

Summary
Feedback inhibition of the PI3K-Akt pathway by the mammalian target of rapamycin complex 1 (mTORC1) has emerged as an important signaling event in tumor syndromes, cancer, and insulin resistance. Cells lacking the tuberous sclerosis complex (TSC) gene products are a model for this feedback regulation. We find that, despite Akt attenuation, the Akt substrate GSK3 is constitutively phosphorylated in cells and tumors lacking TSC1 or TSC2. In these settings, GSK3 phosphorylation is sensitive to mTORC1 inhibition by rapamycin or amino-acid withdrawal, and GSK3 becomes a direct target of S6K1. This aberrant phosphorylation leads to decreased GSK3 activity and phosphorylation of downstream substrates and contributes to the growth factor-independent proliferation of TSC-deficient cells. We find that GSK3 can also be regulated downstream of mTORC1 in a HepG2 model of cellular insulin resistance. Therefore, we define conditions in which S6K1, rather than Akt, is the predominant GSK3 regulatory kinase.
Introduction
The ser/thr kinase Akt (also known as PKB) is activated downstream of growth factor-stimulated PI3K activity and phosphorylates a variety of downstream substrates. The best-characterized Akt targets are the Forkhead box O (FoxO) family of transcription factors (Brunet et al., 1999) and glycogen synthase kinase 3 (GSK3) (Cross et al., 1995). FoxO proteins (FoxO1, FoxO3a, FoxO4) regulate diverse processes through transcriptional effects on a large number of gene targets, such as those encoding metabolic enzymes, pro-apoptotic factors, and cell-cycle inhibitory proteins (Accili and Arden, 2004). GSK3 (GSK3α and GSK3β) regulates an equally diverse array of cellular processes by directly phosphorylating many substrates, including metabolic enzymes, transcription factors, cell-cycle regulatory proteins, and cytoskeletal proteins (Cohen and Goedert, 2004; Jope and Johnson, 2004). Akt’s phosphorylation of both FoxO and GSK3 is inhibitory (Brunet et al., 1999; Cross et al., 1995). Therefore, under conditions where the PI3K-Akt pathway is inactive (e.g., growth factor withdrawal), FoxO proteins and GSK3 are active and, in a cell context-dependent manner, help drive cell-cycle arrest and/or apoptosis.
A more recently identified Akt substrate is the TSC2 protein (also called tuberin), which is a tumor suppressor that forms a heterodimeric complex with the TSC1 protein (also called hamartin). Mutations in TSC1 or TSC2 give rise to the hamartoma syndrome tuberous sclerosis complex (TSC) and the proliferative lung disorder lymphangioleiomyomatosis (LAM). The TSC1-2 complex negatively regulates the mammalian target of rapamycin (mTOR)-raptor complex (mTORC1). TSC2 is a GTPase-activating protein (GAP) for the Ras-related small G protein Rheb, which somehow activates mTORC1 kinase activity when in its GTP-bound active form (reviewed in Sarbassov et al., 2005a). While the molecular mechanism is not fully understood, Akt-mediated multi-site phosphorylation of TSC2 (Inoki et al., 2002; Manning et al., 2002) inhibits its ability to act as a GAP towards Rheb within cells, thereby allowing Rheb-GTP to activate mTORC1 signaling.
mTOR activation is downstream of PI3K-Akt signaling in this pathway, however there is ample evidence that mTOR reciprocally regulates the growth-factor responsiveness of PI3K and Akt. When mTOR is associated with rictor in the mTOR complex 2 (mTORC2), it directly phosphorylates Akt on S473, thereby contributing to Akt activation (Sarbassov et al., 2005b). However, mTORC1-specific inhibitors, such as rapamycin, have been found to enhance PI3K-Akt activation in many cell lines (e.g., O’Reilly et al., 2006; Sun et al., 2005; Tremblay et al., 2005), demonstrating the existence of a basal mTORC1-dependent feedback mechanism. This feedback regulation has garnered significant interest over the past few years, as it could have widespread implications for the pathology and treatment of common human diseases, such as cancer and diabetes, as well as TSC and LAM (reviewed in Harrington et al., 2005; Manning, 2004; Sabatini, 2006)). However, the precise mechanism and downstream consequences of mTORC1 feedback inhibition of PI3K-Akt signaling are, as yet, poorly defined.
Interestingly, cells and tumors lacking a functional TSC1-2 complex exhibit constitutively high levels of mTORC1 activity and, consequently, are strongly defective in growth factor-stimulated PI3K-Akt signaling (Jaeschke et al., 2002; Kwiatkowski et al., 2002; Manning et al., 2005; Radimerski et al., 2002; Zhang et al., 2003). Studies on TSC-deficient cells have revealed multiple mTORC1-dependent feedback mechanisms, as Akt is less responsive to insulin, IGF1, PDGF, and even serum, and prolonged exposure to rapamycin can restore Akt activation in these cells (Harrington et al., 2004; Shah and Hunter, 2006; Shah et al., 2004). Therefore, loss of the TSC1-2 complex provides mTORC1 gain of function evidence to support the many studies that have used rapamycin to implicate mTORC1 signaling in the feedback inhibition of PI3K and Akt. Given the robust and genetically defined nature of systems lacking TSC1 or TSC2, they are ideal for studying the molecular effects of this mTORC1-driven feedback loop.
In a previous study, we found that Akt signaling to its downstream targets FOXO1 and FOXO3a was defective in Tsc2-deficient cells and tumors due to feedback inhibition (Manning et al., 2005). In the study described here, we were surprised to find that in these same cells, GSK3 phosphorylation, on the site normally phosphorylated by Akt, occurs in a growth factor-independent manner and is elevated in both mouse and human TSC tumors. We find that loss of TSC1-2 complex function leads to constitutive phosphorylation and inhibition of GSK3 due to aberrant S6K1 activity. This alternative regulation of GSK3 leads to misregulation of GSK3 substrates and contributes to the ability of TSC-deficient cells to proliferate in the absence of growth factors. Finally, we provide evidence that mTORC1-S6K1 signaling can regulate GSK3 under other conditions of cellular insulin resistance. The mTORC1-mediated inhibition of GSK3 in these settings has clear implications for TSC and LAM and, potentially, for cancer and type-2 diabetes.
Results
Aberrant GSK3 phosphorylation in TSC-deficient cells and tumors
As stated above, cells lacking either TSC1 or TSC2 are defective in growth factor-mediated activation of Akt. This is reflected in a diminished phosphorylation of Akt and its substrate FOXO3a in Tsc2−/− MEFs relative to littermate-derived wild-type cells, both basally and in response to serum, IGF1, or PDGF (Figure 1A). Surprisingly, a second Akt substrate, GSK3β, is highly phosphorylated in these cells, even under serum starvation conditions, and is not further stimulated by growth factors. This constitutive phosphorylation of GSK3β on the inhibitory Akt site (S9) parallels that of S6K1 phosphorylation on T389, which reflects the high levels of growth factor-independent mTORC1 signaling typical of TSC null cells.
Figure 1. GSK3 phosphorylation is constitutive in Tsc2 null cells and is elevated in TSC tumors.
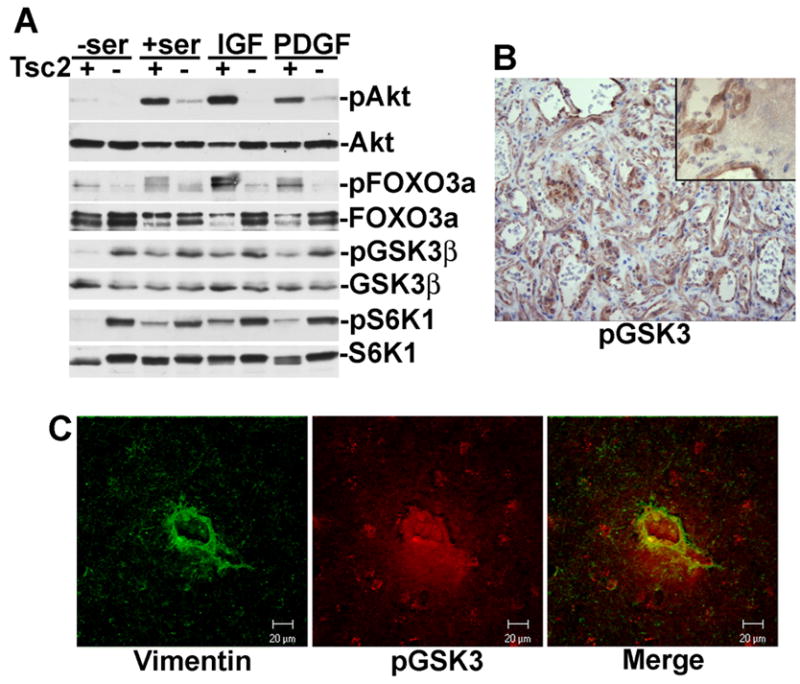
(A) Differential regulation of the Akt substrates FOXO3a and GSK3 in Tsc2−/− MEFs. Littermate-derived Tsc2+/+ (+) and Tsc2−/− (−) MEFs were cultured in the absence (−ser) or presence (+ser) of serum for 16 h, or serum starved then stimulated for 15 min with 50 ng/ml IGF1 or 5 ng/ml.
