

Abstract
Arsenic exposure has been shown to exacerbate atherosclerosis, beginning with activation of the endothelium that lines the vessel wall. Endothelial barrier integrity is maintained by proteins of the adherens junction (AJ) such as vascular endothelial cadherin (VE-cadherin) and β-catenin and their association with the actin cytoskeleton. In the present study, human aortic endothelial cells (HAECs) were exposed to 1, 5 and 10 μM sodium arsenite [As(III)] for 1, 6, 12 and 24 h, and the effects on endothelial barrier integrity were determined. Immunofluorescence studies revealed formation of actin stress fibers and non-uniform VE-cadherin and β-catenin staining at cell-cell junctions that were concentration and time-dependent. Intercellular gaps were observed with a measured increase in endothelial permeability. In addition, concentration-dependent increases in tyrosine phosphorylation (PY) of β-catenin and activation of protein kinase Cα (PKCα) were observed. Inhibition of PKCα restored VE-cadherin and β-catenin staining at cell-cell junctions and abolished the As(III) induced formation of actin stress fibers and intercellular gaps. Endothelial permeability and PY of β-catenin were also reduced to basal levels. These results demonstrate that As(III) induces activation of PKCα, which leads to increased PY of β-catenin downstream of PKCα activation. Phosphorylation of β-catenin plausibly severs the association of VE-cadherin and β-catenin, which along with formation of actin stress fibers, results in intercellular gap formation and increased endothelial permeability. To the best of our knowledge, this is the first report demonstrating that As(III) causes a loss of endothelial monolayer integrity, which potentially could contribute to the development of atherosclerosis.
Keywords: sodium arsenite, stress fibers, VE-cadherin, β-catenin, vascular permeability, PKCα
Introduction
Arsenic, an element present in the earth's crust, has been widely distributed in the environment by human activities such as mining, smelting, manufacturing and pesticide application. Epidemiological studies analyzing the correlation between cardiovascular diseases and arsenic exposure, have linked arsenic to atherosclerosis (Wang et al., 2002), ischemic heart disease (Tseng et al., 2003) and hypertension (Rahman et al., 1999). Drinking water is a significant source of arsenic exposure to humans. Using mouse models of atherosclerosis, we and others have demonstrated that chronic exposure to arsenic through drinking water exacerbates atherosclerotic plaque formation (Bunderson et al., 2004, Simeonova et al., 2003). Atherosclerosis can lead to serious clinical manifestations such as heart attack and stroke.
There are numerous regulatory points in the vascular system that may be disrupted and lead to atherogenesis. The integrity of the vascular endothelium is maintained by proteins of the adherens junctions (AJ), which includes the transmembrane protein VE-cadherin linked to the cytoplasmic protein β-catenin which in turn is linked to the actin cytoskeleton through α-catenin (Ivanov et al., 2001). This association is critical to maintaining cell-cell adhesion and structural integrity. The tyrosine phosphorylation (PY) of AJ proteins can alter their interaction with each other and the cytoskeleton. Endogenous mediators such as histamine have been shown to increase PY of both β-catenin and VE-cadherin, resulting in dissociation of VE-cadherin from the actin cytoskeleton with a subsequent increase in vascular permeability (Andriopoulou et al., 1999). The PY of β-catenin has been shown to cause a fivefold reduction in its affinity for E-cadherin (Lilien and Balsamo, 2005, Roura et al., 1999). Accordingly, vascular endothelial growth factor (VEGF) induced PY of VE-cadherin and β-catenin has been implicated in the loosening of cell-cell contacts and increased vascular permeability (Esser et al., 1998).
Bidirectional signaling between the cadherin-catenin complex and the actin cytoskeleton has been observed. Inhibition of VE-cadherin function induces stress fibers and conversely, stress fibers regulate VE-cadherin localization (Hordijk et al., 1999). Stress fibers consist of proteins such as myosin, tropomyosin and α-actinin intermittently associated with the actin filaments (Byers et al., 1984). Stress fibers are contractile elements and have been shown to develop in vivo during endothelial cell adaptation to unfavorable or pathological situations such as atherosclerosis and hypertension (van Nieuw Amerongen and van Hinsbergh, 2001, White et al., 1983, Wong et al., 1983). It has been shown that the apolipoprotein(a) component of lipoprotein(a), a risk factor for atherosclerotic disorders, induces stress fiber formation and VE-cadherin dispersion accompanied by cell retraction, intercellular gap formation and increased endothelial permeability (Pellegrino et al., 2004). Thus, development of stress fibers in endothelial cells adversely affects monolayer integrity. The effects of arsenic on the proteins involved in maintaining endothelial monolayer integrity remain unknown.
