

Abstract
High-resolution mapping of quantitative trait loci (QTL) in animals has proved to be difficult because the large effect sizes detected in crosses between inbred strains are often caused by numerous linked QTLs, each of small effect. In a study of fearfulness in mice, we have shown it is possible to fine map small-effect QTLs in a genetically heterogeneous stock (HS). This strategy is a powerful general method of fine mapping QTLs, provided QTLs detected in crosses between inbred strains that formed the HS can be reliably detected in the HS. We show here that single-marker association analysis identifies only two of five QTLs expected to be segregating in the HS and apparently limits the strategy's usefulness for fine mapping. We solve this problem with a multipoint analysis that assigns the probability that an allele descends from each progenitor in the HS. The analysis does not use pedigrees but instead requires information about the HS founder haplotypes. With this method we mapped all three previously undetected loci [chromosome (Chr.) 1 logP 4.9, Chr. 10 logP 6.0, Chr. 15 logP 4.0]. We show that the reason for the failure of single-marker association to detect QTLs is its inability to distinguish opposing phenotypic effects when they occur on the same marker allele. We have developed a robust method of fine mapping QTLs in genetically heterogeneous animals and suggest it is now cost effective to undertake genomewide high-resolution analysis of complex traits in parallel on the same set of mice.
Most phenotypes of medical importance can be measured quantitatively, and in many cases the genetic contribution is substantial, accounting for 40% or more of the phenotypic variance. Considerable efforts have been made to isolate the genes responsible for quantitative genetic variation in human populations, but with little success, mostly because genetic loci contributing to quantitative traits (quantitative trait loci, QTL) have only a small effect on the phenotype (1). Association studies have been proposed as the most appropriate method for finding the genes that influence complex traits (2). However, family-based studies may not provide the resolution needed for positional cloning, unless they are very large, whereas environmental or genetic differences between cases and controls may confound population-based association studies (3).
These difficulties have led to the study of animal models of human traits. Studies using experimental crosses between inbred animal strains have been successful in mapping QTLs with effects on a number of different phenotypes, including behavior, but attempts to fine map QTLs in animals often have foundered on the discovery that a single QTL of large effect was in fact caused by multiple loci of small effect positioned within the same chromosomal region (4). A further potential difficulty with detecting QTLs between inbred crosses is the significant reduction in genetic heterogeneity compared with the total genetic variation present in animal populations: a QTL segregating in the wild need not be present in the experimental cross.
In an attempt to circumvent the difficulties encountered with inbred crosses, we have been using a genetically heterogeneous stock (HS) of mice for which the ancestry is known. The heterogeneous stock was established from an eight-way cross of C57BL, BALB/c, RIII, AKR, DBA/2, I, A, and C3H/2 inbred strains (5). Since its foundation 30 years ago, the stock has been maintained by breeding from 40 pairs and, at the time of this experiment, was in its 60th generation. Thus each chromosome from an HS animal is a fine-grained genetic mosaic of the founder strains, with an average distance between recombinants of 1/60 or 1.7 cM.
Theoretically, the HS offers at least a 30-fold increase in resolution for QTL mapping compared with an F2 intercross (6, 7). The high level of recombination means that fine mapping is possible by using a relatively small number of animals; for QTLs of small to moderate effect, mapping to under 0.5 cM is possible with fewer than 2,000 animals. The large number of founders increases the genetic heterogeneity, and in theory one can map all QTLs that account for progenitor strain genetic differences. Potentially, the use of the HS offers a substantial improvement over current methods for QTL mapping.
However, for HS mapping to achieve widespread use, we need to establish its limitations and provide a robust statistical method of analysis. In this paper we describe a multipoint method capable of detecting small-effect QTLs in the HS; we evaluate both its power of QTL detection and the expected degree of QTL resolution. The utility of the method is demonstrated by fine mapping five QTLs for fearfulness in HS mice, only two of which were detectable by single-marker (SM) association.