(B) GSK3 phosphorylation is elevated in liver hemangiomas from Tsc2+/− mice. A representative example from five tumors obtained from five different Tsc2+/− mice aged 12 months is shown at 200X magnification; inset with adjacent normal tissue is shown at 1000X. Phospho-GSK3 is depicted in brown; nuclei are counterstained in blue.
(C) GSK3 phosphorylation is elevated in cortical tuber giant cells from a TSC patient. A representative giant cell from a cortical tuber resection is shown stained with vimentin (green) and phospho-GSK3 (red). Bar: 20 μM.
As we had previously demonstrated defects in Akt activation and signaling to FOXO1 in the liver hemangiomas of a mouse model of TSC (Manning et al., 2005), we next examined GSK3 phosphorylation in these lesions. As in TSC null MEFs, phosphorylated GSK3 was detected in the liver hemangiomas arising in Tsc2+/− mice (Figure 1B) and was substantially higher in the cells comprising the tumor than in adjacent normal tissue (Figure 1B, inset). Therefore, these tumors, which contain elevated mTORC1 signaling concomitant with Akt attenuation (Manning et al., 2005), exhibit high levels of GSK3 phosphorylation.
Tumors classified as cortical tubers are a hallmark of the TSC brain and contain abnormally large cells, referred to as “giant cells”, which express both neuronal and glial markers (Crino, 2004). These cells, which are readily detected by vimentin staining (Hirose et al., 1995), display elevated mTORC1 signaling (Crino, 2004). We found that the majority (77.4%) of vimentin-positive giant cells (>30 μm in diameter) within surgical resections from a TSC patient exhibited high levels of GSK3 phosphorylation (representative cell shown in Figure 1C). Therefore, like markers of mTORC1 activation, GSK3 phosphorylation is elevated in TSC-deficient cells and tumors.
PI3K-independent/mTORC1-dependent phosphorylation of GSK3 in cells lacking the TSC1-2 complex
To determine the mechanism of alternative GSK3 regulation in TSC-deficient cells, we compared the sensitivities of GSK3 phosphorylation to the PI3K inhibitor wortmannin and the mTORC1 inhibitor rapamcyin in littermate-derived pairs of wild-type and Tsc1 or Tsc2 null MEFs. In wild-type MEFs, the phosphorylation of Akt and its downstream targets FOXO3a and GSK3α/β are stimulated by IGF1, inhibited by wortmannin, and completely resistant to rapamycin (Figure 2A-Tsc1+/+, 2B-Tsc2+/+). However, in both Tsc1−/− and Tsc2−/− MEFs, Akt and FOXO3a are not phosphorylated, while GSK3α and β are constitutively phosphorylated in a manner that is resistant to wortmannin and sensitive to rapamcyin (Figure 2A-Tsc1−/−, 2B-Tsc2−/−). In the TSC-deficient cells, the sensitivity of GSK3 phosphorylation to these drugs is identical to that of S6 phosphorylation, while in wild-type cells S6 phosphorylation is sensitive to both wortmannin and rapamycin.
Figure 2. Loss of the TSC1-2 complex leads to wortmannin-resistant rapamycin-sensitive phosphorylation of GSK3.
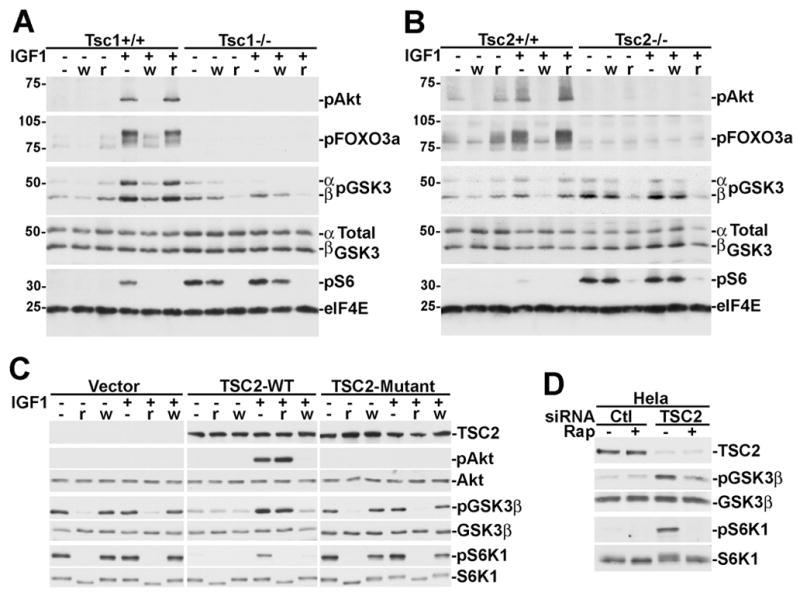
(A) Littermate-derived Tsc1+/+ and Tsc1−/− MEFs were serum starved for 16 h and then pretreated for 15 min with 100 nM wortmannin (w) or 20 nM rapamycin (r) then stimulated for 15 min with 25 ng/ml IGF1, where indicated. Lysates were then immunoblotted with the indicated phospho-specific antibodies or antibodies to total GSK3 and eIF4E as loading controls.
(B) Cells were treated as described in A, but using littermate-derived Tsc2+/+ and Tsc2−/− MEFs.
(C) Rescue of the alternative regulation of GSK3 in Tsc2−/− MEFs with wild-type human TSC2. Retrovirus-infected pools of Tsc2−/− MEFs stably expressing empty vector, human TSC2, or a patient-derived mutant of TSC2 (P419S) were treated as described in A.
(D) TSC2 knockdown in HeLa cells leads to constitutive rapamycin-sensitive GSK3 phosphorylation. 48 hours post-transfection with control or TSC2 siRNAs, HeLa cells were serum starved for 16 h and treated for 15 min with 20 nM rapamycin, where indicated.
In order to further demonstrate that this misregulation of GSK3 results from loss of TSC1-2 complex function, we examined GSK3 phosphorylation in Tsc2−/− MEFs reconstituted with empty vector, wild-type TSC2, or a TSC patient-derived mutation of TSC2. Wild-type TSC2, expressed at levels similar to endogenous (Figure S1A), restored growth factor-responsive, wortmannin-sensitive, rapamycin-resistant phosphorylation of Akt and GSK3 (Figure 2C). However, TSC patient mutant-reconstituted cells were identical to empty vector, as both displayed Akt attenuation and constitutive rapamycin-sensitive phosphorylation of GSK3. As expected, wild-type TSC2, but not the patient mutant, also restored growth-factor dependent S6K1 phosphorylation that was sensitive to both wortmannin and rapamycin.
To determine whether the aberrant phosphorylation of GSK3 requires chronic loss of the TSC1-2 complex or whether transient loss is sufficient, we knocked down TSC2 expression in HeLa cells using siRNAs. Upon efficient knock down of TSC2, growth factor-independent phosphorylation of both S6K1 and GSK3 is evident following 16 or 24 h serum starvation, and both are acutely sensitive to rapamycin (Figures 2D and S1B). Therefore, functional inactivation of the TSC1-2 complex leads to constitutive and rapamycin-sensitive GSK3 phosphorylation.