Several signaling intermediates have been implicated in the regulation of endothelial barrier function. In particular, the various protein kinase C (PKC) isotypes have a host of cellular substrates and are involved in numerous biological processes. PKCs are serine/threonine kinases and have been grouped into 3 subfamilies: classical, novel and atypical PKCs (Newton, 2001). The classical PKCs (cPKC), which consists of α, β and γ subtypes, are activated in a calcium-dependent manner and require diacylglycerol and phosphatidylserine. Amongst the cPKCs, endothelial cells express α and β isotypes (Mattila et al., 1994). The cPKCs have been implicated in the modulation of endothelial barrier function (Yuan, 2003). Activation of PKCα by thrombin has been shown to induce actin stress fibers and increase endothelial permeability (Sandoval et al., 2001). Conflicting data have been published regarding the effects of arsenic on the modulation of intracellular calcium levels and cPKCs; effects that are dependent on the cell line studied and the duration of arsenic treatment (Chen et al., 2000, Liu and Huang, 1997).
Various mechanisms for arsenic-induced atherosclerosis have been proposed including activation of the endothelium that is characterized by up-regulation of adhesion molecules and inflammatory mediators (Simeonova and Luster, 2004). In particular, in situ arsenic exposure through intradermal injections on the dorsal surface of rats has been shown to induce vascular permeability (Chen et al., 2004). However, the mechanism of increased vascular permeability remains unknown. The endothelium maintains a semipermeable barrier and damage to the endothelial lining can result in increased permeability. This could lead to subsequent recruitment and migration of monocytes and oxidized low density lipoproteins (oxLDL) into the intima of the blood vessel, thereby promoting the development of atherosclerotic lesions. Hence, the objective of this study was to determine the effects of arsenic on endothelial barrier integrity and characterize the pathways for arsenic-induced increases in vascular permeability.
Materials and Methods
Cell culture
Human aortic endothelial cells (HAECs) were purchased from Cambrex Bio Science (Walkersville, MD) and were grown in the supplier's formulated medium: EBM-2 medium supplemented with EGM-2 SingleQuots (0.04% hydrocortisone, 0.4% hFGF-B, 2% FBS and 0.1% each of VEGF, R3-IGF-1, ascorbic acid, heparin, hEGF and GA-1000). The cells were incubated at 37°C under a humidified atmosphere containing 5% CO2. Cells were used between passages 2 and 10.
Immunofluorescence
HAECs were grown on 18 mm round coverslips inserted into 12-well plates and coated with 0.1 mg/ml solution of Poly-D-Lysine hydrobromide (Sigma-Aldrich, Saint Louis, MO). HAECs were seeded at a density of 2×105-3 ×105 cells/well and were grown to confluence for 48-72 h. Cells were treated with As(III) (GFS Chemicals, Columbus, OH) and the cPKC inhibitor, Gö 6976 (Calbiochem, San Diego, CA), was added 1 h prior to and during the As(III) treatment wherever applicable. Cells were fixed with 1% paraformaldehyde/ phosphate-buffered saline (PBS) for 30 min at room temperature (RT), permeabilized using 0.1% saponin solution/ PBS for 10 min at RT and then blocked with 5% goat serum/PBS (Sigma-Aldrich, Saint Louis, MO) for 30 min at RT. Cells were then incubated with 2 μg/ml rabbit anti-VE-cadherin polyclonal antibody (Cayman Chemicals, Ann Arbor, MI), 2 μg/ml rabbit anti-β-catenin polyclonal antibody (Cayman Chemicals, Ann Arbor, MI) or 0.24 μg/ml rabbit antibody against cleaved caspase-3 (Cell Signaling Technology, Danvers, MA) in PBS for 2 h at RT or 24 h at 4°C followed by incubation with 10 μg/ml Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (Molecular Probes, Inc. Eugene, OR) for 1 h at RT in the dark. For detection of stress fibers, HAECs were incubated with 16.5 nM Alexa Fluor 568-conjugated phalloidin (Molecular Probes, Inc. Eugene, OR) for 25 min at RT in the dark. Cells were counterstained with Hoechst stain for 5 min at RT, coverslipped with Fluorosave (EMD Biosciences, San Diego, CA) and stored in the dark until imaged. Slides were photographed with a Nikon Digital Eclipse DXM1200 camera attached to a Nikon E-800 microscope using a 60X oil immersion lens.