Materials and Methods
Open-field behavioral testing, genotyping, mapping and generation of markers was performed as described in ref. 8. The following microsatellites were generated: chromosome 1 markers, 103.37 ATAGAACCTGGTGCCTGTGG, TCCCCAGGAGAAGACACAAG and 103.64B AAGGGTTCTGAGGTGCAGAA, TAGTGGTGCACATCTGCA; and chromosome 12 markers, 419.2 TCCAGATCTCCCCACAGTTC, CCACACTCCAGGAAAGGATC, 419.19 GGCAGTGGTAATCAGGATGTG, TCCCTTCTCCTGGTTGTTGT, and 419.21 TCACTGGGCTCTAACCTTGG, GTAAAATGGTGGCAGTGGTG.
Statistical Theory
Failure of SM Association Analysis.
It has been noted in association studies in human populations that SM association analysis may fail to detect QTLs expected to be segregating (1). We encountered the same problem in a study (8) of open-field behaviors of HS mice, a validated animal model of susceptibility to anxiety (9). We typed a total of 67 markers approximately 1 cM apart on 750 HS mice, over five regions where previous F2 intercrosses had detected QTLs (refs. 10 and 11; Table 1). We expected to confirm QTLs in all five regions because the strains that were used in the F2 detection experiments were among the founders of the HS.
Table 1.
QTLs detected in F2 intercrosses and corresponding SM ANOVA logP of HS mice
| Closest marker | Position, cM | LOD | % var. | Cross | SM logP |
|---|---|---|---|---|---|
| D1Mit150 | 84.4 | 13.4 | 9.2 | b | 0.5 |
| D1Mit116 | 80.2 | 7.1 | 6.3 | a | 6.8 |
| D10Mit237 | 76.0 | 8.8 | 8.3 | a | 1.8 |
| D12Mit47 | 31.5 | 4.3 | 4.4 | b | 4.5 |
| D15Mit63 | 44.0 | 11.0 | 8.1 | b | 1.4 |
We used SM analysis of variance to map the QTLs. At each marker the animals were grouped according to their genotype and one-way ANOVA was used to test for significant differences between the group means. Marker-QTL association was indicated by a significant F-statistic in the ANOVA. We confirmed and fine mapped QTLs in only two of the five regions (Table 1). On chromosome 1 a QTL accounting for 6% of the phenotypic variance was mapped into an interval of 0.8 cM, so in some circumstances SM association works well. We therefore sought an explanation for the three failures.
One possible reason is that genetic drift in the HS has resulted in allele fixation, but computer simulations of the HS breeding protocol indicate that only 5% of the genome should be fixed, consistent with the observed level of marker homozygosity. In fact, the explanation is that alleles of the same size are descended from different strains. SM association analysis does not use information about the founder haplotypes or from neighboring markers and cannot distinguish between strains having different QTL effects but identical alleles at a nearby marker. At most markers there are only two or three alleles, so one cannot determine from which of the eight strains a single allele has descended.
For example, consider two markers (D1Mit100, D1Mit496), less than 500 kb apart, near the QTL at position 64 cM on chromosome 1 (Fig. 1A). SM ANOVA yielded a logP of 5.35 at D1Mit100, but gave a logP of 0.04 at D1Mit496 (all significance levels are given here as logP values, i.e., log10p, so e.g., P-value 10−4 corresponds to logP 4). The proximity of the markers rules out recombination as a reason for the difference in significance. Rather, the important difference between D1Mit496 and D1Mit100 is that strain RIII can be distinguished from A/J, C3H, I at D1Mit100.
Figure 1.

SM (green) and DP (red) analyses of regions of chromosomes (Chr) 1 (A and B), chromosome 10 (C), chromosome 12 (D), and chromosome 15 (E). Distances along each chromosome are given in cM (x axis). The y axis measures logP values. DP thresholds (blue) are the empirical 0.1% logP thresholds derived by permuting the genotypes 1,000 times. Selected markers are labeled.
A Multipoint Model Using Progenitors.
To incorporate information from flanking markers and the progenitor haplotypes, we developed a multipoint method that determines the probability of each founder strain being the ancestor of a given allele in the HS. QTLs then are detected by testing for differences between the genetic effects of the progenitor haplotypes rather than by association at each locus. Note that it would not help to reconstruct the haplotypes of the HS at the generation we tested, as this would not determine whether (in the example) an allele at D1Mit496 was derived from RIII or from one of the other strains. The critical issue is to calculate the probability that an allele is descended from one of the eight progenitors, which is different from standard interval mapping (12) or interval mapping with marker cofactors (13–15).
