

Abstract
APOBEC3G and APOBEC3F are cytidine deaminase with duplicative cytidine deaminase motifs that restrict HIV-1 replication by catalyzing C-to-U transitions on nascent viral cDNA. Despite 60% protein sequence similarity, APOBEC3F and APOBEC3G have a different target consensus sequence for editing, and importantly, APOBEC3G has 10-fold higher anti-HIV activity than APOBEC3F. Thus, APOBEC3F and APOBEC3G may have distinctive characteristics that account for their functional differences. Here, we have biochemically characterized human APOBEC3F and APOBEC3G protein complexes as a function of the HIV-1 life cycle. APOBEC3G was previously shown to form RNase-sensitive, enzymatically inactive, high molecular mass complexes in immortalized cells, which are converted into enzymatically active, low molecular mass complexes by RNase digestion. We found that APOBEC3F also formed high molecular mass complexes in these cells, but these complexes were resistant to RNase treatment. Further, the N-terminal half determined RNase sensitivity and was necessary for the high molecular mass complex assembly of APOBEC3G but not APOBEC3F. Unlike APOBEC3F, APOBEC3G strongly interacted with cellular proteins via disulfide bonds. Inside virions, both APOBEC3F and APOBEC3G were found in viral cores, but APOBEC3G was associated with low molecular mass, whereas APOBEC3F was still retained in high molecular mass complexes. After cell entry, both APOBEC3F and APOBEC3G were localized in low molecular mass complexes associated with viral reverse transcriptional machinery. These results demonstrate that APOBEC3F and APOBEC3G complexes undergo dynamic conversion during HIV-1 infection and also reveal biochemical differences that likely determine their different anti-HIV-1 activity.
The replication of the human immunodeficiency virus, type 1 (HIV-1)2 is seriously impaired in human primary lymphocytes if the viral protein Vif is not present. Vif counteracts a series of host factors that belong to the cytidine deaminase family known as APOBEC proteins (apolipoprotein B mRNA-editing catalytic polypeptide). This family consists of APOBEC1 (A1); AID (activation-induced deaminase); APOBEC2 (A2); a subgroup of APOBEC3 (A3) including A3A, A3B, A3C, A3DE, A3F, A3G, and A3H; and APOBEC4 (A4) proteins in humans (1–4). All of these proteins contain one or two Zn2+-binding motifs (HXEX23–28PCX2–4C), which could be the cytidine deaminase active sites (5, 6). For example, A1, AID, A2, A3A, A3C, A3H, and A4 have one cytidine deaminase motif, and A3B, A3DE, A3F, and A3G have two motifs located in their Nor C-terminal domains, respectively. A1 or AID can target cytosines and change them to uracils (C-to-U) on specific RNA or DNA targets resulting in regulation of protein metabolism or immune responses (7, 8). Using a similar mechanism, all A3 proteins except A3H have been found to be able to mutate genomes of various retroviral elements and block their replication (9). Particularly, A3B, A3DE, A3F, and A3G proteins have been shown to block HIV-1 replication (10–15).
A key step for A3s in blocking retroviral replication is their encapsidation from viral producer cells. For A3B, A3F, and A3G, their incorporation into HIV-1 is due to their interactions with Gag proteins in the nucleocapsid region, and this interaction is mediated by cellular or viral genomic RNAs (16–25). Following entry, the virus is uncoated, and reverse transcription occurs resulting in synthesis of viral minus strand cDNA. During this process, A3s deaminate deoxycytidines to form deoxyuridines on the nascent viral cDNAs, resulting in G-to-A mutations in the plus strand DNA and abortive replication (26–29). Nonetheless, the lentiviral protein Vif counters the antiviral activity of A3DE, A3F, and A3G by blocking their encapsidation. Vif specifically binds to A3DE, A3F, and A3G but not A3B (11, 14, 18, 30) and also interacts with a protein complex containing ElonginB, ElonginC, and Cullin5 (Cul5), which are the core subunits of the Cul5-based E3 ubiquitin ligases, such as SCF (31). SCF belongs to the RING (really interesting new gene) family of E3s, which normally contain RING finger proteins such as Rbx1 (Ring box1) to recruit E2 ubiquitin ligases. In fact, Vif has a BC-Box motif (Ser144-Leu145-Gln146) that binds to ElonginC (32, 33) and a HCCH motif (Cys114/Cys133) that binds to Cul5 (34–36). Thus, Vif is able to bridge the Cul5-based E3 ligase complex to A3DE, A3F, and A3G and trigger their degradation in the 26 S proteasome (15, 30, 37–39). As a consequence, viral replication is secured because of insufficient A3 protein presence.
It was previously shown that A3G resides in two different protein complexes in different cells with distinctive enzymatic activity (40). In immortalized or activated primary T cells, it resides in > 700-kDa high molecular mass (HMM) complexes, whereas in the resting primary T cells, it resides in ~100 kDa low molecular mass (LMM) complex (40). The HMM complex contains cellular RNAs and is therefore RNase-sensitive. Upon RNase A treatment, the HMM complex disassembles into the LMM complex. In addition, only the LMM A3G complex exhibits the cytidine deaminase activity in vitro, which suggests that HMM complex may contain an inhibitory protein and/or RNA to prevent A3G from functioning. Whether the other A3 proteins form similar protein complexes and how these complexes are associated with viral life cycle remains unknown.
On the other hand, although A3F and A3G share ~60% protein sequence similarity and block HIV-1 replication by the same mechanism, they can be functionally differentiated. First, they have different target consensus sequences for editing. During cytidine deamination of the viral genome, A3G specifically recognizes deoxythymidines, whereas A3F recognizes deoxycytidines at positions −1 and −2. The C-terminal cytidine deaminase motif determines this target consensus sequence specificity (41). Second, A3G mutates HIV-1 genome at much higher efficiency than A3F, and indeed, A3G anti-HIV-1 activity is 10-fold higher than A3F (42). The delineation of the biochemical basis of these functional differences is critical to the understanding of the molecular mechanism of regulation of antiretroviral activities.
In this report, we biochemically characterized A3F and A3G protein complexes in the HIV-1 life cycle. Using sucrose gradient equilibrium density centrifugation and fast performance liquid chromatography (FPLC), we found that although A3F also formed HMM complexes, they were resistant to RNase treatment. Further, A3F and A3G had different determinants for the HMM complex assembly. Interestingly, inside virions, A3G was in the LMM form, whereas A3F was retained in the HMM complex. In addition, unlike A3F, A3G strongly interacted with cellular proteins via disulfide bonds. Taken together, these results uncovered several aspects of the biochemical differences between A3F and A3G, which could contribute to their different antiretroviral activity.
