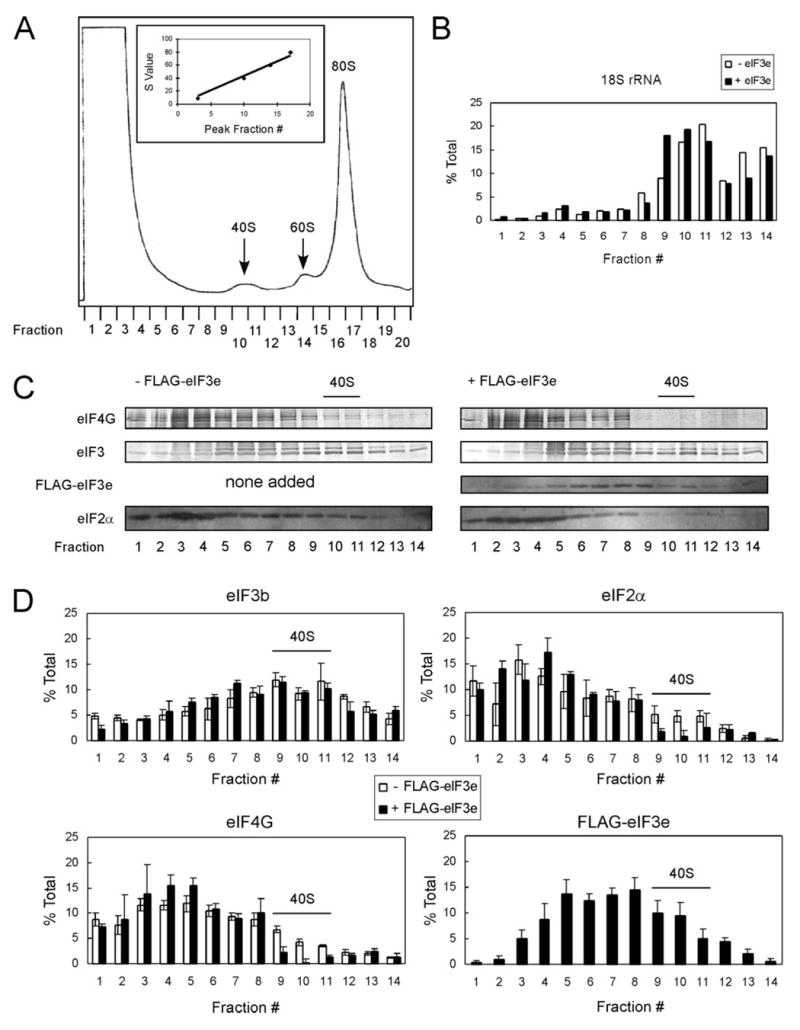
In vitro translation reactions (100 μl) with or without 0.05 μM FLAG-eIF3e were incubated for 15 min, layered on 10 –35% sucrose gradients, and centrifuged at 38,000 rpm for 5 h at 4 °C. Fractions (500 μl) were collected, and absorbance at 260 nm was monitored. A, typical absorbance profile indicating distribution of 40 S, 60 S, and 80 S complexes. Inset, plot of S values versus fraction number. B, real time PCR of 18 S rRNA present in fractions 1–14 with (filled bars) or without (open bars) FLAG-eIF3e. Data are plotted as the percent total 18 S rRNA present in each gradient. C, representative Western blots of trichloroacetic acid-precipitated protein from fractions 1 to 14 for eIF4G, eIF3, FLAG-eIF3e, and eIF2α in reactions lacking (left panel) or containing (right panel) FLAG-eIF3e. eIF4G and eIF3 blots were probed with secondary antibodies coupled to alkaline phosphatase, whereas FLAG-eIF3e and eIF2α were probed with secondary antibodies coupled to horseradish peroxidase and developed with ECL+. D, experiments similar to that in C were performed in triplicate, immunological signals quantitated using ImageQuant software, and average values plotted as the percent of total protein in fractions 1–14. The eIF3b band was used for quantitation of eIF3. Peak fractions of the 40 S ribosomal subunit, as determined in B, are indicated in C and D.