Because the number of possible ancestral haplotype reconstructions increases exponentially with the number of markers, it is impossible to calculate the probability of each haplotype separately. However a dynamic programming (DP) algorithm greatly reduces the complexity. DP was first used by Lander and Green (16) in a different context to reconstruct haplotypes from pedigrees. Our method does not use pedigree information. The analysis is in two stages: ancestral haplotype probability reconstruction using dynamic programming followed by hypothesis testing using linear regression.
We assume that at a QTL locus, L, a chromosome originating from the progenitor strain s, contributes an unknown additive amount Ts to the phenotype, so that the expected genetic effect for a diploid individual with ancestral alleles labeled s, t at the trait locus is Ts + Tt; a test for a QTL is equivalent to testing for differences between the Tss. The DP method computes the probability FLi(s, t) that individual i has the ancestral alleles s, t at L. Then the expected phenotype is
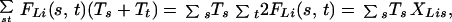 |
1 |
say, and the Tss are estimated by a linear regression of the observed phenotypes across all individuals using the design matrix XL, followed by an ANOVA to test whether the progenitor estimates differ significantly. The method's effectiveness depends on the ability to distinguish ancestral haplotypes across the interval; clearly the power will be lower where all markers have the same noninformative allele distribution, but markers share information where there is a mixture.
This problem can be thought of as a Hidden Markov model, where the hidden states are the progenitor haplotypes and the observed data the genotypes. Define Pmi(s,t) to be the probability that for a certain individual i, the progenitor haplotypes are s,t at marker m, given (i) the genotypes for the ordered markers numbered 1–m, (ii) the founder strain haplotypes, expressed as the probability πm(s|a) that the ancestral state at marker m on a particular chromosome is s given the allele observed at that locus is a, (iii) the genetic distances dm between markers m, m + 1. Ignoring interference and nonrandom mating effects (i.e., pedigree information), the number of recombinants between markers is distributed as a Poisson random variable with mean Gdm, where G is the number of generations since the HS was founded. Consequently the prior probability that on a certain chromosome locus m + 1 is in state s given locus m is in state is σ is
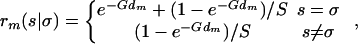 |
2 |
where S is the number of strains. The prior probability of each of the S progenitor strains is 1/S at any locus, and missing data are treated as an allele with equal probability in the founder strains. Conditional on the genotype a, b for individual i at marker m + 1 and the ancestral haplotypes at m being σ,τ the transition probability that the haplotypes at m + 1 are s, t is:
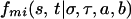 |
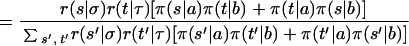 |
3 |
(the subscript m has been dropped from r,π for clarity). As the phase of the genotypes is unknown we must consider both possibilities. Therefore the total probability that that the haplotypes at m + 1 are s, t can be expressed as a DP recurrence relation
 |
4 |
summed over all possible haplotypes σ,τ at m. Pmi(s, t) is computed iteratively across the chromosome, starting at the first marker. Similarly, we can find Qm+1i(s, t), the probability that locus m + 1 is in state s, t given all information from markers m + 1 through M by running the algorithm backward from the terminal marker. Analysis of N individuals, M markers, and S strains requires space proportional to NMS2 and time proportional to NMS4.
QTL Detection.
Suppose the QTL at locus L is between markers m, m + 1 at an unknown distance cdm from m. The probability FLi(s, t) that the haplotypes are s, t at L in individual i will depend on the flanking marker distributions and the pattern of recombination in the interval. Fixing on one chromosome, the locus must either be linked to both markers, or just the left marker, or just the right, or be unlinked, with respective probabilities
 |
5 |
A diploid individual's chromosomes need not be linked the same way. By integrating over c we obtain the intervalwide prior probability that the joint linkage state for the the QTL is XY, as pXY =∫01 pX(c)pY(c) dc. Then, dropping the subscripts for clarity, the probability that the founder alleles are s, t at the QTL L is found by summing over all possible linkage states XY:
 |
6 |
where the probability an unlinked locus is in any given state = 1/S, and P(⋅, t) = Σs Pmi(s, t), Q(⋅, t) = ΣsQm+1i(s, t), etc. For example, the term P(⋅, t)Q(s, t)pRB is the probability that chromosome 1 of the QTL is in state s and linked just to the right-hand marker, and chromosome 2 is in state t and linked to both left and right markers.