EXPERIMENTAL PROCEDURES
Plasmids and Cell Lines
The HIV-1 proviral construct pNL4-3Δvif and pNL-LucΔvif and the mammalian expression vectors for human A3A, A3F, A3G, and A3G-GST fusion were described before (11). A construct expressing A3F-GST fusion was generated similarly as that of A3G-GST. The original constructs expressing chimerical A3F and A3G proteins (A3F+G and A3G+F) were a generous gift from R. S. Harris (41), which were subsequently recloned into pcDNA3.1D/V5-His-TOPO (Invitrogen) and ligated to a GST tag. All of these constructs contain a V5 tag to be expressed at the C terminus of the target proteins. In addition, constructs expressing A3F and A3G with a C-terminal tandem HA/FLAG tag were also generated. Eight cytidine deaminase motif mutants A3FHXE1, A3FPCXXC1, A3FHXE2, A3FPCXXC2, A3GHXE1, A3GPCXXC1, A3GHXE2, and A3GPCXXC2 were generated by the QuikChange XL mutagenesis kit (Stratagene, La Jolla, CA). 293 cell lines stably expressing A3F and A3G were described before (11).
Viral Production
HIV-1 virions were produced from 293T cells by the standard calcium phosphate transfection. Typically, 20 μg of total plasmid DNA was used for each transfection in a 100-mm culture dish with 40% confluence of the cell. The ratio between the proviral construct and A3G expression vectors were kept at 1:1. In some cases, HIV-1 was pseudotyped with vesicular stomatitis virus glycoprotein (VSV-G) protein by co-transfection of 5 μg of VSV-G expression vector. The production of HIV-1 was quantified by p24Gag capture enzyme-linked immunosorbent assay. HIV-1 viruses were then used to infect GHOST-R3/X4/R5 cells.
Isolation of HMM and LMM Complex by Equilibrium Density Centrifugation and FPLC
293 cells expressing various A3 proteins were lysed in a buffer containing 50 mM HEPES-KOH (pH 8.0), 100 mM KCl, 2 mM EDTA, 0.1% (w/v) Nonidet P-40, 2 mM dithiothreitol, 1 × Sigma protease inhibitor mixture, 10 mM NaF, 0.25 mM NaOVO3, 5 nM okadaic acid, 5 nM calyculin A, and 50 mM β-glycerophosphate. After removing the nuclei, the cytosolic fraction was loaded on top of the 4–40% sucrose gradient in a tube of Beckman SW-41 rotor and spun for 6 h at 41,000 rpm. Typically, 12 fractions were collected from each tube. The proteins were then precipitated by trichloroacetic acid and analyzed by Western blot. In addition, high density A3F and A3G complexes were subject to FPLC apparatus using a calibrated HiPrep 16/60 Sephacryl S-300 HR gel filtration column.
To purify HIV-1 virions, virions were first concentrated from filtered culture medium of 293T cells transfected with HIV pro-viral construct by spinning through 20% sucrose cushion in tubes of the Beckman SW-28 rotor at 28,000 rpm for 1 h. Virions were resuspended in STE buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA), loaded on a linear 16–65% sucrose gradient, and spun at 100,000 × g for 16 h in a tube of Beckman SW-41 rotor. Twelve fractions were collected from each tube, and virions were precipitated by trichloroacetic acid, and viral proteins were analyzed by Western blot. To purify viral core, viral pellets were loaded on the top of a 1% Triton X-100 layer followed by a 16–65% sucrose gradient as described before (43) and spun at the same condition as viral purification. To determine A3G or A3F protein complex in HIV-1 virion, complete viral lysates were prepared by incubating viral pellets with 1% Triton X-100 at 37 °C for 3.5 h. Viral lysates were then loaded on the 16–65% sucrose gradient and spun at the same conditions as viral purification. The fractions were collected and analyzed similarly. A simplified schematic description for this protocol was presented in Fig. 5.
FIGURE 5. A3FandA3GproteincomplexinHIV-1virion.
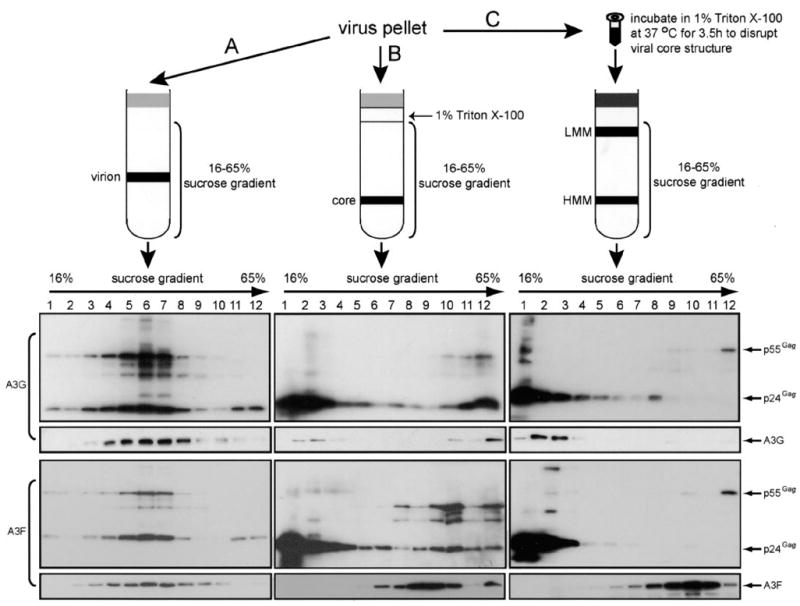
The top section provides a schematic description for three protocols to isolate virion-associated A3G or A3F complexes. Virus pellets were enriched by spinning through 20% sucrose cushion of the culture medium from 293T cells transfected with pNL4–3Δvif and A3G or A3F expression vector and used to determine A3G or A3F protein complexes in purified virion where virus pellets were directly loaded on the top of a16–65% sucrose gradient (A), purified viral core where virus pellets were loaded on the top of a 1% Triton X-100 layer followed with a 16–65% sucrose gradient (B), or complete viral lysates where virus pellets were first incubated with 1% Triton X-100 at 37 °C for 3.5 h and then loaded on a 16–65% sucrose gradient (C). These samples were then subjected to ultracentrifugation, and twelve fractions were collected. After trichloroacetic acid precipitation, the proteins were analyzed by Western blot.