DISCUSSION
In our initial experiments, multiple eIF3 subunits were found to interact with eIF4G by several commonly used techniques for detection of protein-protein interactions. Although it is conceivable that eIF4G simultaneously interacts with multiple eIF3 subunits, those results could also reflect nonspecific binding. Similarly, the subunits identified by the photoaffinity cross-linking approach (eIF3a, eIF3b/c, eIF3d/l, and eIF3e) may bind eIF4G directly or may only be within cross-linking distance of the authentic eIF3-binding subunit. The latter possibility is strengthened by the observations that both mammalian eIF3a (10) and eIF3e (12) bind to eIF3b, and similarly, eIF3l binds to eIF3e (3). These subunits may form part of an eIF4G-proximal surface on eIF3. The cryo-EM structure of an eIF4G·eIF3·40 S complex has recently been solved, demonstrating that a very small region of eIF3 contacts eIF4G (39). This argues against a model for multiple eIF3 subunits binding to eIF4G simultaneously and supports the idea that eIF3e is solely responsible for mediating this interaction.
The notion that multiple eIF3 subunits interact with eIF4G is nonetheless intriguing, especially because some eIF3 subunits can affect the translation of specific mRNAs (59–61). Different eIF3 subunits may interact with eIF4G under specific cellular conditions to alter the spectrum of mRNAs translated. This notion is attractive in light of a recent study by Zhou et al. (4). Different eIF3 complexes could be isolated from S. pombe, containing either eIF3e or eIF3m, and these were able to associate with different subsets of mRNAs. Our results suggest that one mechanism whereby this could be accomplished would be by differential association with eIF4G, i.e. eIF3e-containing complexes associate with eIF4G, whereas eIF3m-containing complexes do not, leading to translation of either eIF4G-dependent or eIF4G-independent mRNAs, respectively. A specific role for eIF3e in highly eIF4G-dependent translation (i.e. mRNAs containing high secondary structure in the 5′-untranslated region) is also attractive because deletion of eIF3e from S. pombe has little effect on global protein synthesis but causes specific defects (see below) (62). An analogous situation occurs with rapamycin, which inhibits highly cap-dependent translation completely, e.g. that of c-Myc mRNA, at concentrations where there is little effect on global protein synthesis (63). The regulated association of subunits with eIF3 is also attractive in light of the recent observation that insulin controls eIF3-eIF4G association through mTOR (25, 26). Although a specific eIF3 subunit has not been reported to be the target of mTOR, it is possible that eIF3e, or another subunit that regulates the binding of eIF3e, may be responsible. Interestingly, eIF3e binds eIF3l in a phosphorylation-specific manner (3).
Even if other eIF3 subunits are able to specifically interact with eIF4G, the one with the highest affinity appears to be eIF3e, based on the observation that only its binding can be detected after partial trypsinolysis of HeLa eIF3. There are several ways that additional specific eIF4G-binding eIF3 subunits could have been missed by this approach. (i) They may have been among the multiple subunits binding only at early time points of trypsinolysis in Fig. 1 but were unable to be distinguished from those that bound indirectly. (ii) Their affinity for eIF4G may have been lost because of cleavage at or near an active binding site. (iii) Their characteristic tryptic peptides were not ionized by MALDI. It is also possible that association of eIF4G with the tryptic fragment representing the N-terminal two-thirds of eIF3e identified in this experiment is nonspecific, because of the fact that it is not present within the native eIF3 complex. However, additional experimental results argue against this, including the similarities of KI value for inhibition of cap-dependent translation by FLAG-eIF3e (Figs. 3–5) and the KD value for the eIF3-eIF4G interaction (23). Also, the binding of eIF3 to S-eIF4G(1015–1118) is inhibited ~50% at equimolar FLAG-eIF3e (Fig. 2C). These findings suggest that FLAG-eIF3e interferes with the same site that is required for binding of eIF3 to eIF4G.
The peptide matching and mapping algorithms developed to identify the eIF4G-binding subunit were essential for this overall approach, but they also have general utility. Manual matching of peaks to theoretical peptides and displaying their origins on the polypeptide chain is laborious and time-consuming, even when there are only a few peaks. In our case, there were thousands of peaks and theoretical peptides, so manual processing was virtually impossible. Yet the matching algorithms delivered the maps for all ~210 gel slices within minutes. A critical feature of the processing algorithm was the ability to vary parameters, like the number of missed cleavages and the match window between actual and theoretical masses. In initial analyses, we could not distinguish signal from noise, which led us to refine the accuracy of mass determination and narrow the match window. The results of changing each parameter could be visualized almost immediately. Furthermore, displaying matches to all subunits simultaneously allowed us to detect multiple subunits with similar migrations on SDS-PAGE in a single slice as well as portions of subunits. These are general algorithms that could be applied to any similar problem of identifying proteins or portions of proteins in complex but defined mixtures from MALDI-TOF-MS data.