These data strongly suggest that GSK3 phosphorylation is independent of PI3K but downstream of mTORC1 in TSC-deficient cells. However, in some of our treatments, we detected a slight sensitivity of GSK3 phosphorylation to wortmannin, albeit much less than wild-type cells. As PI3K-Akt signaling is defective in these cells, it is likely that this effect is due to sensitivity of the PI3K-related kinase mTOR to wortmannin (Brunn et al., 1996), or to contributions from the class III PI3K, hVPS34 (Nobukuni et al., 2005). Therefore, we compared the sensitivity of GSK3 and S6K1 phosphorylation to increasing doses of wortmannin (15-min pretreatment) in wild-type versus Tsc2−/− MEFs. In wild-type cells, 50-nM wortmannin greatly reduces and 100 nM eliminates the phosphorylation of Akt, GSK3, and S6K1 (Figure 3A), indicative of PI3K inhibition. However, in Tsc2−/− cells these doses have only a slight effect on both GSK3 and S6K1, while rapamycin eliminates their phosphorylation. At higher doses, wortmannin is known to directly inhibit mTOR kinase activity (Brunn et al., 1996), and this is reflected in both GSK3 and S6K1 phosphorylation being inhibited at doses of 500 nM and above in the Tsc2 null cells.
Figure 3. mTORC1-dependent phosphorylation of GSK3 in Tsc2 null cells.
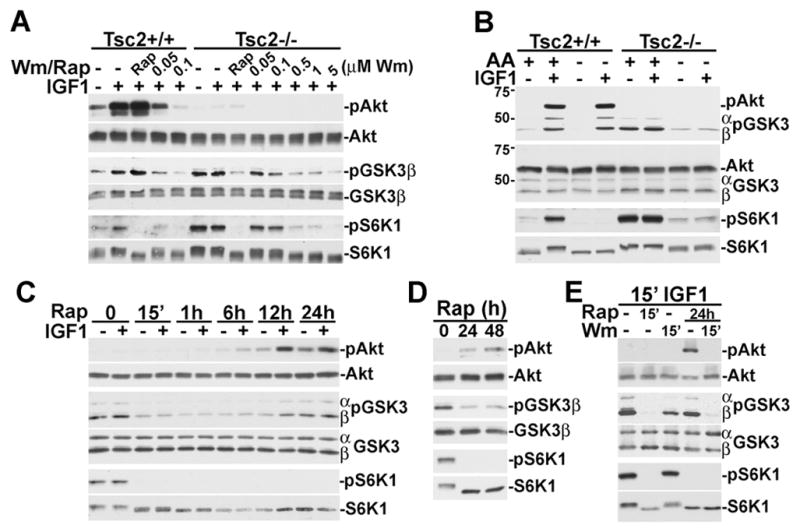
(A) S6K1 and GSK3 phosphorylation are more resistant to wortmannin in Tsc2−/− cells. Tsc2+/+ and Tsc2−/− MEFs were serum starved for 16 h and then pretreated for 15 min with 20 nM rapamycin or increasing doses of wortmannin (50 nM up to 5 μM) then stimulated for 15 min with 50 ng/ml IGF1.
(B) Amino acid-dependent phosphorylation of GSK3 in Tsc2−/− cells. Tsc2+/+ and Tsc2−/−MEFs were serum starved for 16 h and then put in fresh media with or without amino acids for an additional 2 hours prior to 15 min stimulation with 50 ng/ml IGF1, where indicated. Immunoblots for phospho and total Akt and GSK3 were performed together.
(C) Growth factor-responsive phosphorylation of Akt and GSK3 is restored to Tsc2−/−cells after prolonged rapamycin treatment. Tsc2−/− MEFs were serum starved for 24 h in the presence of 20 nM rapamycin for the specified duration prior to 15 min stimulation with 25 ng/ml IGF1, where indicated.
(D) Prolonged rapamycin reduces basal GSK3 phosphorylation in Tsc2−/− cells. Tsc2−/−MEFs were serum starved for 48 h in the presence of 20 nM rapamycin for the specified duration.
(E) Prolonged rapamycin treatment restores wortmannin-sensitive GSK3 phosphorylation to Tsc2−/− cells. Tsc2−/− MEFs were serum starved for 24 h in the presence or absence of 20 nM rapamycin, were pretreated for 15 min with 20 nM rapamycin or 100 nM wortmannin, where indicated, and then stimulated for 15 min with 25 ng/ml IGF1.
In order to further confirm that GSK3 phosphorylation is driven by mTORC1 signaling in TSC-deficient cells, we examined its sensitivity to the presence of amino acids, an obligate stimulus of mTORC1 activity (Hara et al., 1998). As found previously for S6K1 (Nobukuni et al., 2005), in Tsc2−/− cells the constitutive phosphorylation of GSK3 is dependent on amino acids (Figure 3B). However, unlike S6K1, in wild-type cells the IGF1-stimulated phosphorylations of Akt and GSK3 are not sensitive to amino acid withdrawal. These data further demonstrate that upon loss of the TSC1-2 complex there is a switch from growth factor-responsive Akt-dependent GSK3 phosphorylation to nutrient-sensitive mTORC1-dependent GSK3 phosphorylation.
Akt signaling is attenuated in TSC-deficient cells by a rapamycin-sensitive feedback mechanism, and full restoration of growth-factor responsive Akt activation requires prolonged exposure to rapamycin (Harrington et al., 2004; Shah et al., 2004). Therefore, we tested the effects of different durations of rapamycin treatment on GSK3 phosphorylation in Tsc2−/− cells. GSK3 phosphorylation is eliminated within 15 min, as in other experiments, and remains low after 1-h and 6-h treatments, with IGF1 having no stimulatory effect (Figure 3C). However, after 12 and 24-h treatments, GSK3 phosphorylation is responsive to IGF1, as is Akt. While basal phosphorylation of GSK3 is also increased by prolonged rapamycin, it is still significantly lower than the constitutive phosphorylation in untreated TSC-deficient cells (Figure 3D). Consistent with Akt phosphorylating GSK3 after prolonged rapamycin, the restored IGF1-stimulated GSK3 phosphorylation is completely sensitive to wortmannin (Figure 3E).
S6K1 is the GSK3 kinase in TSC-deficient cells
For several reasons, S6K1 was the strongest candidate for the mTORC1-dependent kinase responsible for GSK3 phosphorylation in the absence of Akt signaling. First, S6K1 and S6K2 are the only members of the AGC kinase family, to which Akt belongs, known to be acutely sensitive to mTORC1 inhibition. Second, Akt and S6K1 have similar substrate specificities in vitro (Alessi et al., 1996), and the Akt sites on GSK3α (S21) and β (S9) closely resemble those of the S6K1 sites on eIF4B (S422; Raught et al., 2004) and ribosomal S6 (S236; Flotow and Thomas, 1992) (Figure 4A). Finally, S6K1 was found previously to be capable of phosphorylating GSK3α and β in vitro (Sutherland and Cohen, 1994; Sutherland et al., 1993). Therefore, we first tested whether the constitutively active S6K1 from Tsc2−/− MEFs or HeLa cells with siRNA-mediated knock down of TSC2 could directly phosphorylate GSK3. We found that endogenous S6K1 isolated from serum-starved TSC2-deficient cells, but not control cells, could phosphorylate recombinant GSK3β on S9 in vitro, and this activity is sensitive to pretreatment of the cells with rapamycin (Figure 4B,C).
Figure 4. S6K1 phosphorylates GSK3 in TSC-deficient cells.