Scintillation Proximity Assay for Cytidine Deamination
As described previously (24), a biotinylated primer (5′-biotin-gtc-agcatcctgaattctacc-3′) was annealed to a deoxyoligonucleotide template containing ten 5′-ccca-3′ repeats and a complementary sequence for the biotinylated primer (5′-cccacccaccca-cccacccacccacccacccacccacccaggtagaattcaggatgctgac-3′). This hybrid was then immobilized on streptavidin-polyvinyltoluene scintillation proximity assay beads (Amersham Biosciences) and incubated with the A3G proteins in a total volume of 64 μl with a buffer containing 50 mM Tris-HCl (pH 8.0), 80 mM KCl, 10 mM MgCl2, 10 mM dithiothreitol, 2.5 mM EGTA, and 0.05% (w/v) Nonidet P-40 at 37 °C for 2 h. Then the polymerization reaction was performed by adding 0.04 mM mixture of dTTP and dGTP, 0.25 μCi of [3H]dATP (Amersham Biosciences), and 0.02 unit of Klenow fragment (Roche Applied Science) for another 40 min. The reaction was stopped by the addition of 100 μl of 120 mM EDTA (pH 8.0). The entire reaction product was then transferred to a scintillation tube with 3 ml of TBE buffer and determined by the LS 6000TA scintillation counter (Beckman Coulter, Fullerton, CA). As a positive control, a parallel experiment was performed using a similar template where the ten 5′-ccca-3′ repeats were replaced by ten 5′-uuua-3′ repeats, and the resulting incorporation of [3H]dATP into this template was referred to as 100% deamination activity. The results were shown as relative values to this positive control.
Interaction of A3F and A3G with Other Proteins via Disulfide Bonds
293T cells were either transfected with an A3-GST construct alone or cotransfected with an A3 nonfusion construct. After 48 h, the cells were lysed. Cytosolic fractions were incubated with GSH-Sepharose beads, and bound proteins were eluted with 1 mM glutathione (Sigma) in phosphate-buffered saline. Eluted samples were treated with N-ethylmaleimide (NEM) (Pierce) at 8 mM final concentration for 2 h at room temperature. After separation by nonreducing SDS-PAGE, the samples were detected by Western blot using anti-V5 or FLAG antibodies.
Western Blot
Horseradish peroxidase-conjugated anti-V5 or FLAG antibodies (Invitrogen) were used to directly detect the expression of A3s or their GST fusions. HIV-1 p17MA, p24Gag, and p32IN were detected by antibodies from National Institutes of Health AIDS Research and Reference Reagent Program. Horseradish peroxidase-conjugated anti-rabbit or mouse IgG secondary antibodies were obtained commercially (Pierce). Detection of the horseradish peroxidase-conjugated antibody was performed using an enhanced chemiluminescence detection kit (Amersham Biosciences Bioscience).
RESULTS
Comparison of A3F and A3G Protein Complexes in Cells
As noted above, A3G forms a RNase-sensitive, enzymatically inactive HMM complex in immortalized cells. Because the formation of this complex regulates the function of A3G, we wished to determine whether A3F is also subject to similar modulation. Previously, A3G HMM and LMM complexes were isolated by FPLC (40). To further maximize for the yield in the isolation, which is particularly important for detection of A3 proteins inside HIV-1 virions (see below), we employed equilibrium density centrifugation to segregate A3G protein complexes. Cytosolic fractions were prepared from 293 cells stably expressing human A3G and subjected to 4–40% sucrose velocity gradient ultracentrifugation. As shown in Fig. 1A, A3G was solely found in high density fractions (including fractions 7–12). In contrast, human A3A was only found in low density fractions (fractions 3 and 4). In addition, preincubation of cell lysates with RNase A resulted in a shift of A3G from high to the low density fractions. These results indicate that the A3G found in the high density fractions could be the A3G HMM complexes and that A3G in the low density fractions could be the A3G LMM complexes. To further confirm this, A3G complexes in different densities were subject to FPLC analysis. As presented in Fig. 1B, the molecular mass of A3G in high density fractions (A3Ghigh density) was higher than 700 kDa, and most of the A3G in the low density fractions (A3Glow density) was lower than 160 kDa. We further compared the deaminase activity of A3G found in these low and high density fractions (Fig. 1C). As controls, total cell lysates of 293 cells without A3G expression did not exhibit any activity (lane 1). Cell lysates of 293 cells expressing A3G exhibited marginal deaminase activity (lane 2), and treatment with RNase A resulted in a 5-fold increase in activity (lane 3). As expected, A3G in the high density fraction exhibited limited deaminase activity, which was only comparable with that of RNase-treated total cell lysates (lane 4), and A3G in the low density fraction exhibited at least 10-fold higher activity than that in this high density fraction (lane 5). Thus, we concluded that the density centrifugation-isolated high density A3G complexes are indeed A3G HHM complexes, and those in the low density fractions are LMM complexes.
FIGURE 1. A3 HMM and LMM complexes in 293T cells.
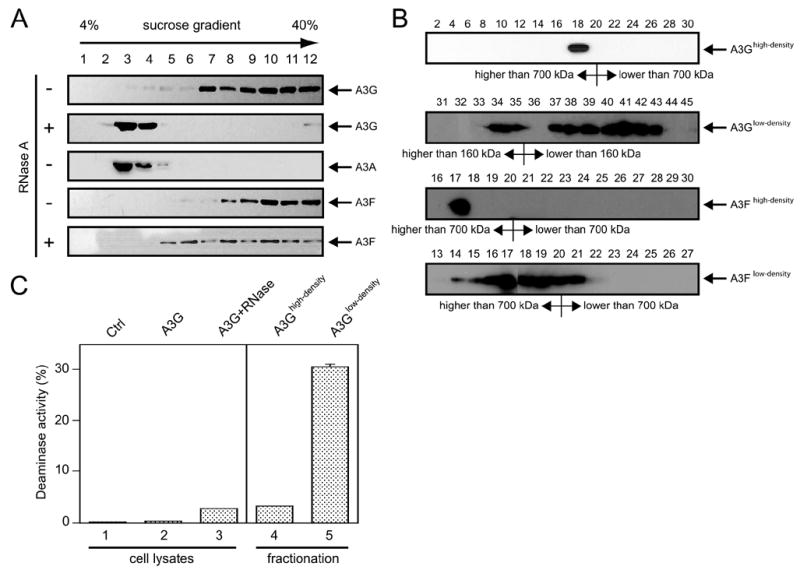
A, isolation of A3A, A3F, and A3G HMM and LMM complexes by equilibrium density centrifugation. Cytosolic fractions were prepared from 293 cells expressing human A3A, A3F, and A3G. After RNase A or mock treatment, they were subject to 4 – 40% sucrose velocity gradient ultracentrifugation. Twelve fractions were collected from each tube. The proteins from each fraction were precipitated by trichloro-acetic acid and analyzed by Western blot. B, molecular mass of A3F and A3G complexes in high or low density fractions. Samples in these fractions were further subject to FPLC analysis. In total, 60 fractions from each analysis were collected for Western blot, and detected proteins with marked molecular mass were presented. C, A3G cytidine deaminase assay. Samples from high density, low density, and total cell lysate with or without RNase A treatment of 293 cells stably expressing A3G were subjected to an in vitro cytidine deamination assay as described under “Experimental Procedures.” Control (Ctrl) was the total cell lysate of the parent 293 cell. The error bars represent the standard deviations in at least three independent experiments.