We found that greatest sensitivity to detect a QTL occurs when the generations G is set substantially higher that the true number. Likely reasons for this phenomenon are that the distances of nearby markers may be inaccurate, and the presence of erroneous genotypes that create false recombinant events. On a 450-Mhz Pentium III running redhat linux 2.2, 750 mice, 45 markers, and eight strains can be analyzed (i.e., DP plus linear regression) in 73 central processor unit s using a C-program, happy.
We test for a QTL in the intervals between adjacent markers rather than at each marker locus; DP logP values refer to marker intervals and SP logP values to markers at the interval endpoints. In Fig. 1, DP logP values are plotted as step functions that are constant over each interval. It is possible to generate pointwise logP values but they do not differ significantly from the interval-wise values.
Results
Significance Levels and Resolution.
We examined a 10-cM region around each of the five QTLs identified in the F2 intercrosses (Table 1), placing markers on the radiation hybrid map and, where possible, the European Collaborative Interspecific Backensi genetic map to provide accurate marker positions necessary for the method. The results are shown in Fig. 1.
To check the accuracy of the tabulated ANOVA significance levels, we permuted the phenotypes between animals and repeated the ANOVA 1,000 times, thereby taking into account the large number of markers, the fact that the tests are no longer independent, and that the phenotypes may not be normally distributed. At each marker interval the logP values were ranked, and the 5%, 1%, and 0.1% significance levels were defined as the corresponding percentiles. They are slightly less than their theoretical values, so the use of logP derived from a tabulated F distribution is reliable and conservative. Fig. 1 shows the 0.1% significance levels. Additionally, the most significant permuted logP in each region was close to the reciprocal of the number of intervals, so the tests may be treated as independent. Therefore, to establish significance levels appropriate for any mapping experiment, we need only divide the individual regression P-value by the number of intervals. We analyzed a total of 63 intervals, so the 1.0% and 0.1% logP thresholds are 3.8 and 4.8, respectively. All of the QTLs we have detected exceed the 1% level, and only one (near D15Mit134, logP 3.95) fails to exceed the 0.1% level.
We used a bootstrap procedure to determine mapping resolution. A data set was created by sampling the animals with replacement and the regression analysis repeated in the neighborhood of each QTL. The number of times that each marker interval contained the most significant logP was recorded in 500 iterations. Table 2 gives the intervals with the highest percentage of QTL locations. On chromosome 10, 99% of bootstraps placed the QTL into a 0.5-cM interval; however, on chromosome 12 we found that almost 20% of bootstraps indicated a second location for the QTL. The bootstrap-defined range for chromosomes 1 and 15 agree with an independent high-resolution haplotype and recombinant inbred segregation test carried out on the BALB/cJ and C57BL/6 crosses (17) (see Table 2). Consequently, we conclude that the QTLs have been replicated in the HS.
Table 2.
Bootstrap estimates of QTL locations compared with positions determined by RIST analysis of BALBc/J X C57BL/6 (17)
| Chromosome | DP range | Bootstrap probability | RIST range |
|---|---|---|---|
| Chr1 | 64.0–65.0 | 0.92 | |
| 82.3–83.8 | 0.96 | 80.0–84.3 | |
| Chr10 | 69.5–70.0 | 0.99 | |
| Chr12 | 31.0–32.0 | 0.80 | |
| 32.5–33.0 | 0.20 | ||
| Chr15 | 42.0–43.5 | 0.91 | 43.0–47.2 |
The bootstrap probability is the proportion of times the highest logP value in the neighborhood of the QTL fell in the specified DP range. RIST, recombinant inbred segregation test.
Table 3 documents the DP effect sizes and the T statistics associated for the three QTLs where SM analysis failed, together with the ancestral alleles of the two flanking marker loci. This confirms that the DP method succeeds over SM when QTLs with opposite effects are associated with the same allele.
Table 3.