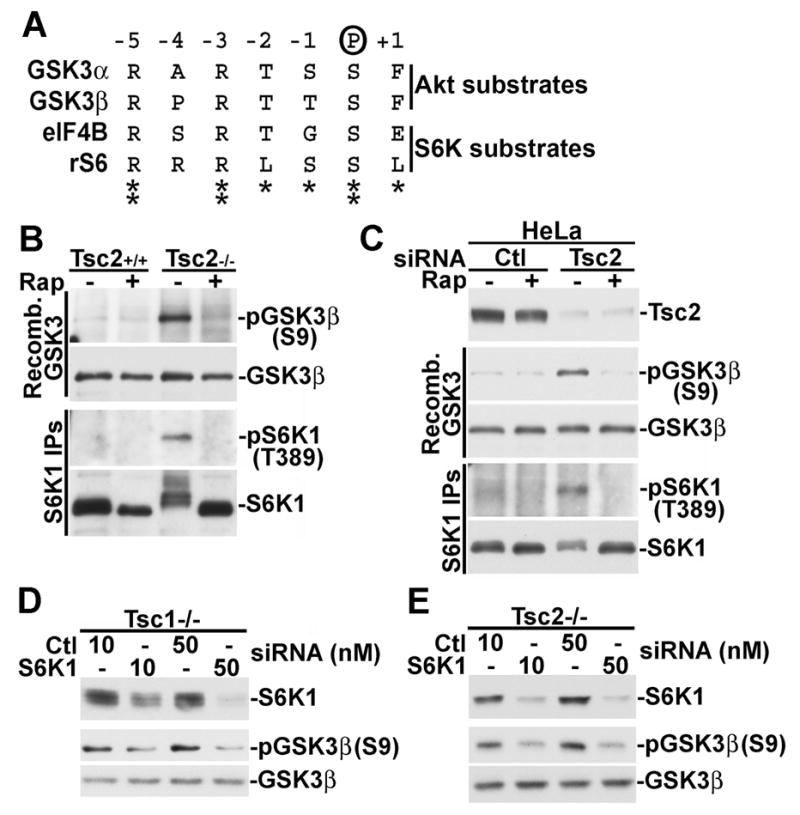
(A) The corresponding phosphorylation sites on GSK3 and the S6K1 substrates eIF4B and S6 are similar. The residues flanking GSK3α-S21, GSK3β-S9, eIF4B-S422, and ribosomal S6-S236 are aligned.
(B) S6K1 from serum-starved Tsc2−/− MEFs, but not wild-type MEFs, can phosphorylate GSK3β-S9 in vitro. Tsc2+/+ and Tsc2−/− MEFs were serum starved for 16 h and then treated for 15 min with 20 nM rapamycin, where indicated. Immunoprecipitated S6K1 was used in an in vitro kinase assay with bacterially produced GSK3β as the substrate. GSK3 phosphorylation on S9 was detected using a phospho-specifc antibody.
(C) S6K1 from serum-starved HeLa cells treated with TSC2 siRNAs, but not control siRNAs, can phosphorylate GSK3β-S9 in vitro. 24 h post-transfection with control or TSC2-targetting siRNAs, HeLa cells were treated as described in B.
(D) S6K1 knockdown in Tsc1−/− cells blocks the constitutive phosphorylation of GSK3. 24 hours post-transfection with the indicated doses of control or S6K1-targetting siRNAs, Tsc1−/− MEFs were serum starved for 16 hours.
(E) Tsc2−/− MEFs were treated as described in D.
To test whether S6K1 is the in vivo GSK3 kinase in TSC null cells, we used S6K1-specific siRNAs to reduce S6K1 levels in both Tsc1−/− and Tsc2−/− MEFs. GSK3 is highly phosphorylated under serum starvation conditions in these TSC-deficient cells, and knockdown with S6K1 siRNAs, but not control siRNAs, nearly abolish this constitutive phosphorylation in both cell lines (Figure 4D,E). Interestingly, siRNAs targeting S6K2, alone or in combination with S6K1 siRNAs, had little effect on GSK3 phosphorylation in these cells, despite significantly decreasing S6 phosphorylation (Figure S2). Therefore, constitutive S6K1 activity in cells lacking the TSC1-2 complex leads to growth-factor independent phosphorylation of GSK3 on the same site that Akt phosphorylates in wild-type cells.
Decreased GSK3 kinase activity and activation by rapamycin in TSC-deficient cells
GSK3 is an unusual kinase in that it is activated by growth-factor withdrawal due to deactivation of its inhibitory kinase Akt. Our finding that GSK3 is constitutively phosphorylated by S6K1 in TSC-deficient MEFs suggests that GSK3 remains inhibited in these cells under serum-starvation conditions. To test this, we compared endogenous GSK3 kinase activity in immunoprecipitates from serum-starved littermate-derived Tsc2+/+ and Tsc2−/− MEFs. Consistent with the inhibitory phosphorylation of GSK3 under these conditions, GSK3 activity was significantly lower in the Tsc2−/− cells relative to the wild-type cells (Figure 5A). This ~40% decease in activity is similar to that reported for insulin-stimulated GSK3 inhibition, which is caused by Akt-mediated phosphorylation (Cross et al., 1995). Importantly, while a 30-min treatment with rapamycin had only a minor effect on GSK3 activity in wild-type cells, it strongly activated the kinase in Tsc2−/−cells, consistent with rapamycin blocking the inhibitory phosphorylation in these cells (Figure 5A).
Figure 5. GSK3 activity is decreased in Tsc2 null cells and reactivated by rapamycin.
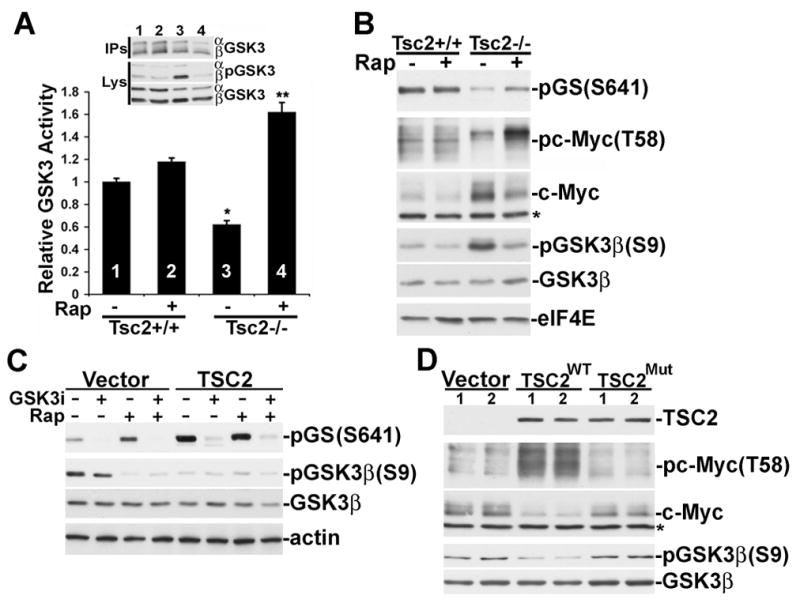
(A) GSK3 kinase activity is lower in serum-starved Tsc2−/− cells and is activated by rapamycin treatment. Tsc2+/+ and Tsc2−/− MEFs were serum starved for 16 h and then treated for 30 min with 20 nM rapamycin (Rap), where indicated. The activity of immunoprecipitated GSK3 was assayed using a primed phospho-peptide derived from glycogen synthase. Data are presented as mean ± SEM activity relative to untreated wild-type cells. *1 versus 3, P<0.01; **3 versus 4, P<0.001.
(B) Phosphorylation of the GSK3 sites on glycogen synthase and c-Myc are decreased in Tsc2−/− cells and restored by rapamycin. Tsc2+/+ and Tsc2−/− MEFs were serum starved for 6 h and then treated for 30 min with Rap, where indicated. Total levels of eIF4E are provided as a loading control.
(C) Rescue of GSK3-dependent glycogen synthase phosphorylation in Tsc2−/− MEFs by human TSC2. Retrovirus-infected pools of Tsc2−/− MEFs were serum starved for 4 h and treated with 10 μM SB216763 and/or Rap, where indicated. Total levels of actin are provided as a loading control.
(D) Rescue of c-Myc phosphorylation and degradation in Tsc2−/− MEFs by human TSC2. Two independent samples each of retrovirus-infected pools of Tsc2−/− MEFs stably expressing empty vector, human TSC2, or a patient-derived mutant of TSC2 (P419S) were serum starved for 4 h prior to lysis. *Indicates cross-reacting band in both B and D.