Next, we examined A3F protein complexes. A cytosolic fraction from 293 cells stably expressing human A3F was prepared and subjected to equilibrium density centrifugation. A3F was found in high density fractions 9–12 (Fig. 1A, lower panels). Surprisingly, RNase A treatment only partially shifted A3F from high density to lower density (fraction 5) and resulted in a rather broad distribution in the gradient fractions. Next, we determined the molecular mass of A3F in these fractions (Fig. 1B, lower two panels). The molecular mass of A3F in the high density fractions (A3Fhigh density) was higher than 700 kDa, and most of the RNase A-treated A3F fractions (A3Flow density) remained around 700 kDa. Thus, we concluded that A3F also resides in the HMM complex in 293 cells, and unlike A3G the A3F HMM complex is insensitive to RNase treatment.
Mapping the Determinants for A3 HMM Complex RNase Sensitivity
Having discovered that there is a difference in RNase sensitivity between A3F and A3G HMM complexes, we next mapped the determinant for this difference. Because both A3F and A3G have duplicative cytidine deaminase motifs, we took advantage of published constructs expressing chimerical proteins in which the N- and C-terminal halves of A3F were swapped for those of A3G, respectively (41). A3F+G chimerical protein expresses the N-terminal half of A3F and C-terminal half of A3G, and A3G+F chimerical protein expresses the N-terminal half of A3G and C-terminal half of A3F (Fig. 2A). The chimerical proteins were expressed in 293T cells, and the protein-associated complexes were prepared using equilibrium density centrifugation as above. Like the wild type A3F and A3G, both A3F+G and A3G+F were able to assemble HMM complexes (Fig. 2B). However, the complexes fell into a broader range of density as compared with those of wild type A3F or A3G proteins. Interestingly, when these HMM complexes were treated with RNase A, only those of A3G and A3G+F, but not A3F and A3F+G, were sensitive to the treatment. These results indicate that the N-terminal half of A3F and A3G proteins determines their HMM complex RNase sensitivity.
FIGURE 2. Mapping A3G RNase-sensitive region.
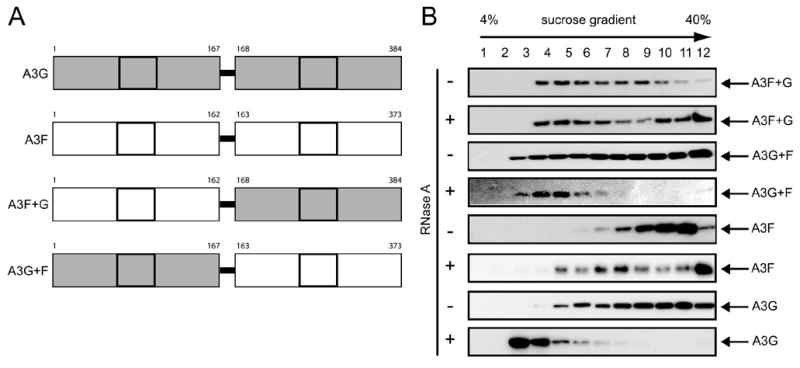
A, a schematic representation of A3G, A3F, and their recombinant forms A3F+G and A3G+F. The cytidine deaminase motif was squared, and the boundary for generation of A3F and A3G recombinant proteins was positioned. B, RNase sensitivity of A3F+G and A3G+F HMM complexes. A3F, A3G, A3F+G, and A3G+F protein complexes were prepared as in Fig. 1A and subject to Western blot.
Mapping the Determinants for A3F and A3G HMM Complex Assembly
Having demonstrated that both A3F and A3G can assemble HMM complexes in cells, we next investigated the sequence determinant for this assembly. It has been reported that the N-terminal cytidine deaminase motif has much higher RNA affinity than the C-terminal cytidine deaminase motif (44). Because A3G HMM complexes contain cellular RNA, we decided to mutate the N-terminal cytidine deaminase motif of both A3F and A3G by generating two kinds of mutants, HXE1 and PCXXC1, where the first HXE and PCXXC motifs were changed to AXS and LSXXS, respectively. As controls, we also mutated the C-terminal cytidine deaminase motif of both A3F and A3G by generating mutants of HXE2 and PCXXC2 where the second HXE and PCXXC motifs were changed to AXS and LSXXS, respectively (Fig. 3A). As presented in Fig. 3B, all of these mutants proteins were expressed at similar levels to their wild type proteins, and these mutations seriously disrupted A3F or A3G anti-HIV-1 activities, although the ability of A3FPCXXC2 was less compromised, proving the success of the introduction of these mutations.
FIGURE 3. Mapping the key determinant for A3F and A3G HMM complex assembly.
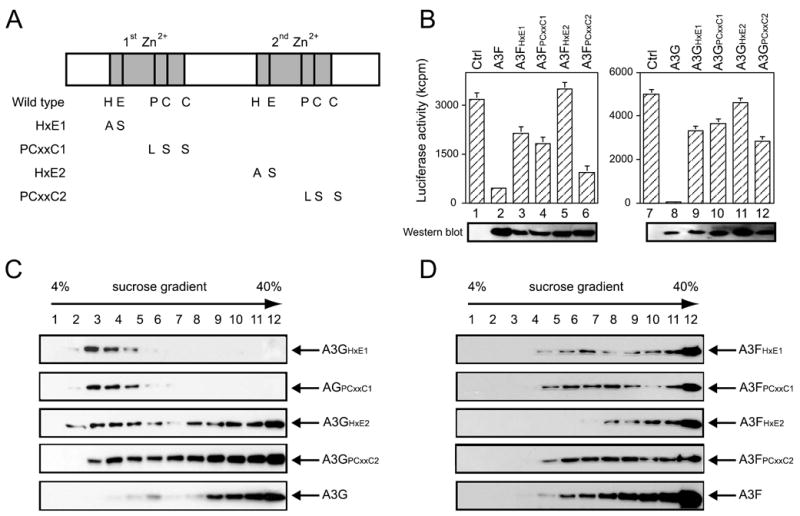
A, schematic representation of mutations introduced to the cytidine deaminase motif in A3F and A3G. Four mutants were generated for each of A3F and A3G, which were designated as A3FHXE1, A3FPCXXC1, A3FHXE2, A3FPCXXC2, A3GHXE1, A3GPCXXC1, A3GHXE2, and A3GPCXXC2. B, anti-HIV-1 activities of these A3F and A3G mutants. pNL-LucΔvif was co-transfected with these mutant constructs into 293T cells, and viral infectivity was analyzed in GHOST cells. Ctrl, control plasmid. C and D, A3G and its mutants (C) or A3F and its mutants (D) were expressed in 293T cells, and their protein complexes were isolated and analyzed similarly as in Fig. 1A with the exception of RNase A treatment.