The moderate effect of FLAG-eIF3i and FLAG-eIF3j on competition of native eIF3 binding to eIF4G and inhibition of cap-dependent translation may reflect the biological role of eIF3j rather than nonspecific interactions. It is likely that the decrease in eIF3 binding to eIF4G in the presence of FLAG-eIF3j is because of an indirect effect on eIF3, because FLAG-eIF3j binds only weakly to S-eIF4G(1015–1118) and only in the presence of eIF3. If our preparations of eIF3 are substoichio-metric for eIF3j, FLAG-eIF3j may be incorporated into native eIF3, and this may weaken the eIF3-eIF4G interaction through an indirect effect. Interestingly, eIF3j is present in binary complexes with 40 S ribosomal subunits and also in reconstituted 43 S complexes but absent in reconstituted 48 S complexes containing eIF3, eIF2·GTP·Met-tRNAi, eIF1, eIF1A, and mRNA (64), suggesting that the presence of eIF3j and eIF4G is mutually exclusive in eIF3-containing complexes. The moderate inhibition of HCV IRES-mediated translation by both FLAG-eIF3e and FLAG-eIF3j may be due to a similar mechanism. FLAG-eIF3e and FLAG-eIF3j may affect the affinity of other eIF3 subunits for the HCV IRES or the 40 S ribosomal subunit. In fact, eIF3a, eIF3b, eIF3d, and eIF3f have all been shown to specifically interact with both the HCV and CSFV IRESes (65), as has the 40 S ribosomal subunit (41). Because eIF3e and eIF3j both bind eIF3b (10,12), eIF3b binds eIF3a (10, 66), and eIF3j binds the 40 S ribosomal subunit (10), it is plausible that FLAG-eIF3e or FLAG-eIF3j could alter the binding affinity of other subunits or the 40 S ribosomal subunit for these IRESes. Considering that the CSFV IRES is more potent translationally (57), these IRES-binding subunits may have higher affinity for the CSFV IRES compared with the HCV IRES, explaining why it is more refractory to inhibition by FLAG-eIF3e and FLAG-eIF3j (Fig. 5). The in vitro competition seen with FLAG-eIF3i may be a result of an indirect mechanism because it binds eIF3b (67), which also binds eIF3e (12).
The results presented in Fig. 6 suggest that FLAG-eIF3e has several effects in a complete translation system. First, it is able to be incorporated into eIF3, both bound and unbound to the 40 S subunit, because it is present both in fractions expected to contain the multifactor complex (~20 S) and in 40 S initiation complexes. Second, it plays a role in eIF4G binding, because it displaces eIF4G from the 40 S regions. Third, it negatively affects eIF2 binding to the same complexes. It has been suggested that eIF3e plays some role in eIF2 binding to eIF3 because of the ability of the viral stress-inducible translational inhibitor p56 to bind eIF3e (in humans) and prevent stimulation of eIF2·GTP·Met-tRNAi complex formation by eIF3 (68). The destabilization of this complex could also explain the limited inhibitory effect of FLAG-eIF3e observed on HCV and CSFV IRES-dependent translation.
The identification of a non-core eIF3 subunit as a direct binding partner of eIF4G is somewhat unexpected. As noted in the Introduction, the conserved core of S. cerevisiae can function with mammalian factors. However, because S. cerevisiae lacks at least six non-core subunits, including eIF3e, it is difficult to understand the roles of these non-core subunits in translation initiation. Interaction of eIF3 and eIF4G in S. cerevisiae has not been demonstrated, so it is possible that direct binding of eIF4G to eIF1 and/or eIF5 (69) mediates an indirect association of eIF4G with eIF3, as previously proposed for eIF5 (70). Another explanation is that additional eIF3 subunits bind eIF4G but were not identified in our experiments and that these mediate the eIF3-eIF4G interaction in S. cerevisiae. Interestingly, a PCI homology domain is present in eIF3e, and this motif also occurs in eIF3a (2), eIF3c (2), eIF3k (71), eIF3l (3), and eIF3m (4). Because this domain was left partially intact in the N-terminal tryptic fragment of eIF3e that binds to eIF4G, it is possible one or more of these subunits can functionally compensate for eIF3e in binding to eIF4G if this domain is involved in interaction with eIF4G.