We also examined the status of GSK3-specific phosphorylation sites on well-characterized substrates within these cells. Following 6 hours of serum starvation, phosphorylation of the GSK3 sites on both glycogen synthase (GS; S641) and c-Myc (T58) were significantly lower in the Tsc2−/− cells than the wild-type cells (Figure 5B). Importantly, short-term rapamycin treatment (30 min) increased the phosphorylation of both GS and c-Myc in the Tsc2−/− cells but had no effect on the wild-type cells. GSK3-mediated phosphorylation of c-Myc on T58 targets it for degradation (Sears et al., 2000), thereby contributing to c-Myc instability upon growth-factor withdrawal. In agreement with this mode of regulation, after 6 h serum starvation c-Myc protein levels are higher in the Tsc2−/− cells than the wild type cells.
To demonstrate that these effects on GSK3 signaling are due to loss of TSC2, we examined Tsc2−/− MEFs reconstituted with vector or human TSC2. After four hours of serum starvation, the TSC2-reconstituted MEFs showed a loss of GSK3 phosphorylation and elevated levels of GSK3-dependent (i.e., SB216763-sensitive) GS phosphorylation (Figure 5C). Furthermore, rapamycin had no effect on GS phosphorylation in the presence of wild-type TSC2, whereas it enhanced GSK3-mediated GS phosphorylation in the vector control cells. We also examined c-Myc phosphorylation and protein levels in these reconstituted MEFs following four hours of serum withdrawal. Compared to vector-control cells, those cells reconstituted with wild-type TSC2 have greatly elevated levels of c-Myc T58 phosphorylation and a corresponding decrease in total c-Myc protein levels (Figure 5D, replicates shown). Finally, Tsc2−/− MEFs reconstituted with the patient-derived mutation are similar to the vector control cells in that they contain low levels of c-Myc phosphorylation and increased levels of total c-Myc. Therefore, the ability of GSK3 to properly regulate downstream targets is defective in cells lacking a functional TSC1-2 complex.
Constitutive GSK3 inhibition contributes to the growth factor-independent proliferation of TSC-deficient cells
We next tested whether defects in GSK3 regulation might affect the proliferation properties of cells lacking the Tsc1-2 complex. In full serum, we find that Tsc2−/− MEFs proliferate slightly slower than their wild-type counterparts (Figure 6A), perhaps due to their decreased responsiveness to serum (Figure 1A; Zhang et al., 2003). However, under serum starvation conditions, the Tsc2−/− cells continue to divide, whereas the wild-type cells arrest (Figure 6B). This serum-free proliferation of Tsc2-deficient cells is dependent on mTORC1 activity, as rapamycin treatment leads to a growth arrest comparable to untreated wild-type cells. Since rapamycin treatment of TSC-deficient cells blocks the constitutive phosphorylation of GSK3 (Figures 2 and 3) and leads to its activation (Figure 5), we hypothesized that GSK3 activation could contribute to the cytostatic effects of rapamycin on these cells. To test this, we cultured Tsc2−/− MEFs for 48 hours in serum-free media in the presence of rapamycin or rapamycin plus one of two structurally distinct GSK3 inhibitors, LiCl or SB216763 (Coghlan et al., 2000; Cohen and Goedert, 2004; Stambolic et al., 1996). Interestingly, co-treatment with either GSK3 inhibitor partially, but significantly, rescues the Tsc2 null cells from the cytostatic effects of rapamycin, as detected by both cell counts (Figure 6C) and BrdU incorporation (Figure 6D). Tsc1−/− MEFs behave in a similar manner under low serum conditions (Figure S3A). However, GSK3 inhibitors have no effect on the proliferation of wild-type cells under these conditions (Figure S3B) nor do they affect the ability of rapamycin to inhibit mTORC1 signaling (data not shown). To assure that the effect of these compounds on Tsc2−/− cells is due specifically to GSK3 inhibition, we knocked down both GSK3α and β using siRNAs (Figure 6E). Compared to the pharmacological inhibitors, this efficient genetic ablation of GSK3 expression even more strongly rescues Tsc2−/−cells from rapamycin-induced growth arrest, while having only a mild effect on vehicle-treated cells (Figure 6F).
Figure 6. Aberrant regulation of GSK3 contributes to the serum-free proliferation property of TSC-deficient cells.
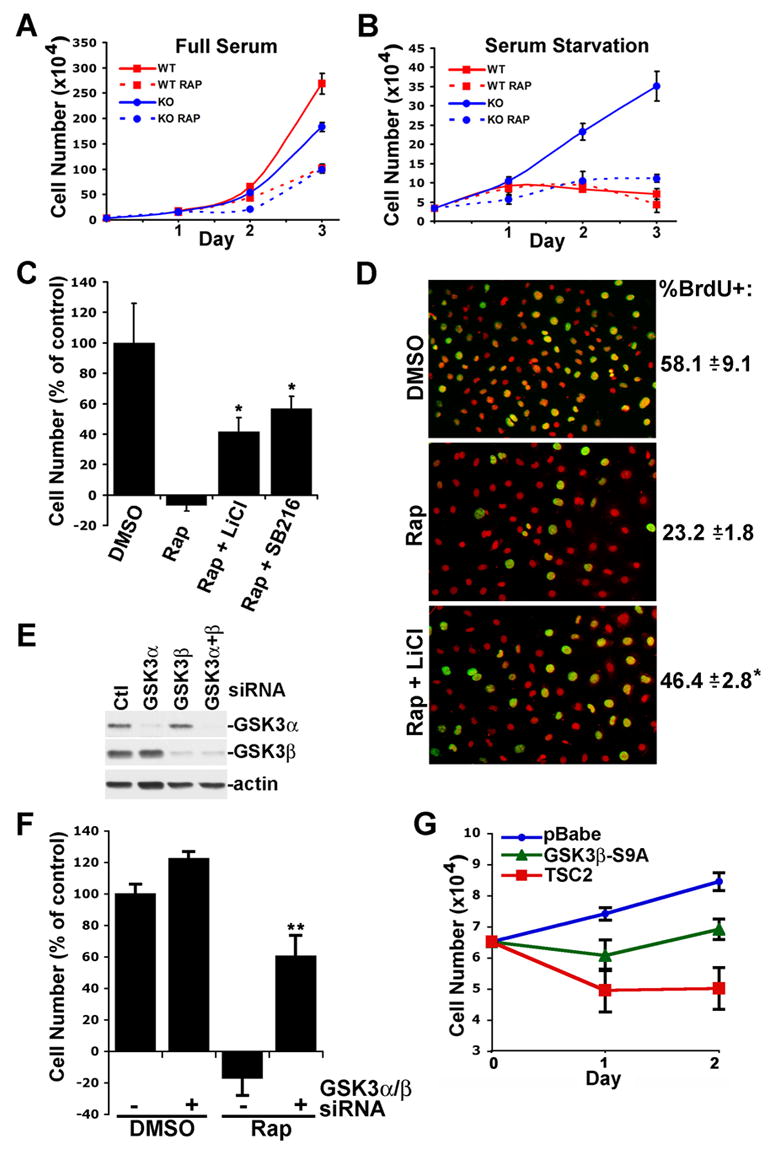
(A) Proliferation of littermate-derived Tsc2+/+ (WT) and Tsc2−/− (KO) MEFs in full serum. Cells were cultured in 10% fetal bovine serum with fresh nutrients and serum provided daily in the presence of 0.1% DMSO (vehicle) or 20 nM rapamycin (Rap). Data are presented as mean ± SEM.
(B) Cells were cultured as in A but under serum starvation conditions.
(C) Pharmacological inhibition of GSK3 partially blocks the effects of rapamycin on the serum-free proliferation property of Tsc2−/− cells. Tsc2−/− MEFs were cultured for 48 h in serum free media in the presence of vehicle, Rap, or Rap plus either 10 mM LiCl or 10 μM SB216763. Data are presented as the mean ± SEM percentage of cells present relative to vehicle control, after subtracting the original cell number plated. *Rap versus Rap + LiCl or Rap + SB216763, P<0.05.
(D) Cells were cultured as described in C but with BrdU added for the final 24 h. Data are presented as the mean ± SEM percentage of DAPI-stained nuclei (red) also positive for BrdU (yellow/green). *Rap versus Rap + LiCl, P<0.05.
(E) SiRNA knockdown of GSK3α and β in Tsc2−/− MEFs.