Permeability Assay
HAECs (1×105-1.5 ×105) were seeded on 0.4 μm pore polyester membrane transwell inserts (Corning Incorporated, Acton, MA) coated with 0.1 mg/ml solution of Poly-D-Lysine hydrobromide in 12 well plates. Cells were grown to confluence for 48-72 h. The volume of medium in the Transwell was 500 μl with 1.5 ml in the plate well at all times. The As(III) treatments were added to the Transwells and the cPKC inhibitor, Gö 6976, was added 1 h prior to and during the As(III) treatment wherever applicable. At the final 1 h of respective treatments, fluorescein isothiocyanate (FITC)-labeled dextran (average molecular weight 40,000, final concentration of 1 mg/ml; Sigma-Aldrich, Saint Louis, MO) was added to each Transwell including control wells. After completion of the treatment period, the fluorescence intensity of the medium in the plate well was analyzed using a SpectraMax GeminiXS fluorescence plate reader (Molecular Devices Corporation, Sunnyvale, CA). For each sample, 5 aliquots of 200 μl each were analyzed using an excitation wavelength of 490 nm and emission wavelength of 520 nm.
Immunoprecipitation and Western blot analysis
The PY of VE-cadherin and β-catenin was analyzed by immunoprecipitation followed by Western blot. HAECs were seeded at a density of 7.5×105 cells/ 25 cm2 cell culture flask and allowed to reach confluence for 48-72 h. Confluent cells were treated with As(III) and the cPKC inhibitor, Gö 6976 was added 1 h prior to and during the As(III) treatment wherever applicable. After completion of treatment, cells were rinsed with cold PBS containing phosphatase inhibitors (1 mM β-glycerophosphate, 1 mM Sodium pyrophosphate and 2.5 mM Sodium orthovanadate). The sodium orthovanadate was activated for maximal inhibition of protein tyrosine phosphatases (activation method obtained from the list of protocols provided by Upstate Cell Signaling Solutions). Cells were harvested using cold 1X RIPA buffer (20 mM Tris, pH 7.5, 5 mM EDTA, 2 mM EGTA, 1% Igepal, 0.1% SDS and 0.5% deoxycholic acid) containing protease inhibitors (Complete protease inhibitor cocktail tablet; Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitors. Cells were homogenized by sonication and centrifuged at 10,000 rpm for 15 min at 4°C. The supernatant was sequentially incubated with 5 μg/ml each of mouse anti-VE-cadherin monoclonal antibody followed by mouse anti-β-catenin monoclonal antibody (Abcam, Cambridge, MA) for 24 h at 4°C. Immune complexes were precipitated using immobilized protein G (Pierce, Rockford, IL). After incubation for 2 h at 4°C, the agarose bound immune complexes were washed three times with cold 1X RIPA buffer containing protease and phosphatase inhibitors and then heated at 70°C for 10 min in 1X solution of reducing agent and sample buffer (Invitrogen Life Technologies, Carlsbad, CA). The proteins were resolved using 4-12% NuPAGE Novex Bis-Tris gel (Invitrogen Life Technologies, Carlsbad, CA), transferred to a polyvinylidenedifluoride (PVDF) membrane, blocked with 5% BSA/ TBST (TBST: 1.57 gm/l Tris HCl (pH=8), 8.76 gm/l NaCl, 500 μl/l Tween 20) for 1 h at RT and incubated with 2 μg/ml mouse anti-phosphotyrosine monoclonal antibody (BD Transduction Laboratories, San Jose, CA) overnight at 4°C in 1% BSA/ TBST solution. This was followed by incubation of the membrane with 1 μg/ml peroxidase labeled horse anti-mouse secondary antibody (Vector laboratories Inc., Burlingame, CA) for 1 h at RT in 1% BSA/ TBST solution. The protein bands were visualized by ECL Western blot detection system (Amersham, Buckinghamshire, England). To quantitate PY, the total protein content of immunoprecipitated VE-cadherin and β-catenin was determined as follows. After analysis of phosphotyrosine content, the membranes were stripped at 60°C for 1 h using stripping buffer (100 mM β-mercaptoethanol, 2% SDS and 62.5 mM Tris HCL, pH 6.8), blocked with 5% non-fat milk/ TBST for 1 h at RT and reprobed for VE-cadherin and β-catenin protein using 1.25 μg/ml rabbit anti-VE-cadherin and 1 μg/ml rabbit anti-β-catenin polyclonal antibody (Cayman Chemicals, Ann Arbor, MI) overnight at 4°C in 0.4% non-fat milk/ TBST. This was followed by incubation with 5 μg/ml peroxidase labeled goat anti-rabbit secondary antibody (Vector laboratories Inc., Burlingame, CA) for 1 h at RT in 0.4% non-fat milk/ TBST, and the protein bands were visualized as previously described.