A number of observations have been reported regarding eIF3e function. Absence of eIF3e in S. pombe results in loss of cell membrane integrity, inefficient chromosome segregation, and ubiquitin-mediated proteolysis (72). eIF3e is one of several subunits (the others being eIF3a, -c, -f, -h, -i, -k, -l, and -m) that contain motifs (PCI, MPN, and WD40) homologous to those in proteins known to be associated with the COP9 signalosome and 19 S proteasome lid (4, 71). In fact, eIF3e directly associates with the COP9 signalosome and the 26 S proteasome (73, 74). eIF3e re-localizes to cytoplasmic foci (containing rRNA and eIF4E) during osmotic and thermal stresses of S. pombe (75). Also, eIF3e binds the interferon-inducible p56 protein in humans (68) (as opposed to eIF3c in mice (76)), co-localizes with PML protein in nuclear promyelocytic leukemia bodies (77), interacts with human T-cell leukemia virus type 1 Tax oncoprotein (77), and contains putative nuclear localization and nuclear export signals (78). Because our results indicate that eIF3e directly interacts with eIF4G and affects cap-dependent translation, it would be interesting to re-examine these various observations for possible links to translation.
Several eIF3 subunits, including eIF3e, are dysregulated in certain cancers. eIF3a is overexpressed in some human mammary, esophageal, and lung carcinomas (79), and eIF3h is increased by gene amplification in some mammary and prostate cancers (80). The most striking example is the involvement of eIF3e in mouse mammary tumor virus (MMTV)-mediated malignancy. The gene encoding eIF3e, Int-6, is a common site of integration of the MMTV genome, resulting in generation of C-terminally truncated forms of eIF3e (81). Although the role of such truncated eIF3e polypeptides in oncogenesis by MMTV is not understood, interaction with eIF4G could be one possible route by which this is achieved. Mammalian cells transfected with C-terminally truncated eIF3e, but not full-length eIF3e, display properties of malignantly transformed cells, including resistance to apoptosis and anchorage-independent growth (82, 83). In addition, cells expressing full-length eIF3e exhibit a slow growth phenotype, suggesting an inhibition of translation consistent with our in vitro observations. This correlates with decreased levels of eIF3e expression in some breast and lung carcinomas (84). A model has been presented that full-length eIF3e is a negative regulator of translation (85), based on its proposed roles in MMTV- (81, 83) and HTLV-1-induced (77) malignancy. Our results suggest different models involving alterations in cap-dependent translation via an interaction between C-terminally truncated eIF3e and eIF4G.
Acknowledgments
We thank Tatyana Pestova for pXL.HCV(40–373).NS′ and pXL.CSFV(1–442).NS′, Ewa Grudzien for radiolabeled m7G-Luc-A60 and many helpful discussions, and the Louisiana State University Health Sciences Center-Shreveport Research Core Facility for subsidizing mass spectrometry experiments.
The abbreviations used are
- eIF
eukaryotic initiation factor
- BSA
bovine serum albumin
- CSFV
classical swine fever virus
- HCV
hepatitis C virus
- IRES
internal ribosome entry site
- MALDI-TOF-MS
matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- Mnk
mitogen-activated protein kinase-interacting serine/threonine kinase
- PABP
poly(A)-binding protein
- RRL
rabbit reticulocyte lysate
- DTT
dithiothreitol
- SASD
sulfosuccinimidyl-2-[p-azidosalicylamido]ethyl-1,3′-dithiopropionate
- MMTV
mouse mammary tumor virus
Footnotes
This work was supported by National Institutes of Health Grants GM20818 (to R. E. R.) and GM22135 (to J. W. B. H.).
A unified nomenclature system for eIF3 subunits assigns the same letter to homologous subunits across all eukaryotic organisms (2). The subunits are named in order of highest to lowest molecular mass (e.g., eIF3a is 170 kDa, eIF3b is 116 kDa, etc.), although subunits discovered after establishment of this nomenclature system do not follow this rule (e.g. eIF3l is 69 kDa (3) and eIF3m is 42 kDa (64)).
There are both multiple genes for eIF4G (47, 86) and multiple protein iso-forms encoded by a single gene (87, 88). The recombinant proteins used in this work are all derived from human eIF4G-1, and the amino acid numbers all refer to the longest form, eIF4G-1f (1600 amino acids) (88). The nomenclature system recommended by an ad hoc committee appointed by the IUBMB is used for eIF4G (89). Forms of eIF4G-1 containing amino acids 653–1118, 1015–1118, and 1118 –1600 plus N-terminal His6 and S-peptide tags are referred to as S-eIF4G(653–1118), S-eIF4G(1015–1118), and S-eIF4G(1118 –1600), respectively.