(F) SiRNA knockdown of GSK3α and β significantly blocks the effects of rapamycin on Tsc2−/− cells. 24 hours post-transfection with control or GSKα and β-targetting siRNAs, equal numbers of Tsc2−/− MEFs were reseeded and serum starved in the presence of vehicle or Rap for 24 h. Data are presented as in C. **control siRNAs + Rap versus GSK3α/β siRNAs + Rap, P<0.001.
(G) GSK3β-S9A can suppress the growth factor-independent proliferation of Tsc2−/− cells. Retrovirus-infected pools of Tsc2−/− MEFs stably expressing empty vector (pBabe-puro) or pBabe-puro encoding GSK3β-S9A or TSC2 were plated in equal numbers and cultured in the absence of serum for 48 h.
In a reciprocal line of experiments, we tested the effects of a mutant of GSK3 that lacks the inhibitory phosphorylation site (GSK3β-S9A) on the proliferation properties of Tsc2−/− MEFs. Expression of human TSC2 completely blocks the serum-free proliferation of these cells, thereby demonstrating the specificity of this phenotype for loss of TSC2 (Figure 6G). Interestingly, expression of GSK3β-S9A also suppresses this growth factor-independent proliferation, albeit not to the extent of TSC2 expression. Therefore, these collective data demonstrate that constitutive phosphorylation and inhibition of GSK3 is a contributing factor to the mTORC1-dependent serum-free proliferation property of TSC-deficient cells.
mTORC1-dependent phosphorylation of GSK3 under other conditions of cell intrinsic insulin resistance
Our findings that GSK3 falls under control of mTORC1 signaling when Akt activation is attenuated in TSC-deficient cells suggest that this alternative mode of GSK3 regulation might occur in other models of cellular insulin resistance. It is widely known that chronic exposure of insulin-responsive cells to insulin desensitizes PI3K-Akt signaling to further insulin stimulation and, like TSC-deficient cells (Harrington et al., 2004; Shah and Hunter, 2006; Shah et al., 2004), this insulin resistance has been shown to involve mTORC1-dependent downregulation of the IRS1 and IRS2 proteins (reviewed in Harrington et al., 2005; Manning, 2004). Therefore, we tested whether GSK3 phosphorylation can be regulated by mTORC1 signaling in insulin resistant HepG2 cells. As expected, 16-h pretreatment with insulin significantly lowered the responsiveness of Akt phosphorylation to acute (15 min) insulin (Figure 7A, compare lanes 3 and 6). While chronic insulin decreased basal Akt and FOXO3a phosphorylation, it had very little effect on the basal state of GSK3 and S6K1 phosphorylation (compare lanes 1 and 4). Interestingly, in addition to inhibiting S6K1 phosphorylation, short-term (15 min) rapamycin treatment significantly lowered the basal phosphorylation of GSK3 under these conditions, while it slightly increased phosphorylation of Akt and FOXO3a (lane 5). The opposing effects of rapamycin on Akt and GSK3 phosphorylation under this state of insulin resistance were quantified using an infrared imaging system (Figure 7B). This approach demonstrates a significant increase in Akt phosphorylation concomitant with a significant decrease in GSK3α and β phosphorylation in response to rapamycin. Therefore, these data suggest that mTORC1-S6K1 regulation of GSK3 might extend to other settings in which the PI3K-Akt pathway is subjected to feedback inhibition, such as during conditions of insulin resistance.
Figure 7. mTORC1-dependent phosphorylation of GSK3 under conditions of cellular insulin resistance.
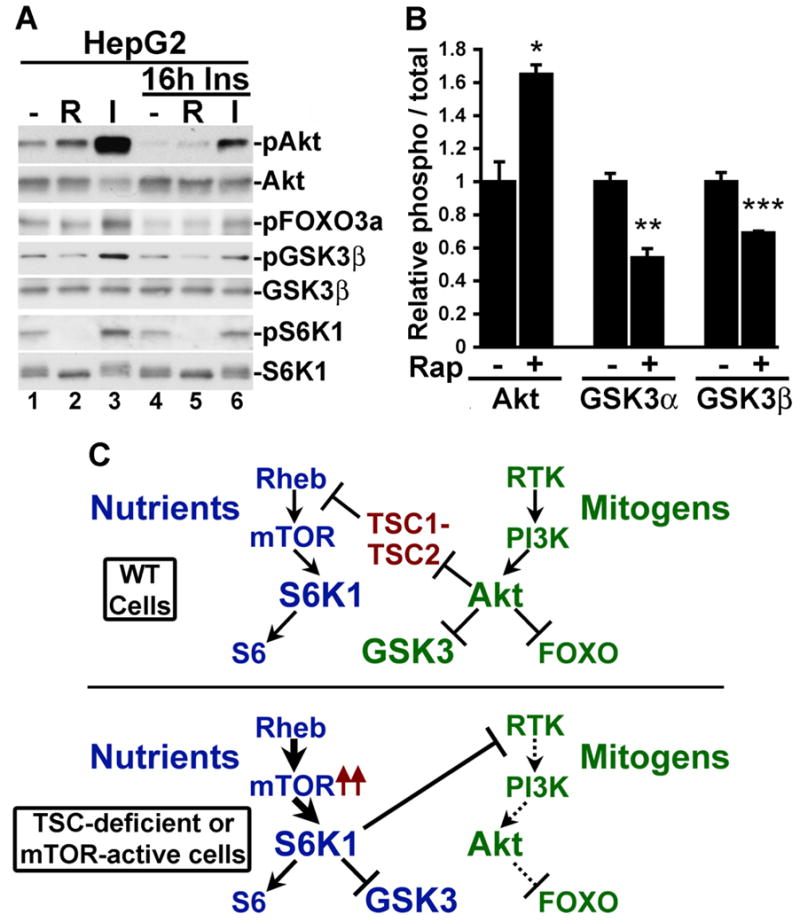
(A) GSK3 phosphorylation is sensitive to rapamycin in insulin-resistant HepG2 cells. HepG2 cells were serum starved for 16 h in the absence or presence of 100 nM insulin and were treated for 15 min with 20 nM rapamycin (R) or 100 nM insulin (I), as indicated.
(B) Rapamycin increases Akt phosphorylation but decreases GSK3 phosphorylation in insulin-resistant HepG2 cells. Cells were treated for 16 h with 100 nM insulin and then treated for 15 min with 20 nM rapamycin. Three independent lysates from each condition were then immunoblotted for phospho-Akt-S473, total Akt, phospho GSK3α/β-S21/S9, and total GSK3α/β, and the data were quantified using an infared imaging system. The data are expressed as the mean +/− SEM relative ratio of phospho-protein to total protein between untreated and rapamycin-treated samples. *p=0.0105; **p=0.0001; ***p=0.0439
(C) Differential regulation of GSK3 by the mitogen-stimulated PI3K-Akt pathway (green) and the nutrient-sensitive mTOR-S6K1 pathway (blue).
Discussion
S6K1 as a bona fide GSK3 kinase
In addition to Akt, several members of the AGC kinase family can phosphorylate GSK3 on its amino-terminal regulatory site in vitro (e.g., Goode et al., 1992; Sutherland and Cohen, 1994; Sutherland et al., 1993), and some have suggested roles in GSK3 regulation in vivo. For instance, there are data demonstrating that stimulus-specific activation of PKA, PKC, or RSK isoforms can lead to GSK3 phosphorylation in some settings (e.g., Eldar-Finkelman et al., 1995; Fang et al., 2002; Li et al., 2000). In the early 90’s, in vitro data from the Cohen laboratory lead to speculation that S6K1 was the GSK3 kinase downstream of insulin (Sutherland and Cohen, 1994; Sutherland et al., 1993). However, based on seminal studies from the same laboratory, it is now widely believed that Akt is the relevant GSK3 kinase downstream of receptor tyrosine kinase signaling (Cross et al., 1995) (Figure 7C, top). Our data demonstrate that under conditions where growth factor stimulation of Akt is attenuated, S6K1 can directly phosphorylate and inhibit GSK3 in vivo (Figure 7C, bottom). This raises an interesting question with regard to signaling specificity of AGC family kinases, many of which share overlapping features in their substrate specificities. Under normal growth factor-stimulated conditions, undefined temporal or spatial factors, perhaps combined with differences in kinase activity thresholds, are likely to dictate that Akt rather than S6K1 is the predominant regulatory kinase for GSK3. Interestingly, exogenous amino acids have been shown to transiently inhibit GSK3 and activate glycogen synthase in muscle cells without affecting Akt, and this effect is blocked by rapamcyin, thereby implicating mTORC1 signaling (Armstrong et al., 2001; Peyrollier et al., 2000). Our data show that GSK3 phosphorylation is dependent on amino acids in the absence of TSC1-2 complex function. Therefore, in certain settings, GSK3 is regulated by the availability of nutrients through mTORC1-S6K1 signaling (Figure 7C, bottom). In this context, GSK3 would be active when nutrients are sparse, thereby allowing it to contribute to inhibition of macromolecular synthesis through phosphorylation of specific substrates, such as glycogen synthase and eIF2B (Cohen and Goedert, 2004; Jope and Johnson, 2004).