To determine the protein content of VE-cadherin without immunoprecipitation, cells were rinsed with PBS and harvested with 1X RIPA buffer containing protease inhibitors. Cells were homogenized with 18 and 25 gauge needles and boiled for 10 min. Protein concentrations were determined by using the Bradford protein assay (Biorad, Hercules, CA). The proteins were resolved using 4-12% NuPAGE Novex Bis-Tris gel, transferred to PVDF membrane, blocked with 5% non-fat milk/ TBST for 2 h at RT and probed for VE-cadherin protein using 1.25 μg/ml rabbit anti-VE-cadherin polyclonal antibody for 2 h at RT or overnight at 4°C in 0.4% non-fat milk/ TBST. This was followed by incubation with 5 μg/ml peroxidase labeled goat anti-rabbit secondary antibody for 1h at RT in 0.4% non-fat milk/ TBST. The protein bands were visualized by ECL Western blot detection system. Protein loading was normalized using β-actin as loading control. β-actin protein was detected using 0.24 μg/ml mouse anti-β-actin monoclonal antibody (Abcam, Cambridge, MA) and 1 μg/ml peroxidase labeled horse anti-mouse as the secondary antibody.
To determine the activation of PKCα, HAECs were seeded at a density of 7.5×105 cells in 25 or 75 cm2 cell culture flasks and after 3-5 days, confluent cells were treated with As(III). Cells were rinsed with cold PBS, harvested using homogenizing buffer (50 mM Tris HCl, pH 7.5, 1 mM EDTA and protease inhibitors) and sonicated. To separate the membrane and cytosolic fractions, the lysates were centrifuged at 100,000× g for 1 h at 4°C. The supernatant was collected as the cytosolic fraction. The pellet was dissolved in 1X RIPA buffer containing protease inhibitors, sonicated and centrifuged again at 100,000× g for 1 h at 4°C. The supernatant was the membrane fraction. Protein concentrations were determined by using the Bradford protein assay. The proteins were resolved using 4-12% NuPAGE Novex Bis-Tris gels. To detect good quality protein bands, different amounts of cytosolic and membrane fraction proteins were loaded; 10-20 μg protein for the cytosolic fraction and 40-55 μg protein for the membrane fraction. After separation, the proteins were transferred to PVDF membranes, blocked with 5% non-fat milk/ TBST for 1 h at RT and probed for PKCα using 0.4 μg/ml mouse anti-PKCα monoclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) overnight at 4°C in 0.4% non-fat milk/ TBST. This was followed by incubation with 1 μg/ml horse anti-mouse secondary antibody conjugated with horseradish peroxidase for 1 h at RT in 0.4% non-fat milk/ TBST. Protein bands were visualized by ECL Western blot detection system. Na+/K+ ATPase was used as loading control for the membrane fraction using 0.4 μg/ml mouse anti-Na+/K+ ATPase monoclonal antibody (Upstate Cell Signaling Solutions, Lake placid, NY) and GAPDH was used as loading control for the cytosolic fraction using 1 μg/ml mouse anti-GAPDH monoclonal primary antibody (Chemicon International, Inc., Temecula, CA). In both cases, 1 μg/ml horse anti-mouse secondary antibody was used.
Statistical analysis
Results are presented as means ± SE. Statistical analysis was performed using one-way ANOVA followed by Newman-Keuls multiple comparison test. Differences were considered significant at p < 0.05.