We then determined the influence of these mutations on A3G HMM complex formation. These mutant proteins were expressed in 293T cells, and protein complexes were prepared as above. When compared with the wild type A3G, both A3GHXE1 and A3GPCXXC1 lost their ability to assemble the HMM complexes, whereas both A3GHXE2 and A3GPCXXC2 still retained such ability (as presented in Fig. 3C). Interestingly, none of these mutations in A3F (A3FHXE1, A3FPCXXC1, A3FHXE2, and A3FPCXXC2) had any impact on the assembly of A3F HMM complex (Fig. 3D). Thus, we concluded that: 1) the N-terminal cytidine deaminase motif of A3G determines its ability to assemble the HMM complex and 2) none of A3F cytidine deaminase motifs is responsible for its HMM complex assembly.
A3G Interacts with Other Cellular Proteins via Disulfide Bonds
Differential HMM complex RNase sensitivity and assembly between A3F and A3G suggests the existence of a remarkable biochemical difference between these two proteins, which stimulated us to investigate whether they interact with other cellular proteins differently. Because both A3F and A3G proteins contain many cysteines, we examined their potential involvement in interactions with other cellular proteins. To test this possibility, A3F-GST, A3G-GST, A3F+G-GST, and A3G+F-GST fusion proteins were expressed in 293T cells and purified by GSH-Sepharose beads. The proteins were then treated with NEM, a thiol-activating reagent that reacts with sulfhydryls and triggers disulfide-linked protein complex formation. After NEM treatment and resolution by SDS-PAGE under nonreducing conditions, we clearly saw a supershift band for A3G and A3G+F only (Fig. 4A, lanes 3 and 7 marked by *). This result suggests that cysteines in A3G, precisely in the N-terminal half of, are more exposed and easily subject to protein-protein interactions than those in A3F. To distinguish this supershift was due to A3G-A3G or A3G with other cellular protein interactions, we co-expressed A3G-GST or A3F-GST with their nonfusion forms, respectively. These two kinds of proteins are distinguishable by Western blot because of the existence of a V5 tag at the end of A3F-GST or A3G-GST and a FLAG tag at the end of A3F or A3G. Indeed, the supershift was only observed by anti-V5 antibody but not by anti-FLAG antibody in A3G-GST and A3G co-expressing cells (Fig. 4B, lane 4). Thus, the disulfide bond-mediated protein-protein interactions are caused by interactions of A3G with other cellular proteins but not A3G multimerization.
FIGURE 4. Interaction of A3G with cellular proteins by disulfide bond.
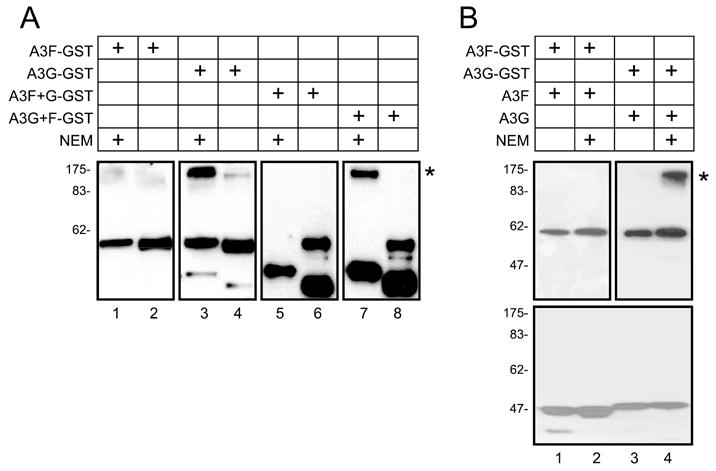
A, detection and mapping of labile disulfide bond in A3G proteins. A3F-GST, A3G-GST, A3F+G-GST, and A3G+F-GST fusion proteins were expressed in 293T cells, and the proteins were purified by GSH-Sepharose beads. The samples were then treated with 8 mM NEM and analyzed by nonreducing SDS-PAGE followed by Western blot with anti-V5 antibody. B, absence of A3G-A3G interaction via disulfide bond. A3F-GST or A3G-GST was co-expressed with A3F or A3G, respectively, and a similar experiment was performed as in A. After nonreducing SDS-PAGE resolution, the proteins were analyzed by Western blot with anti-V5 antibody for A3-GST fusions or anti-FLAG antibody for A3 only. Those supershift bands were marked with *.