Implications for human disease
Our finding that GSK3 is aberrantly regulated by S6K1 upon loss of the TSC1-2 complex has important implications for the pathogenesis of the TSC and LAM diseases. Elevated mTORC1 signaling is clearly a driving force behind the neoplastic lesions in these diseases. However, this increased mTORC1 signaling also leads to feedback inhibition of Akt (Harrington et al., 2004; Radimerski et al., 2002; Shah et al., 2004), perhaps contributing to the benign nature of tumors lacking TSC gene function (Manning et al., 2005). Inhibition of tumor promoting signals from the PI3K-Akt pathway could, in theory, block tumorigenesis altogether. As GSK3 inhibition contributes to the effects of PI3K-Akt signaling on cell proliferation, growth, and survival, the constitutive inhibition of GSK3 described here is likely to be an important factor in TSC tumor development. Importantly, we found that pharmacological or genetic inhibition of GSK3 was not sufficient to stimulate growth factor-independent proliferation of wild-type cells (not shown). This suggests that, while aberrant GSK3 inhibition is a key factor, other signaling events triggered by loss of the TSC1-2 complex, including mTORC1-dependent increases in protein translation, also contribute to this property in TSC-deficient cells.
GSK3 has a large number of substrates that, when misregulated, could contribute to the location and unusual characteristics of many TSC lesions (Cohen and Goedert, 2004; Jope and Johnson, 2004). For instance, tumors within the TSC brain are comprised of neurons with aberrant morphology (Crino, 2004), and TSC patients display a number of neuropsychiatric disorders (Prather and de Vries, 2004), both of which could, at least partially, be explained by misregulation of the many known neuronal targets of GSK3. In addition, the characteristic accumulation of glycogen within TSC cardiac rhabdomyomas (Gomez et al., 1999) could be explained by increased glycogen synthase activity in TSC-deficient cardiomyocytes due to constitutive inhibition of GSK3. It is intriguing that GSK3 activity has recently been found to be required for proper localization of the polycystic kidney disease protein polycystin-2 (or PKD2) within kidney cells (Streets et al., 2006), potentially explaining the occurrence of kidney cysts in TSC patients. Furthermore, GSK3 inhibition has also been shown to enhance smooth muscle cell survival, even under hypoxic conditions (Loberg et al., 2002), a property that is likely to contribute to the pathogenesis of LAM. Therefore, in addition to the known downstream targets of mTORC1 signaling, we must now consider that many of the targets and processes downstream of GSK3 are also likely to be misregulated upon functional loss of the TSC1-2 complex.
A basal mTORC1-dependent inhibition of Akt signaling has recently been recognized in a number of cancer cell lines and in tumor biopsies (O’Reilly et al., 2006; Sun et al., 2005). It will be interesting to determine whether Akt substrates are differentially affected by this feedback mechanism and, more specifically, whether GSK3 is regulated downstream of mTORC1 in these settings. In this regard, a recent study of breast cancer cell lines demonstrated that rapamycin treatment could activate GSK3 and that this contributed to the ability of rapamycin to enhance apoptosis in response to paclitaxel (Dong et al., 2005). However, no effect on GSK3 phosphorylation was observed, and the mechanism remains unknown. As there are a number of PI3K-independent pathways that affect mTORC1 and S6K1 signaling (Sarbassov et al., 2005a), it is possible that S6K1-mediated inhibition of GSK3 could also contribute to tumorigenesis downstream of oncogenic events that activate such pathways.
In addition to TSC-deficient cells, we found that another model of cell intrinsic insulin resistance displays mTORC1-dependent phosphorylaton of GSK3. In mammals, insulin-stimulated inhibition of GSK3 by Akt plays a role in maintaining blood glucose levels by increasing glycogen synthesis in insulin-responsive tissues, such as liver and skeletal muscle (Cline et al., 2002; Coghlan et al., 2000; McManus et al., 2005). Activation of mTORC1-S6K1 signaling in liver and muscle has been linked to insulin resistance (e.g., Khamzina et al., 2005; Tremblay et al., 2005; Um et al., 2004), especially under states of obesity. Whether mTORC1 and S6K1 can regulate GSK3 under such conditions is currently unknown, but this could represent a compensatory mechanism to partially alleviate the consequences of insulin resistance in some tissues. Analogous to our findings here, it is possible that GSK3 would come under control of dietary amino acids in these tissues.
Knowledge of alternative wiring of signaling pathways triggered by loss of tumor suppressors or under specific pathological or physiological states is critical to our complete understanding of the consequences of such perturbations. The study described here adds to the complexity of signaling crosstalk between the PI3K and mTOR pathways, which can be either interdependent or independent depending on cell intrinsic and extrinsic factors. As detailed above, our finding that GSK3 is controlled by the nutrient-sensitive mTORC1 pathway in certain cellular contexts has the potential to help explain clinical features and predict clinical outcomes in a diverse array of human diseases.
Experimental Procedures
Cell culture, constructs, and siRNAs
HeLa cells and MEF lines were maintained in Dulbecco’s modified Eagle medium with 4.5 g/L glucose (DMEM) containing 10% fetal bovine serum (FBS). Immortalized littermate-derived pairs of wild-type and null Tsc1 (3T3 immortalized) and Tsc2 (p53−/−) MEFs were provided by D.J. Kwiatkowski (Brigham and Women’s Hospital, Boston MA) and were described previously (Kwiatkowski et al., 2002; Zhang et al., 2003). For amino acid starvation, cells were incubated in Eagle’s minimum essential medium (MEM) overnight, washed once in PBS, and then incubated for 2 hours in the presence (DMEM) or absence (Dulbecco’s PBS) of amino acids, both containing 1 g/l glucose. For insulin resistance experiments, HepG2 cells were cultured for 16 h in MEM plus 0.1% FBS with or without 100 nM insulin, and prior to the final indicated treatments, the medium on all samples was changed to fresh MEM plus 0.1% FBS without insulin.
Retroviral IRES-hygro constructs expressing human TSC2 or a patient-derived TSC2 missense (Figures 2 and 5) were provided by D.J. Kwiatkowski. Retroviral pBabe-puro-TSC2 (Figure 6G) was described previously (Brugarolas et al., 2004). pBabe-puro-GSK3β-S9A was generated by standard site-directed mutagenesis from a cDNA clone of human GSK3β.
All RNAi experiments were carried out with SMARTpool siRNAs (Dharmacon) and Lipofectamine 2000 (invitrogen) according to the manufacturers’ instructions using the following concentrations or the same concentrations of a non-targeting pool of control siRNAs: 80 nM human TSC2, 10 or 50 nM mouse S6K1, 80 nM mouse GSK3α and β.
For proliferation assays, equal numbers of cells (3.5–7 × 104) were seeded into 6-well plates and incubated with or without 10% FBS in the presence of vehicle (0.1% DMSO) or the indicated inhibitors. The appropriate fresh medium was provided each day. Each growth condition was carried out in triplicate, and cells were counted every 24 h using a hemacytometer. For BrdU incorporation, cells were cultured on coverslips in serum-free media in the presence of the indicated inhibitors for 48 hours, with 10 μM BrdU added for the final 24 h. BrdU incorporation was detected using a fluorescein-conjugated anti-BrdU antibody (Roche) according to the manufacturer’s instructions. Each condition was carried out in triplicate.