DP effect sizes, T statistics, and alleles at three marker intervals containing QTLs undetected by SM analysis
| Strain | Chromosome 1
|
Chromosome
10
|
Chromosome 15
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Effect | T | D1Mit541 | D1Mit115 | Effect | T | D10Mit103 | D10Mit271 | Effect | T | D15Mit29 | D15Mit134 | |
| A | −1.39 | −4.67 | 122 | 146 | 4.31 | 4.48 | 148 | 111 | −1.29 | −2.76 | 151 | 145 |
| AKR | 0.11 | 0.56 | 118 | 122 | 5.02 | 4.31 | 142 | 103 | 1.93 | 2.76 | 151 | 145 |
| BALB/cJ | −2.84 | −2.10 | 126 | 142 | −3.81 | −0.39 | 148 | 111 | −1.29 | −2.76 | 151 | 145 |
| C3H | 2.84 | 2.10 | 126 | 142 | −3.21 | −3.40 | 144 | 103 | −1.29 | −2.76 | 151 | 145 |
| C57BL/6 | 1.61 | 2.14 | 126 | 148 | −0.24 | −0.31 | 148 | 115 | 1.93 | 2.76 | 151 | 145 |
| DBA | −1.71 | −2.31 | 114 | 148 | 1.74 | 2.04 | 144 | 115 | 0.88 | 4.65 | 151 | 145 |
| I | 1.79 | 2.37 | 114 | 142 | 0.30 | 0.46 | 142 | 109 | −0.04 | −0.09 | 183 | 139 |
| RIII | 0.84 | 1.22 | 122 | 122 | −4.21 | −3.90 | 142 | 113 | 4.20 | −2.09 | 177 | 145 |
Simulations.
We also compared the DP and SM methods by simulating the HS breeding protocol for 60 generations. The large number of variables involved means that is not feasible to simulate all possible combinations of effect size and progenitor allele distributions, so instead we used the DP-estimated progenitor effects and the observed progenitor alleles for the loci on chromosomes 1, 10, 12, and 15, which captures the QTL phase association derived from the DP analysis. QTL effect sizes were scaled so that the QTL accounted for 5% of the total phenotypic variance in the final generation. In about 5% of simulations the QTL alleles went to fixation and these results were discarded. The percentages of successful detections for the two methods are given in Table 4. DP was always more efficient than SM. The efficiencies vary between loci, which we attribute to the different phase relationships between alleles and QTLs. Additional simulations using other phase associations confirm a marked variation in detection rates. We also performed 500 computer simulations where there was no QTL present. After taking into account the number of simulations made, no false positives were found.
Table 4.
Estimated probabilities of detecting a QTL accounting for 5% of phenotypic variance modeled on observed allele and trait effects from Fig. 1 and Table 3
| Chr1 | Chr1 | Chr10 | Chr12 | Chr15 | |
|---|---|---|---|---|---|
| DP | 0.92 | 0.80 | 0.86 | 0.50 | 0.62 |
| SM | 0.89 | 0.69 | 0.65 | 0.23 | 0.38 |
Five hundred simulations were performed in each case. A 0.1% significance level on 63 intervals defined a successful detection. Chr, chromosome.
Discussion
We have shown that using DP generally improves QTL detection and fine mapping. The failure of SM methods appears to be because of different QTL alleles occurring on similar haplotypes. In human populations a similar phenomenon has been observed, where numerous mutations for thalassaemia have been found on apparently identical haplotypes (18). In particular, the oscillatory behavior of SM analysis in Fig. 1A is similar to that often observed in studies of linkage disequilibrium. Presumably the relationship between QTLs and ancestral human chromosomes is equally complex, reducing the power of genomewide association studies unless ancestral chromosomes can be reconstructed (19).
DP analysis of HS animals provides a fast, robust, and cost-effective strategy for high-resolution analysis of complex quantitative traits. There are many possible applications of the method.
Genomewide Scans.
Multipoint DP analysis of HS mice seamlessly combines data from any marker type [especially single nucleotide polymorphism (SNPs) and microsatellites] thus making it ideal for high-resolution genomewide scans, as has become possible with the publication of a first-generation set of high-density SNPs for the mouse (20). We used simulation of the HS breeding protocol to estimate the power of our method for a whole genome scan with SNPs. We simulated 50 diallelic markers spaced at 1-cM intervals, with a QTL that explained 2.5%, 5%, and 10% of the phenotypic variance placed midway between the two central markers, in populations sizes of 500 and 1,000 animals. To establish significance levels appropriate for a genome scan, we divide the individual P-value by the number of intervals (3,000) analyzed. In Table 5 we show the probabilities of successful QTL detections obtained for three genomewide significance levels (5%, 1% and 0.1%, corresponding to log P-values of 4.38, 5.08, 6.08).