Results
Dose response of HAECs to cPKC inhibitor Gö 6976
Indolocarbazole Gö 6976 has been shown to be a potent and selective inhibitor of cPKCs via interference with binding of ATP to the kinase (Martiny-Baron et al., 1993). Preliminary experiments at 5 min, 15 min, 30 min, 1 h, 1.5 h, 2 h and 6 h revealed that 10 μM As(III) induces intercellular gaps beginning at 1 h of treatment (data not shown) that were identified by immunofluorescence staining of VE-cadherin. To determine the effective concentration of Gö 6976 to be used in our experiments, cells were exposed to 10 μM As(III) alone or to 10 nM, 50 nM, 100 nM, 250 nM, 500 nM and 1 μM Gö 6976 in the presence of 10 μM As(III) as depicted in Fig. 1. The duration of As(III) treatment was 1 h and the respective Gö 6976 treatments were applied 1 h prior to and during As(III) treatment. As analyzed by immunofluorescence staining of VE-cadherin, 250 nM-1 μM concentrations of Gö 6976 (Fig.1 F-H) were able to inhibit the As(III) induced gap formation. Therefore, in all the further experiments, 250 nM Gö 6976 was used to inhibit the activity of cPKCs. Also, the effect of Gö 6976 on cell viability was assessed using the MTT assay (Bunderson et al., 2002) and no loss of viability was observed at concentrations 10 nM-500 nM up to 24 h (data not shown).
FIG. 1.

Dose response of cPKC inhibitor Gö 6976. Confluent HAEC monolayers were either untreated (A) or treated with 10 μM As(III) (B) for 1 h and the intercellular gaps (indicated with arrows) were visualized by immunofluorescence staining of VE-cadherin (green). The nuclei were stained with Hoechst (blue). Pretreating the cells with increasing concentration of Gö 6976 for 1 h prior to and during 10 μM As(III) treatment inhibited the gap formation in a concentration-dependent manner: 10 μM As(III) + 10 nM Gö 6976 (C), 10 μM As(III) + 50 nM Gö 6976 (D), 10 μM As(III) + 100 nM Gö 6976 (E), 10 μM As(III) + 250 nM Gö 6976 (F), 10 μM As(III) + 500 nM Gö 6976 (G) and 10 μM As(III) + 1 μM Gö 6976 (H). Magnification=60X. The images are representative of 3 independent experiments performed.
Effect of As(III) exposure on VE-cadherin localization and intercellular gap formation
Based on results from preliminary experiments, the concentration and time-dependent effects of As(III) on VE-cadherin localization and intercellular gap formation were analyzed by immunofluorescence staining of VE-cadherin. Cells were treated with 1, 5 and 10 μM As(III) for 1, 6, 12 and 24 h. At the 1 h time point (Fig. 2), the frequency of gap formation observed with 10 μM As(III) treatment (Fig. 2D) was much greater than 5 μM As(III) (Fig. 2C) and only a few gaps were observed with 1 μM As(III) treatment (Fig. 2B) as compared to the untreated cells (Fig. 2A). Cells appeared to be pulling apart from the adjacent cells. Some areas of undamaged monolayer could be observed. Also, non-uniform staining of VE-cadherin was observed where the gaps appeared. To corroborate the fact that the observed effects were not due to cells undergoing apoptosis, immunofluorescence staining for cleaved caspase-3 was performed. Figures 2E and F demonstrate little cleaved caspase-3 staining for either untreated or 10 μM As(III) treated cells, respectively. Also, MTT assay showed no loss of cell viability at any of the As(III) concentrations used up to 48 h (data not shown). The cPKC inhibitor, Gö 6976, inhibited gap formation at all concentrations of As(III) treatment and uniform distribution of VE-cadherin could be observed at cell-cell junctions. The images of 250 nM Gö 6976 only treatment (Fig. 2G) and 10 μM As(III) + 250 nM Gö 6976 treatment (Fig. 2H) are shown.
FIG. 2.
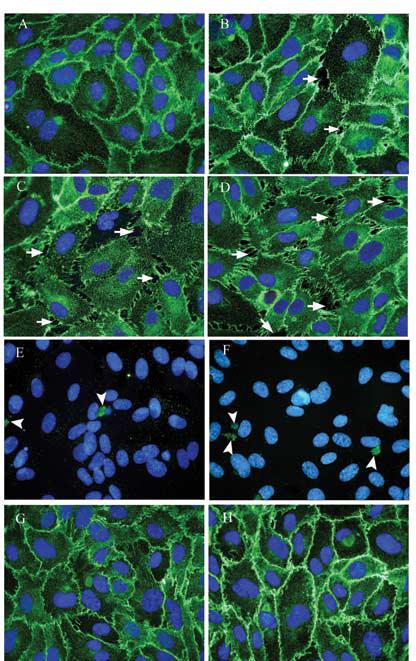
Effect on VE-cadherin localization and intercellular gap formation with As(III) exposure for 1 h. Confluent HAEC monolayers were either untreated (A) or treated with 1 μM As(III) (B), 5 μM As(III) (C) and 10 μM As(III) (D) for 1 h and the intercellular gaps (indicated with arrows) induced with As(III) treatment were visualized by immunofluorescence staining of VE-cadherin (green). The nuclei were stained with Hoechst (blue). Cleaved caspase-3 staining (green) for untreated (E) and 10 μM As(III) (F) treated cells is indicated by arrowheads. Pretreatment with 250 nM Gö 6976 for 1 h prior to and during 10 μM As(III) treatment (H) inhibited the As(III) induced gaps whereas 250 nM Gö 6976 only treatment (G) did not have any effect on the endothelial monolayer. Magnification=60X. The images are representative of 4 independent experiments performed.