Immunoblots and kinase assays
Cell lysates and immunoblots were prepared as described previously (Manning et al., 2002). All IP-kinase assays were adaptations of a previously described protocol (Manning et al., 2002); the specific details are provided with the supplemental materials. The antibodies used in this study were obtained from Cell Signaling Technologies. The phospho-specific antibodies recognize Akt-S473, FOXO3a-T32, GSK3α-S21, GSK3β-S9, S6K1-T389, S6-S236, GS-S641, and c-Myc-T58. For quantification purposes, an Odyssey Infrared Imaging System (LI-COR) was employed according to the manufacturer’s protocol, and the relative intensities of protein bands were measured using the Odyssey version 1.2 software.
Immunohistochemistry
Formalin-fixed paraffin-embedded liver hemangioma tissues from 12-month old Tsc2+/−mice were obtained in a previous study (Manning et al., 2005). Immunohistochemistry with a phospho-GSK3α/β-S21/S9 antibody (1:100) was performed according to the antibody manufacturer’s instructions and detected using Evision+ (DakoCytomatation).
Cortical tuber resections from epilepsy surgery on a TSC patient (performed at Children’s Hospital, Boston) were fixed in 4% paraformaldehyde and sectioned serially at 50-μm thickness. Sections were incubated with monoclonal anti-vimentin (1:500) and polyclonal anti-phospho-GSK3β-S9 antibodies (1:200) and were subsequently stained with fluorescent-conjugated secondary antibodies. Negative controls included omission of the primary antibody and did not reveal any staining. Images were obtained on Zeiss LSM510 Meta Laser Scanning Confocal Microscope and merged using Zeiss Meta software.
Statistical analyses
Quantitative data for cell proliferation and GSK3 kinase assays were analyzed by one-way analysis of variance using GraphPad InStat version 2.04a. Multiple comparisons were performed using Tukey’s honestly significant difference procedure. Results obtained using the Odyssey Infrared Imaging System were analyzed using a paired two-tailed Student’s t-test.
Supplementary Material
Acknowledgments
We are indebted to Dr. D.J. Kwiatkowski for reagents and to Drs. J. Blenis, R.J. Shaw, and K. Düvel for helpful discussions and/or critical comments on the manuscript. This work was supported by the LAM Foundation (H.H.Z.), the Tuberous Sclerosis Alliance (M.S.), the Leukemia and Lymphoma Society (B.D.M.), the American Diabetes Association (B.D.M.), and NIH grant CA122617 (B.D.M) and T32 training grant ES07155 (C.C.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Caudwell FB, Andejelkovic M, Hemmings BA, Cohen P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996;399:333–338. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- Armstrong JL, Bonavaud SM, Toole BJ, Yeaman SJ. Regulation of glycogen synthesis by amino acids in cultured human muscle cells. J Biol Chem. 2001;276:952–956. doi: 10.1074/jbc.M004812200. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC, Jr, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. Embo J. 1996;15:5256–5267. [PMC free article] [PubMed] [Google Scholar]
- Cline GW, Johnson K, Regittnig W, Perret P, Tozzo E, Xiao L, Damico C, Shulman GI. Effects of a novel glycogen synthase kinase-3 inhibitor on insulin-stimulated glucose metabolism in Zucker diabetic fatty (fa/fa) rats. Diabetes. 2002;51:2903–2910. doi: 10.2337/diabetes.51.10.2903. [DOI] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;7:793–803. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19:716–725. doi: 10.1177/08830738040190091301. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Dong J, Peng J, Zhang H, Mondesire WH, Jian W, Mills GB, Hung MC, Meric-Bernstam F. Role of glycogen synthase kinase 3beta in rapamycin-mediated cell cycle regulation and chemosensitivity. Cancer Res. 2005;65:1961–1972. doi: 10.1158/0008-5472.CAN-04-2501. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Seger R, Vandenheede JR, Krebs EG. Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3T3 cells. J Biol Chem. 1995;270:987–990. doi: 10.1074/jbc.270.3.987. [DOI] [PubMed] [Google Scholar]
- Fang X, Yu S, Tanyi JL, Lu Y, Woodgett JR, Mills GB. Convergence of multiple signaling cascades at glycogen synthase kinase 3: Edg receptor-mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C-dependent intracellular pathway. Mol Cell Biol. 2002;22:2099–2110. doi: 10.1128/MCB.22.7.2099-2110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotow H, Thomas G. Substrate recognition determinants of the mitogen-activated 70K S6 kinase from rat liver. J Biol Chem. 1992;267:3074–3078. [PubMed] [Google Scholar]
- Gomez MR, Sampson JR, Whittemore VH. Tuberous Sclerosis. 3. New York, NY: Oxford University Press; 1999. [Google Scholar]
- Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J Biol Chem. 1992;267:16878–16882. [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005;30:35–42. doi: 10.1016/j.tibs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, Hukee MJ, VandenBerg SR, Charlesworth JC. Tuber and subependymal giant cell astrocytoma associated with tuberous sclerosis: an immunohistochemical, ultrastructural, and immunoelectron and microscopic study. Acta Neuropathol (Berl) 1995;90:387–399. doi: 10.1007/BF00315012. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Jaeschke A, Hartkamp J, Saitoh M, Roworth W, Nobukuni T, Hodges A, Sampson J, Thomas G, Lamb R. Tuberous sclerosis complex tumor suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J Cell Biol. 2002;159:217–224. doi: 10.1083/jcb.jcb.200206108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005;146:1473–1481. doi: 10.1210/en.2004-0921. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, Onda H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in TSC1 null cells. Hum Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol Cell Biol. 2000;20:9356–9363. doi: 10.1128/mcb.20.24.9356-9363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loberg RD, Vesely E, Brosius FC., 3rd Enhanced glycogen synthase kinase-3beta activity mediates hypoxia-induced apoptosis of vascular smooth muscle cells and is prevented by glucose transport and metabolism. J Biol Chem. 2002;277:41667–41673. doi: 10.1074/jbc.M206405200. [DOI] [PubMed] [Google Scholar]
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19:1773–1778. doi: 10.1101/gad.1314605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. Embo J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102:14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrollier K, Hajduch E, Blair AS, Hyde R, Hundal HS. L-leucine availability regulates phosphatidylinositol 3-kinase, p70 S6 kinase and glycogen synthase kinase-3 activity in L6 muscle cells: evidence for the involvement of the mammalian target of rapamycin (mTOR) pathway in the L-leucine-induced up-regulation of system A amino acid transport. Biochem J . 2000;350(Pt 2):361–368. [PMC free article] [PubMed] [Google Scholar]
- Prather P, de Vries PJ. Behavioral and cognitive aspects of tuberous sclerosis complex. J Child Neurol. 2004;19:666–674. doi: 10.1177/08830738040190090601. [DOI] [PubMed] [Google Scholar]
- Radimerski T, Montagne J, Hemmings-Mieszczak M, Thomas G. Lethality of Drosophila lacking TSC tumor suppressor function rescued by reducing dS6K signaling. Genes Dev. 2002;16:2627–2632. doi: 10.1101/gad.239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N, Hershey JW. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. Embo J. 2004;23:1761–1769. doi: 10.1038/sj.emboj.7600193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005a;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005b;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah OJ, Hunter T. Turnover of the Active Fraction of IRS1 Involves Raptor-mTOR- and S6K1-Dependent Serine Phosphorylation in Cell Culture Models of Tuberous Sclerosis. Mol Cell Biol. 2006;26:6425–6434. doi: 10.1128/MCB.01254-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival defects. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Streets AJ, Moon DJ, Kane ME, Obara T, Ong AC. Identification of an N-terminal glycogen synthase kinase 3 phosphorylation site which regulates the functional localization of polycystin-2 in vivo and in vitro. Hum Mol Genet. 2006;15:1465–1473. doi: 10.1093/hmg/ddl070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- Sutherland C, Cohen P. The alpha-isoform of glycogen synthase kinase-3 from rabbit skeletal muscle is inactivated by p70 S6 kinase or MAP kinase-activated protein kinase-1 in vitro. FEBS Lett. 1994;338:37–42. doi: 10.1016/0014-5793(94)80112-6. [DOI] [PubMed] [Google Scholar]
- Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J . 1993;296(Pt 1):15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, Nowotny P, Waldhausl W, Marette A, Roden M. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005;54:2674–2684. doi: 10.2337/diabetes.54.9.2674. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.