A3F and A3G Protein Complexes in Virions
Having examined protein complexes in cells, next we asked whether these A3F and A3G complexes could be identified in virions. Previously, it was shown that A3G exists inside of the HIV-1 core (20). Because the viral core sediments to the high density fraction during equilibrium density centrifugation, it is difficult to distinguish between viral core and A3 HMM complexes unless the viral core structure is completely disrupted. It has been reported that the HIV-1 core is very unstable in vitro and by incubating with 1% Triton X-100 at 37 °C for 3 h, more than 75% of viral cores were disassembled (47). We therefore prepared viral lysates by following this protocol to detect A3 complex (Fig. 5C). As controls, we also purified intact virions (Fig. 5A) and intact core (Fig. 5B) with either A3F or A3G, and their integrity was determined by Western blot for p24Gag and p55Gag after equilibrium density isolation. As seen in Fig. 5A, the peak fraction for virion was found in fraction 6 (first and third panels from top), and as seen Fig. 5B, the peak fraction for viral core was found in fraction 12 (first and third panels from top). These are consistent with the densities for virions (1.15–1.17 g/ml) and core (1.24–1.26 g/ml). A3F and A3G were both detected in these fractions, which confirmed their encapsidation by the virion and localization inside viral core (Fig. 5, A and B, second and fourth panels from top). As previously reported, the HIV-1 core was relatively unstable, and during the process of core purification, most of the viral cores were disassembled (Fig. 5B, first and third panels from top, lanes 1 and 2) (43). Interestingly, along with the disassembly of the viral core, we started to detect the LMM form of A3G (Fig. 5B, second panel from top, lanes 2 and 3), which suggested that A3G LMM complex might be associated with HIV-1 core; in contrast, A3F remained with the HMM fractions under the same conditions (Fig. 5B, fourth panel from top, lanes 5–11), which suggested that A3F HMM complex might be associated with HIV-1 core. This was further confirmed by analysis of complete viral lysates. Western blot of the viral lysates showed that the viral cores were completely disrupted as evident by the lack of p24Gag in the high density fractions and by the presence of p24Gag proteins in the low density fractions (Fig. 5C, first and third panels from top, lanes 1 and 2). Importantly, A3G proteins were only detected in the low density fractions including fractions 2 and 3 (Fig. 5C, second panel from top), whereas the A3F proteins were detected in the high density fractions (Fig. 5C, fourth panel from top). These results indicate that there is a clear difference between virion-associated A3F and A3G: A3F is associated with a HMM complex, whereas A3G is associated with a LMM complex.
A3F and A3G Protein Complexes in Target Cells
As noted above, the HMM complex is enzymatically inactive. To block HIV-1 replication, the complex must be in an active form upon entering into target cells to trigger cytidine deamination on viral cDNA. It is therefore necessary to determine whether A3G stays in the LMM complex and A3F dissociates with the HMM complex in target cells. To do so, we chose GHOST, a fibroblast cell line as target cells for HIV-1 infection. Cells were infected by Vif-deficient HIV-1 particles containing either A3F or A3G. Four hours after infection, A3F and A3G protein complexes were isolated from cells as before. As presented in Fig. 6A, A3G was found in a very broad range of density fractions that spanned from HMM to LMM complexes, and when treated with RNase A, all of these higher density samples were shifted to the low density fractions. Interestingly, as presented in Fig. 6B, the majority of A3F was absolutely detected in the low density fractions, which indicated that they belong to the LMM complexes. Thus, both A3F and A3G exist in the LMM complexes in the target cells. Moreover, we determined whether these A3F and A3G LMM complexes are associated with the viral reverse transcription complex that normally includes p24CA, p17MA, and p32IN viral proteins. The same membranes in Fig. 6 (A and B) were blotted for antibodies against these different viral proteins. Indeed, all of these viral proteins were detected in the low density fractions that included fraction 3 and 4 (Fig. 6, A and B, see Western blot for p24CA, p17MA, and p32IN in the absence of RNase treatment). Thus, not only do both A3F and A3G exist in the LMM complexes, but they are also co-localized with viral reverse transcription machinery so that both proteins can effectively mutate the viral genome and block viral replication.
FIGURE 6. A3F and A3G protein complex in target cells.
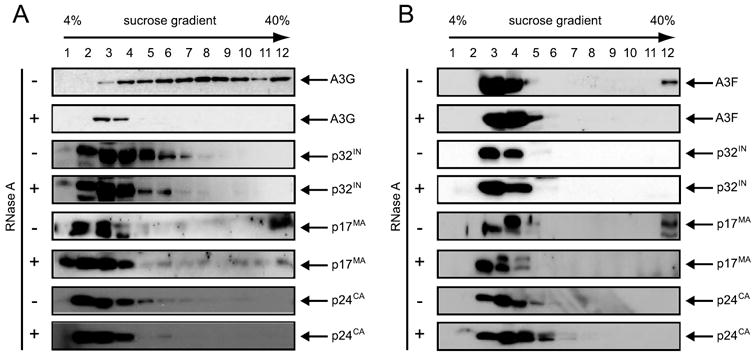
Isolation of A3G (A) and A3F (B) protein complexes in HIV-1 infected cells. HIV-1 virions pseudotyped by VSV-G were produced by co-transfection of pNL4 –3Δvif, A3G or A3F, and VSV-G expression vectors. Viruses were used to infect GHOST cells. Four hours later, the cells were washed extensively, and cytosolic fraction was prepared. After RNase A or mock treatment, the samples were spun through a 4 – 40% sucrose gradient and analyzed by Western blot for A3G or A3F and viral proteins p32IN, p17MA, and p24CA to localize viral reverse transcription machinery.
DISCUSSION
In this report, we compared A3G and A3F protein complexes that are associated with HIV-1 life cycle. Two major observations were made. First, we found a dynamic conversion of A3F and A3G from HMM complexes to LMM complexes in the HIV-1 life cycle, which provides a novel mechanism for understanding how their antiviral activities are regulated. Second, we discovered several important differences between A3F and A3G complexes that are important for understanding their different anti-HIV-1 activities.
Surprisingly, although A3F and A3G share almost 60% sequence similarity on protein level, they differ in several key aspects: 1) although both A3F and A3G assemble the HMM complexes in cells, only the A3G HMM complex is RNase sensitive; 2) they have different requirements for HMM complex assembly in that A3G is dependent on the N-terminal deaminase motif, whereas A3F has an unknown determinant other than a deaminase motif; 3) A3G interacts with other cellular proteins via its N-terminal disulfide bonds; and 4) In HIV-1 virions, A3F resides in HMM complexes, whereas A3G resides in LMM complexes. Undoubtedly, all of these observations direct us to new questions that are potentially critical to HIV infectivity and will facilitate understanding the architecture of A3 protein-associated cytidine deamination machinery.
The difference in HMM complex RNA sensitivity between A3F and A3G provides the first evidence that distinguishes these proteins biochemically. The A3F HMM complex might contain much less cellular RNA than A3G. Alternatively, the A3F HMM complex might have a specific structural component to protect it from RNA degradation. In either case, it is reasonable to speculate that the A3F protein itself must form a distinguishable three-dimensional conformation different from A3G protein. The existence of the labile disulfide bonds in the A3G protein further supports this speculation. Such a conformational difference could result in the assembly of different HMM complexes with different RNA compositions and/or structure. Because A3G is apparently more effective in blocking HIV-1 replication in laboratory assays, it stands to reason that the RNase-sensitive HMM complex causes much more serious damage to the viral genome than the RNase-insensitive HMM complex in these experiments. The RNase-sensitive HMM complexes might exhibit higher affinity to viral genome and therefore become more effective in attacking their targets. Nonetheless, from the difference in RNase sensitivity, we conclude that A3F and A3G assemble distinctive HMM complexes and that such a difference could only be fully demonstrated by understanding each component in these complexes.