Table 5.
Estimated DP probability of detecting a QTL explaining 2.5%, 5%, and 10% of the phenotypic variance at genomewide significance levels of 5%, 1%, and 0.1% using 3,000 diallelic (SNP) markers 1 cM apart and population sizes 500 and 1,000 HS mice
| QTL
|
2.5
|
5
|
10
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Sig | 5 | 1 | 0.1 | 5 | 1 | 0.1 | 5 | 1 | 0.1 |
| 500 | 0.07 | 0.07 | 0.03 | 0.55 | 0.49 | 0.30 | 0.60 | 0.53 | 0.37 |
| 1,000 | 0.18 | 0.12 | 0.05 | 0.92 | 0.88 | 0.85 | 0.97 | 0.95 | 0.89 |
SNP, single nucleotide polymorphism.
Mapping Traits in Parallel.
Not only can the method simultaneously map multiple QTLs of small effect, it also can fine map many traits in parallel, removing the need for separate F2 QTL detection experiments, and avoiding problems where a QTL is present in the HS but not in the F2. The only requirements in trait selection are that the measurements do not interfere with each other and each phenotype has a heritable variance in the founders of the HS. Based on our work required to fine map a single trait, whole-genome mapping would be cost effective when 20 or more traits are mapped in parallel on the same set of mice. A potential disadvantage of this approach would be that the number of tests performed per trait would be about 20 times larger (3,000 markers in a genome scan at 1-cM resolution compared with an F2 detection experiment on 100 markers followed by fine mapping about a further 100), so significance thresholds would be correspondingly higher.
Mapping Modifier Genes.
Numerous mouse models of human disease, either spontaneous or genetically engineered mutants, have been established. These animals are either maintained on an inbred background, or by continually backcrossing onto a F1 between two inbred lines (21). It has been shown that the phenotype of the mutant will vary depending on the genetic background, indicating that modifier genes can have a significant effect. Consequently molecular characterization of modifiers is likely to provide novel insights into pathogenesis.
Mapping modifier loci by crosses between inbred mutant strains is qualitatively similar to and has the same limits of resolution as standard QTL detection experiments. However, it is possible to extend our method to fine map modifier genes. Consider an F1 cross between an HS animal and a mutant. For simplicity we will assume the mutant is on a constant inbred background, so that all modifier loci may be treated as coming from the HS chromosome. Therefore, the expected effect on the observed phenotype of a modifier locus descended from progenitor strain s will be Ts, that is, the analysis resembles that of a haploid genome.
Proceeding analogously to the analysis of a pure HS population, at the marker m, let βm(a) be the probability that a chromosome around marker m is from the inbred background, given the allele observed is a. Let πm(s|a) be the probability that a chromosome around m is HS and derived from ancestral strain s, given a. Then the transition probability that the HS ancestral strain at m + 1 is s given the observed genotype a,b for individual i at m + 1, and the HS is in state σ at m, is
 |
7 |
Consequently the probability Pm+1 i(s) that the HS chromosome is from founder strain s at marker m + 1, conditional on all of the genotypes for markers 1,2 … m + 1 satisfies
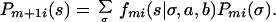 |
8 |
The remaining analysis follows the previous case with the obvious simplifications for a haploid genome and is omitted.
In the case of a HS intercrossed with a backcross, it can be shown that the backcross-derived chromosome will resemble an HS chromsome derived from two founder strains after three generations of breeding. Consequently, we can analyze the data as a cross between two HS of different founders and ages.
QTL Detection and Fine Mapping Using F2 × HS Hybrids.
Consider a hybrid whose parents are an HS and an F1 intercross of two inbreds. The HS-derived chromosome will have a high level of recombination, whereas the other will be an F2 intercross. Such animals can be used for both a genome scan with 100 markers at 20- to 30-cM spacing, using information from just the F2 chromosomes, followed by fine mapping at 1-cM resolution those regions likely to contain a QTL, using the HS chromosomes. The method of analysis is similar to that of a cross between two distinct HS. During the QTL detection phase, the high level of recombination in the HS chromosome means that HS QTLs will probably be unlinked to the markers, so their state cannot be determined and their effects contribute to the residual variance. However, during the fine mapping, the state of the F2 chromosome can in general be determined and is usually constant across each region.