B. Joshi and R. E. Rhoads, unpublished observations.
References
- 1.Hershey JWB, Merrick WC. In: Translational Control of Gene Expression. Sonenberg N, Hershey JWB, Mathews MB, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 33–88. [Google Scholar]
- 2.Browning KS, Gallie DR, Hershey JW, Hinnebusch AG, Maitra U, Merrick WC, Norbury C. Trends Biochem Sci. 2001;26:284. doi: 10.1016/s0968-0004(01)01825-4. [DOI] [PubMed] [Google Scholar]
- 3.Morris-Desbois C, Rety S, Ferro M, Garin J, Jalinot P. J Biol Chem. 2001;276:45988–45995. doi: 10.1074/jbc.M104966200. [DOI] [PubMed] [Google Scholar]
- 4.Zhou C, Arslan F, Wee S, Krishnan S, Ivanov AR, Oliva A, Leatherwood J, Wolf DA. BMC Biol. 2005;3:14. doi: 10.1186/1741-7007-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinnebusch AG. In: Translational Control of Gene Expression. Sonenberg N, Hershey JWB, Mathews MB, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 185–244. [Google Scholar]
- 6.Phan L, Zhang X, Asano K, Anderson J, Vornlocher HP, Greenberg JR, Qin J, Hinnebusch AG. Mol Cell Biol. 1998;18:4935–4946. doi: 10.1128/mcb.18.8.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhoads RE, Dinkova TD, Korneeva NL. The C. elegans Research Community, Wormbook. 2005. WormBook. [DOI] [Google Scholar]
- 8.Burks EA, Bezerra PP, Le H, Gallie DR, Browning KS. J Biol Chem. 2001;276:2122–2131. doi: 10.1074/jbc.M007236200. [DOI] [PubMed] [Google Scholar]
- 9.Asano K, Vornlocher HP, Richter-Cook NJ, Merrick WC, Hinnebusch AG, Hershey JW. J Biol Chem. 1997;272:27042–27052. doi: 10.1074/jbc.272.43.27042. [DOI] [PubMed] [Google Scholar]
- 10.Fraser CS, Lee JY, Mayeur GL, Bushell M, Doudna JA, Hershey JW. J Biol Chem. 2004;279:8946–8956. doi: 10.1074/jbc.M312745200. [DOI] [PubMed] [Google Scholar]
- 11.Mayeur GL, Fraser CS, Peiretti F, Block KL, Hershey JW. Eur J Biochem. 2003;270:4133–4139. doi: 10.1046/j.1432-1033.2003.03807.x. [DOI] [PubMed] [Google Scholar]
- 12.Shalev A, Valasek L, Pise-Masison CA, Radonovich M, Phan L, Clayton J, He H, Brady JN, Hinnebusch AG, Asano K. J Biol Chem. 2001;276:34948–34957. doi: 10.1074/jbc.M102161200. [DOI] [PubMed] [Google Scholar]
- 13.Nielsen KH, Valasek L, Sykes C, Jivotovskaya A, Hinnebusch AG. Mol Cell Biol. 2006;26:2984–2998. doi: 10.1128/MCB.26.8.2984-2998.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asano K, Clayton J, Shalev A, Hinnebusch AG. Genes Dev. 2000;14:2534–2546. doi: 10.1101/gad.831800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valasek L, Nielsen KH, Zhang F, Fekete CA, Hinnebusch AG. Mol Cell Biol. 2004;24:9437–9455. doi: 10.1128/MCB.24.21.9437-9455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valasek L, Nielsen KH, Hinnebusch AG. EMBO J. 2002;21:5886–5898. doi: 10.1093/emboj/cdf563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsen DS, Savner EM, Mathew A, Zhang F, Krishnamoorthy T, Phan L, Hinnebusch AG. EMBO J. 2003;22:193–204. doi: 10.1093/emboj/cdg030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fletcher CM, Pestova TV, Hellen CU, Wagner G. EMBO J. 1999;18:2631–2637. doi: 10.1093/emboj/18.9.2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bandyopadhyay A, Maitra U. Nucleic Acids Res. 1999;27:1331–1337. doi: 10.1093/nar/27.5.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Methot N, Song MS, Sonenberg N. Mol Cell Biol. 1996;16:5328–5334. doi: 10.1128/mcb.16.10.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park HS, Browning KS, Hohn T, Ryabova LA. EMBO J. 2004;23:1381–1391. doi: 10.1038/sj.emboj.7600140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vornlocher HP, Hanachi P, Ribeiro S, Hershey JW. J Biol Chem. 1999;274:16802–16812. doi: 10.1074/jbc.274.24.16802. [DOI] [PubMed] [Google Scholar]