We observed that unlike A3F, A3G is associated with LMM complexes inside virions, which raises another question of how the A3G protein complex is encapsidated by HIV-1 during viral replication. Does HIV-1 specifically encapsidate the A3G LMM complex or the A3G HMM complex that reverts to an LMM complex after viral budding? Indeed, accumulated evidence from other groups and ours strongly supports the second possibility. First, in HIV-producing 293T cells, p55Gag clearly co-fractionated with A3G in the high density fractions, which suggests that they are both in the HMM complex. When treated with RNase A, although A3G converted to the LMM complex, p55Gag still remained in the HMM complex (data not shown). In addition, several groups have published that the interaction between p55Gag and A3G is RNA-dependent (20, 23–25). These results clearly indicate that p55Gag only interacts with A3G HMM but not LMM complexes. Second, results in Fig. 3A show that both A3GHXE1 and A3GPCXXC1 mutants could only assemble the LMM complexes. When they were tested for HIV-1 encapsidation, both A3GHXE1 and A3GPCXXC1 were poorly incorporated into HIV-1 particles, unlike A3GHXE2 and A3GPCXXC2, which could form HMM complexes, and were efficiently incorporated (data not shown). Thus, HIV-1 p55Gag might selectively interact with and only encapsidate A3 HMM complex.
It will be very interesting to know how A3G HMM complexes are converted to the LMM complexes in the mature virions and how this conversion occurs for A3F complexes in the target cells. Apparently, A3F and A3G have different mechanisms. Two possibilities are under consideration: 1) The A3G HMM complex is mechanically disrupted in the virion and 2) The A3F HMM complex is disrupted by virion-associated RNase. To test the first possibility, we incorporated A3G into immature HIV-1 virions bearing inactive viral protease by assuming that maturation of the virion might cause mechanical damage to the HMM complex. However, we were still able to detect the LMM A3G complex in these virions (data not presented), which suggested that the conversion of HMM to LMM complex occurs prior to virion maturation at least in the case of A3G. As to the second possibility, it is known that HIV-1 reverse transcriptase has three enzymatic activities: RNA-dependent DNA polymerase activity to synthesize minus strand viral DNA, DNA-dependent DNA polymerase activity to synthesize the plus strand viral DNA, and RNase H activity. The RNase H activity has multiple functions during reverse transcription: it degrades the viral genome RNA after it has been converted to an RNA-DNA hybrid, it specifically cleaves the viral RNA in the polypurine tract region to generate the RNA primer used for the initiation of the second strand of DNA, and it removes both the tRNA and the polypurine tract RNAs after they have been used to prime DNA synthesis (48). RNase H activity of HIV-1 reverse transcriptase might be responsible for the HMM-to-LMM conversion of A3F complex in the target cell. However, because RNase H specifically hydrolyzes the phosphodiester bonds of RNA that is hybridized to DNA, it is important to know whether A3F HMM complex contains any RNA-DNA hybrids. Moreover, HIV-1 might encapsidate cellular RNases that are activated after viral budding and/or entry, and identification of these enzymes may facilitate understanding of this mechanism.
Another important finding from this investigation is that the N-terminal half of A3G is the determinant for its biochemical differences from A3F. Unlike A3F, not only does A3G N-terminal cytidine deaminase motif determines the assembly of the HMM complexes (Fig. 3A), but also cysteines on A3G N-terminal half interact with other cellular proteins to form protein complexes (Fig. 4A). As noted before, A3G contains two very conserved cytidine deamination motifs separated in their N-and C-terminal regions. Initial studies involving mutational analysis showed that both motifs were required for A3G anti-HIV-1 activity (28, 29). However, more recent studies demonstrated that the function of these two domains could be clearly differentiated. Indeed, although both motifs are capable of binding nuclei acids, the first motif has much higher affinity for nuclei acids than that of the second motif and therefore determines A3G encapsidation into virions. On the other hand, the deaminase activity of A3G is solely associated with the second motif, which has more direct impact on its antiviral activity (44, 49–51). These observations are indeed consistent with our results (Fig. 3A), which provide a further explanation of why the first motif determines A3G encapsidation. Our data show that because of its high affinity for RNA, the first motif of A3G is responsible for its residence in the RNA-dependent HMM complex, which is then specifically encapsidated by HIV-1. Why HIV-1 p55Gag only recognizes the HMM complex of A3G, as well as why A3G resides in the HMM complex with hidden enzymatic activity in dividing cells, needs to be addressed. A simple scenario could be that the enzymatically active A3G attacks host nucleic acids and therefore is toxic to cell proliferation. In addition, it is also possible that the deaminase activity of A3G plays a role in keeping cells in a resting stage by preventing them from dividing. Moreover, an interaction of the A3G N-terminal half with other cellular proteins via disulfide bonds was detected by NEM treatment, indicating that its N-terminal half contains labile cysteines. Although it is generally believed that NEM triggers a stable adduct with protein thiols, the precise mechanism of the NEM-induced reaction is still not clear. Previously, this reagent was used to map the disulfide bonds between surface and transmembrane subunits of murine leukemia virus envelope proteins (45, 46). It was inferred in this case that the NEM adduct could react with a second thiol by displacing the maleimidyl group to form the disulfide bond. Nonetheless, how this disulfide bond-mediated protein interaction contributes to the assembly of the RNase-sensitive HMM complexes is worth further investigation.
In summary, we discovered several biochemical differences between A3F and A3G proteins and their complexes and identified a dynamic conversion between HMM and LMM complexes in the HIV-1 life cycle. Such a conversion explains why A3 proteins could block HIV-1 replication in activated T cells. Our findings therefore open intriguing new directions for further studies of the post-translational regulation of these anti-retroviral host factors.
Acknowledgments
We thank Walter J. Esselman for critical reading and insightful comments on this manuscript. We thank Michael R. Garavito for use of the FPLC and Yi Zeng for technical assistance. We thank Whitney Soltau, Lai Mun Siew, and Alexander Brown for excellent experimental assistance. We thank Reuben S. Harris for A3F+G and A3G+F expression vectors. The following reagents were obtained through the National Institutes of Health AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health: HIV-1 p24CA monoclonal antibody (catalog no. 3537) from Bruce Chesebro and Kathy Wehrly; HIV-1 p17MA polyclonal antibody (catalog no. 4811) from Paul Spearman; and HIV-1 p32IN polyclonal antibody (catalog no. 757) from Duane Grandgenett.