The relative merits of this approach over performing two separate experiments depend on the costs of the animals, phenotyping, and the phenotypic variance between the HS founders. If necessary, the increased residual variance during the QTL detection phase could be offset by increasing the marker density.
Acknowledgments
We thank L. Cardon for helpful discussions. This work was supported by the Wellcome Trust (R.M., C.J.T., and J.F.). The analysis software and data sets are available from http://www.well.ox.ac.uk/happy.
Abbreviations
- QTL
quantitative trait loci
- HS
heterogeneous stock
- SM
single marker
- DP
dynamic programming
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 12389.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.230304397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.230304397
References
- 1.Risch N J. Nature (London) 2000;405:847–856. doi: 10.1038/35015718. [DOI] [PubMed] [Google Scholar]
- 2.Risch N J, Merikangas K. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 3.Roses A D. Nature (London) 2000;405:857–865. doi: 10.1038/35015728. [DOI] [PubMed] [Google Scholar]
- 4.Legare M E, Bartlett F S, Frankel W N. Genome Res. 2000;10:42–48. [PMC free article] [PubMed] [Google Scholar]
- 5.McClearn G E, Wilson J R, Meredith W. In: Contributions to Behavior-Genetic Analysis: The Mouse as a Prototype. Lindzey G, Thiessen D, editors. New York: Appleton Century Crofts; 1970. pp. 3–22. [Google Scholar]
- 6.Darvasi A, Soller M. Genetics. 1995;141:1199–1207. doi: 10.1093/genetics/141.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darvasi A, Soller M. Behav Genet. 1997;27:125–132. doi: 10.1023/a:1025685324830. [DOI] [PubMed] [Google Scholar]
- 8.Talbot C J, Nicod A, Cherny S S, Fulker D W, Collins A C, Flint J. Nat Genet. 1999;21:305–308. doi: 10.1038/6825. [DOI] [PubMed] [Google Scholar]
- 9.Gray J A. The Psychology of Fear and Stress. Cambridge, U.K.: Cambridge Univ. Press; 1987. [Google Scholar]
- 10.Flint J, Corley R, DeFries J C, Fulker D W, Gray J A, Miller S, Collins A C. Science. 1995;269:1432–1435. doi: 10.1126/science.7660127. [DOI] [PubMed] [Google Scholar]
- 11.Gershenfeld H K, Neumann P E, Mathis C, Crawley J N, Li X, Paul S M. Behav Gen. 1997;27:201–210. doi: 10.1023/a:1025653812535. [DOI] [PubMed] [Google Scholar]
- 12.Lander E S, Botstein D. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng Z B. Genetics. 1994;136:1457–1468. doi: 10.1093/genetics/136.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jansen R C. Theor Appl Genet. 1992;85:252–260. doi: 10.1007/BF00222867. [DOI] [PubMed] [Google Scholar]
- 15.Jansen R C, Stam P. Genetics. 1993;135:205–211. doi: 10.1093/genetics/135.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lander E S, Green P H. Proc Natl Acad Sci USA. 1987;84:2363–2367. doi: 10.1073/pnas.84.8.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turri M G, Talbot C J, Radcliffe R A, Wehner J M, Flint J. Mamm Genome. 1999;10:1098–1101. doi: 10.1007/s003359901169. [DOI] [PubMed] [Google Scholar]
- 18.Flint J, Harding R M, Clegg J B, Boyce A J. Hum Genet. 1993;91:91–117. doi: 10.1007/BF00222709. [DOI] [PubMed] [Google Scholar]
- 19.Service S K, Lang D W, Freimer N B, Sandkuijl L A. Am J Hum Genet. 1999;64:1728–1738. doi: 10.1086/302398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindblad-Toh K, Winchester E, Dalyand M J, Wang D G, Hirschhorn J N, Laviolette J P, Ardlie K, Reich D E, Robinson E, Sklar P, et al. Nat Genet. 2000;24:381–386. doi: 10.1038/74215. [DOI] [PubMed] [Google Scholar]
- 21.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies S W, Bates G P. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]