- 23.Korneeva NL, Lamphear BJ, Hennigan FLC, Rhoads RE. J Biol Chem. 2000;275:41369–41376. doi: 10.1074/jbc.M007525200. [DOI] [PubMed] [Google Scholar]
- 24.Jivotovskaya AV, Valasek L, Hinnebusch AG, Nielsen KH. Mol Cell Biol. 2006;26:1355–1372. doi: 10.1128/MCB.26.4.1355-1372.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holz MK, Ballif BA, Gygi SP, Blenis J. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 26.Harris TE, Chi A, Shabanowitz J, Hunt DF, Rhoads RE, Lawrence JC., Jr EMBO J. 2006;25:1659–1668. doi: 10.1038/sj.emboj.7601047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamphear BJ, Kirchweger R, Skern T, Rhoads RE. J Biol Chem. 1995;270:21975–21983. doi: 10.1074/jbc.270.37.21975. [DOI] [PubMed] [Google Scholar]
- 28.Tarun SZ, Sachs AB. EMBO J. 1996;15:7168–7177. [PMC free article] [PubMed] [Google Scholar]
- 29.Imataka H, Gradi A, Sonenberg N. EMBO J. 1998;17:7480–7489. doi: 10.1093/emboj/17.24.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pyronnet S, Imataka H, Gingras AC, Fukunaga R, Hunter T, Sonenberg N. EMBO J. 1999;18:270–279. doi: 10.1093/emboj/18.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imataka H, Sonenberg N. Mol Cell Biol. 1997;17:6940–6947. doi: 10.1128/mcb.17.12.6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Mol Cell Biol. 1999;19:1871–1880. doi: 10.1128/mcb.19.3.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK. Mol Cell. 1999;3:707–716. doi: 10.1016/s1097-2765(01)80003-4. [DOI] [PubMed] [Google Scholar]
- 34.Ptushkina M, von der Haar T, Karimm MM, Hughers JM, McCarthy JEG. EMBO J. 1999;18:4068–4075. doi: 10.1093/emboj/18.14.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korneeva NL, First EA, Benoit CA, Rhoads RE. J Biol Chem. 2005;280:1872–1881. doi: 10.1074/jbc.M406168200. [DOI] [PubMed] [Google Scholar]
- 36.Scheper GC, Parra JL, Wilson M, Van Kollenburg B, Vertegaal AC, Han ZG, Proud CG. Mol Cell Biol. 2003;23:5692–5705. doi: 10.1128/MCB.23.16.5692-5705.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kessler SH, Sachs AB. Mol Cell Biol. 1998;18:51–57. doi: 10.1128/mcb.18.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otero LJ, Ashe MP, Sachs AB. EMBO J. 1999;18:3153–3163. doi: 10.1093/emboj/18.11.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siridechadilok B, Fraser CS, Hall RJ, Doudna JA, Nogales E. Science. 2005;310:1513–1515. doi: 10.1126/science.1118977. [DOI] [PubMed] [Google Scholar]
- 40.Reynolds JE, Kaminski A, Kettinen HJ, Grace K, Clarke BE, Carroll AR, Rowlands DJ, Jackson RJ. EMBO J. 1995;14:6010–6020. doi: 10.1002/j.1460-2075.1995.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pestova TV, Shatsky IN, Fletcher SP, Jackson RJ, Hellen CU. Genes Dev. 1998;12:67–83. doi: 10.1101/gad.12.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korneeva NL, Lamphear BJ, Hennigan FLC, Merrick WC, Rhoads RE. J Biol Chem. 2001;276:2872–2879. doi: 10.1074/jbc.M006345200. [DOI] [PubMed] [Google Scholar]
- 43.Jemielity J, Fowler T, Zuberek J, Stepinski J, Lewdorowicz M, Niedzwiecka A, Stolarski R, Darzynkiewicz E, Rhoads RE. RNA (N Y) 2003;9:1108–1122. doi: 10.1261/rna.5430403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grudzien E, Kalek M, Jemielity J, Darzynkiewicz E, Rhoads RE. J Biol Chem. 2006;281:1857–1867. doi: 10.1074/jbc.M509121200. [DOI] [PubMed] [Google Scholar]