The abbreviations used are
- HIV-1
human immunodeficiency virus, type 1
- A1
APOBEC1
- A2
APOBEC2
- A3
APOBEC3
- A4
APOBEC4
- HMM
high molecular mass
- LMM
low molecular mass
- FPLC
fast performance liquid chromatography
- VSV-G
vesicular stomatitis virus glycoprotein
- NEM
N-ethylmaleimide
- GST
glutathione S-transferase
Footnotes
This work was supported by National Institutes of Health Grant AI063944.
References
- 1.Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. Mol Biol Evol. 2005;22:367–377. doi: 10.1093/molbev/msi026. [DOI] [PubMed] [Google Scholar]
- 2.Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- 3.Rogozin IB, Basu MK, Jordan IK, Pavlov YI, Koonin EV. Cell Cycle. 2005;4:1281–1285. doi: 10.4161/cc.4.9.1994. [DOI] [PubMed] [Google Scholar]
- 4.Wedekind JE, Dance GS, Sowden MP, Smith HC. Trends Genet. 2003;19:207–216. doi: 10.1016/S0168-9525(03)00054-4. [DOI] [PubMed] [Google Scholar]
- 5.MacGinnitie AJ, Anant S, Davidson NO. J Biol Chem. 1995;270:14768–14775. [PubMed] [Google Scholar]
- 6.Yamanaka S, Poksay KS, Balestra ME, Zeng GQ, Innerarity TL. J Biol Chem. 1994;269:21725–21734. [PubMed] [Google Scholar]
- 7.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 8.Teng B, Burant CF, Davidson NO. Science. 1993;260:1816–1819. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- 9.Cullen BR. J Virol. 2006;80:1067–1076. doi: 10.1128/JVI.80.3.1067-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Curr Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 11.Dang Y, Wang X, Esselman WJ, Zheng YH. J Virol. 2006;80:10522–10533. doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liddament MT, Brown WL, Schumacher AJ, Harris RS. Curr Biol. 2004;14:1385–1391. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 13.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 14.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. EMBO J. 2004;23:2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. J Virol. 2004;78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alce TM, Popik W. J Biol Chem. 2004;279:34083–34086. doi: 10.1074/jbc.C400235200. [DOI] [PubMed] [Google Scholar]
- 17.Cen S, Guo F, Niu M, Saadatmand J, Deflassieux J, Kleiman L. J Biol Chem. 2004;279:33177–33184. doi: 10.1074/jbc.M402062200. [DOI] [PubMed] [Google Scholar]
- 18.Doehle BP, Schafer A, Cullen BR. Virology. 2005;339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Douaisi M, Dussart S, Courcoul M, Bessou G, Vigne R, Decroly E. Biochem Biophys Res Commun. 2004;321:566–573. doi: 10.1016/j.bbrc.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Khan MA, Kao S, Miyagi E, Takeuchi H, Goila-Gaur R, Opi S, Gipson CL, Parslow TG, Ly H, Strebel K. J Virol. 2005;79:5870–5874. doi: 10.1128/JVI.79.9.5870-5874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kremer M, Bittner A, Schnierle BS. Virology. 2005;337:175–182. doi: 10.1016/j.virol.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Luo K, Liu B, Xiao Z, Yu Y, Yu X, Gorelick R, Yu XF. J Virol. 2004;78:11841–11852. doi: 10.1128/JVI.78.21.11841-11852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schafer A, Bogerd HP, Cullen BR. Virology. 2004;328:163–168. doi: 10.1016/j.virol.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Svarovskaia ES, Xu H, Mbisa JL, Barr R, Gorelick RJ, Ono A, Freed EO, Hu WS, Pathak VK. J Biol Chem. 2004;279:35822–35828. doi: 10.1074/jbc.M405761200. [DOI] [PubMed] [Google Scholar]
- 25.Zennou V, Perez-Caballero D, Gottlinger H, Bieniasz PD. J Virol. 2004;78:12058–12061. doi: 10.1128/JVI.78.21.12058-12061.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 27.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 28.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marin M, Rose KM, Kozak SL, Kabat D. Nat Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 31.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 32.Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Genes Dev. 2004;18:2861–2866. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Genes Dev. 2004;18:2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. Proc Natl Acad Sci U S A. 2005;102:11444–11449. doi: 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mehle A, Thomas ER, Rajendran KS, Gabuzda D. J Biol Chem. 2006;281:17259–17265. doi: 10.1074/jbc.M602413200. [DOI] [PubMed] [Google Scholar]
- 36.Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu XF. Virology. 2006;349:290–299. doi: 10.1016/j.virol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Liu B, Sarkis PT, Luo K, Yu Y, Yu XF. J Virol. 2005;79:9579–9587. doi: 10.1128/JVI.79.15.9579-9587.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheehy AM, Gaddis NC, Malim MH. Nat Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 39.Stopak K, de Noronha C, Yonemoto W, Greene WC. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 40.Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Nature. 2005;435:108–114. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- 41.Hache G, Liddament MT, Harris RS. J Biol Chem. 2005;280:10920–10924. doi: 10.1074/jbc.M500382200. [DOI] [PubMed] [Google Scholar]
- 42.Bishop KN, Holmes RK, Malim MH. J Virol. 2006;80:8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kotov A, Zhou J, Flicker P, Aiken C. J Virol. 1999;73:8824–8830. doi: 10.1128/jvi.73.10.8824-8830.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Navarro F, Bollman B, Chen H, Konig R, Yu Q, Chiles K, Landau NR. Virology. 2005;333:374–386. doi: 10.1016/j.virol.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 45.Pinter A, Kopelman R, Li Z, Kayman SC, Sanders DA. J Virol. 1997;71:8073–8077. doi: 10.1128/jvi.71.10.8073-8077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinter A, Lieman-Hurwitz J, Fleissner E. Virology. 1978;91:345–351. doi: 10.1016/0042-6822(78)90382-3. [DOI] [PubMed] [Google Scholar]
- 47.Forshey BM, von Schwedler U, Sundquist WI, Aiken C. J Virol. 2002;76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schultz SJ, Zhang M, Champoux JJ. J Biol Chem. 2006;281:1943–1955. doi: 10.1074/jbc.M510504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huthoff H, Malim MH. Virology. 2005;334:147–153. doi: 10.1016/j.virol.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 50.Iwatani Y, Takeuchi H, Strebel K, Levin JG. J Virol. 2006;80:5992–6002. doi: 10.1128/JVI.02680-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Opi S, Takeuchi H, Kao S, Khan MA, Miyagi E, Goila-Gaur R, Iwatani Y, Levin JG, Strebel K. J Virol. 2006;80:4673–4682. doi: 10.1128/JVI.80.10.4673-4682.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]