- 45.Cai A, Jankowska-Anyszka M, Centers A, Chlebicka L, Stepinski J, Stolarski R, Darzynkiewicz E, Rhoads RE. Biochemistry. 1999;38:8538–8547. doi: 10.1021/bi9830213. [DOI] [PubMed] [Google Scholar]
- 46.Sechi S, Chait BT. Anal Chem. 1998;70:5150–5158. doi: 10.1021/ac9806005. [DOI] [PubMed] [Google Scholar]
- 47.Yan R, Rychlik W, Etchison D, Rhoads RE. J Biol Chem. 1992;267:23226–23231. [PubMed] [Google Scholar]
- 48.Meyer LJ, Milburn SC, Hershey JWB. Biochemistry. 1982;21:4206–4212. doi: 10.1021/bi00261a003. [DOI] [PubMed] [Google Scholar]
- 49.Asano K, Kinzy TG, Merrick WC, Hershey JW. J Biol Chem. 1997;272:1101–1109. doi: 10.1074/jbc.272.2.1101. [DOI] [PubMed] [Google Scholar]
- 50.Sorensen P, Farber NM, Krystal G. J Biol Chem. 1986;261:9094–9097. [PubMed] [Google Scholar]
- 51.Wollenweber HW, Morrison DC. J Biol Chem. 1985;260:15068–15074. [PubMed] [Google Scholar]
- 52.Shephard EG, de Beer FC, von Holt C, Hapgood JP. Anal Biochem. 1988;168:306–313. doi: 10.1016/0003-2697(88)90323-5. [DOI] [PubMed] [Google Scholar]
- 53.Altmann M, Schmitz N, Berset C, Trachsel H. EMBO J. 1997;16:1114–1121. doi: 10.1093/emboj/16.5.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vende P, Piron M, Castagne N, Poncet D. J Virol. 2000;74:7064–7071. doi: 10.1128/jvi.74.15.7064-7071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hunter T, Hunt T, Jackson RJ, Robertson HD. J Biol Chem. 1975;250:409–417. [PubMed] [Google Scholar]
- 56.Lodish HF. Nature. 1974;251:385–388. doi: 10.1038/251385a0. [DOI] [PubMed] [Google Scholar]
- 57.Jackson RJ. In: Translational Control of Gene Expression. Sonenberg N, Hershey JWB, Mathews MB, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 127–184. [Google Scholar]
- 58.Pestova TV, Kolupaeva VG, Lomakin IB, Pilipenko EV, Shatsky IN, Agol VI, Hellen CU. Proc Natl Acad Sci U S A. 2001;98:7029–7036. doi: 10.1073/pnas.111145798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park HS, Himmelbach A, Browning KS, Hohn T, Ryabova LA. Cell. 2001;106:723–733. doi: 10.1016/s0092-8674(01)00487-1. [DOI] [PubMed] [Google Scholar]
- 60.Kim TH, Kim BH, Yahalom A, Chamovitz DA, von Arnim AG. Plant Cell. 2004;16:3341–3356. doi: 10.1105/tpc.104.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dong Z, Liu LH, Han B, Pincheira R, Zhang JT. Oncogene. 2004;23:3790–3801. doi: 10.1038/sj.onc.1207465. [DOI] [PubMed] [Google Scholar]
- 62.Bandyopadhyay A, Lakshmanan V, Matsumoto T, Chang EC, Maitra U. J Biol Chem. 2002;277:2360–2367. doi: 10.1074/jbc.M107790200. [DOI] [PubMed] [Google Scholar]
- 63.Mendez R, Myers MG, White MF, Rhoads RE. Mol Cell Biol. 1996;16:2857–2864. doi: 10.1128/mcb.16.6.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Unbehaun A, Borukhov SI, Hellen CU, Pestova TV. Genes Dev. 2004;18:3078–3093. doi: 10.1101/gad.1255704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sizova DV, Kolupaeva VG, Pestova TV, Shatsky IN, Hellen CU. J Virol. 1998;72:4775–4782. doi: 10.1128/jvi.72.6.4775-4782.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Methot N, Rom E, Olsen H, Sonenberg N. J Biol Chem. 1997;272:1110–1116. doi: 10.1074/jbc.272.2.1110. [DOI] [PubMed] [Google Scholar]
- 67.Verlhac MH, Chen RH, Hanachi P, Hershey JW, Derynck R. EMBO J. 1997;16:6812–6822. doi: 10.1093/emboj/16.22.6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hui DJ, Bhasker CR, Merrick WC, Sen GC. J Biol Chem. 2003;278:39477–39482. doi: 10.1074/jbc.M305038200. [DOI] [PubMed] [Google Scholar]
- 69.He H, von der Haar T, Singh CR, Ii M, Li B, Hinnebusch AG, McCarthy JE, Asano K. Mol Cell Biol. 2003;23:5431–5445. doi: 10.1128/MCB.23.15.5431-5445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Asano K, Shalev A, Phan L, Nielsen K, Clayton J, Valasek L, Donahue TF, Hinnebusch AG. EMBO J. 2001;20:2326–2337. doi: 10.1093/emboj/20.9.2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scheel H, Hofmann K. BMC Bioinformatics. 2005;6:71. doi: 10.1186/1471-2105-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.von Arnim AG, Chamovitz DA. Curr Biol. 2003;13:R323–R325. doi: 10.1016/s0960-9822(03)00238-0. [DOI] [PubMed] [Google Scholar]
- 73.Hoareau Alves K, Bochard V, Rety S, Jalinot P. FEBS Lett. 2002;527:15–21. doi: 10.1016/s0014-5793(02)03147-2. [DOI] [PubMed] [Google Scholar]
- 74.Yahalom A, Kim TH, Winter E, Karniol B, von Arnim AG, Chamovitz DA. J Biol Chem. 2001;276:334–340. doi: 10.1074/jbc.M006721200. [DOI] [PubMed] [Google Scholar]
- 75.Dunand-Sauthier I, Walker C, Wilkinson C, Gordon C, Crane R, Norbury C, Humphrey T. Mol Biol Cell. 2002;13:1626–1640. doi: 10.1091/mbc.01-06-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hui DJ, Terenzi F, Merrick WC, Sen GC. J Biol Chem. 2005;280:3433–3440. doi: 10.1074/jbc.M406700200. [DOI] [PubMed] [Google Scholar]
- 77.Desbois C, Rousset R, Bantignies F, Jalinot P. Science. 1996;273:951–953. doi: 10.1126/science.273.5277.951. [DOI] [PubMed] [Google Scholar]
- 78.Guo J, Sen GC. J Virol. 2000;74:1892–1899. doi: 10.1128/jvi.74.4.1892-1899.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pincheira R, Chen Q, Zhang JT. Br J Cancer. 2001;84:1520–1527. doi: 10.1054/bjoc.2001.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nupponen NN, Porkka K, Kakkola L, Tanner M, Persson K, Borg A, Isola J, Visakorpi T. Am J Pathol. 1999;154:1777–1783. doi: 10.1016/S0002-9440(10)65433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marchetti A, Buttitta F, Miyazaki S, Gallahan D, Smith GH, Callahan R. J Virol. 1995;69:1932–1938. doi: 10.1128/jvi.69.3.1932-1938.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rasmussen S, Kordon E, Callahan R, Smith G. Oncogene. 2001;20:5291–5301. doi: 10.1038/sj.onc.1204624. [DOI] [PubMed] [Google Scholar]
- 83.Mayeur GL, Hershey JW. FEBS Lett. 2002;514:49–54. doi: 10.1016/s0014-5793(02)02307-4. [DOI] [PubMed] [Google Scholar]
- 84.Marchetti A, Buttitta F, Pellegrini S, Bertacca G, Callahan R. Int J Oncol. 2001;18:175–179. doi: 10.3892/ijo.18.1.175. [DOI] [PubMed] [Google Scholar]
- 85.Asano K, Merrick WC, Hershey JW. J Biol Chem. 1997;272:23477–23480. doi: 10.1074/jbc.272.38.23477. [DOI] [PubMed] [Google Scholar]
- 86.Gradi A, Imataka H, Svitkin YV, Rom E, Raught B, Morino S, Sonenberg N. Mol Cell Biol. 1998;18:334–342. doi: 10.1128/mcb.18.1.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Byrd MP, Zamora M, Lloyd RE. Mol Cell Biol. 2002;22:4499–4511. doi: 10.1128/MCB.22.13.4499-4511.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bradley CA, Padovan JC, Thompson TL, Benoit CA, Chait BT, Rhoads RE. J Biol Chem. 2002;277:12559–12571. doi: 10.1074/jbc.M111134200. [DOI] [PubMed] [Google Scholar]
- 89.Clark BFC, Grunberg-Manago M, Gupta NK, Hershey JWB, Hinnebusch AG, Jackson RJ, Maitra U, Mathews MB, Merrick WC, Rhoads RE, Sonenberg N, Spremulli LL, Trachsel H, Voorma HO. Biochimie (Paris) 1996;78:1119–1122. [Google Scholar]