At the 6 h time point (Fig. 3), a similar concentration-dependent effect of As(III) was observed but the size and frequency of gaps was greater than that observed at the 1 h time point. Exposure to 1 μM (Fig. 3B) and 5 μM As(III) (Fig. 3C) revealed non-uniform staining of VE-cadherin where the gaps appeared whereas treatment with 10 μM As(III) (Fig. 3D) resulted in loss of VE-cadherin staining at the cell-cell junctions. Immunofluorescence staining of cleaved caspase-3 in untreated (Fig. 3E) and 10 μM As(III) treated cells (Fig. 3F) revealed no differences in apoptosis. Also, Gö 6976 inhibited gap formation at all concentrations of As(III) treatment and restored VE-cadherin staining at the cell-cell junctions. The images of 250 nM Gö 6976 only (Fig. 3G) and 10 μM As(III) + 250 nM Gö 6976 treatment (Fig. 3H) are shown. At the 12 and 24 h time points, concentration-dependent effects of As(III) treatment were similar to those seen at earlier time points (data not shown). However, the frequency and size of the gaps observed were maximal at the 6 h time point.
FIG. 3.
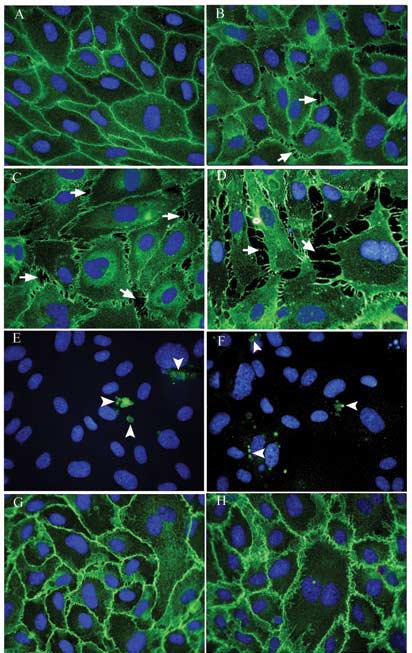
Effect on VE-cadherin localization and intercellular gap formation with As(III) exposure for 6 h. Confluent HAEC monolayers were either untreated (A) or treated with 1 μM As(III) (B), 5 μM As(III) (C) and 10 μM As(III) (D) for 6 h and the intercellular gaps (indicated with arrows) induced with As(III) treatment were visualized by immunofluorescence staining of VE-cadherin (green). The nuclei were stained with Hoechst (blue). Cleaved caspase-3 staining (green) for untreated (E) and 10 μM As(III) (F) treated cells is indicated by arrowheads. Pretreatment with 250 nM Gö 6976 for 1 h prior to and during 10 μM As(III) treatment (H) inhibited the As(III) induced gaps whereas 250 nM Gö 6976 only treatment (G) did not have any effect on the endothelial monolayer. Magnification=60X. The images are representative of 4 independent experiments performed.
Effect of As(III) treatment on VE-cadherin protein content
The immunofluorescence results in Fig. 1-3 showed that VE-cadherin distribution was altered by As(III) exposure. Therefore, the effects of 1, 5, and 10 μM As(III) treatments on the protein levels of VE-cadherin at the 24 h time point were determined. Western blotting analysis revealed a concentration-dependent effect of As(III) on VE-cadherin protein levels in the HAECs (Fig. 4A and B). Approximately 8, 15 and 30% reductions in VE-cadherin protein were observed with 1, 5 and 10 μM As(III) treatment, respectively, compared to the control (untreated) HAECs (Fig. 4B).
FIG. 4.
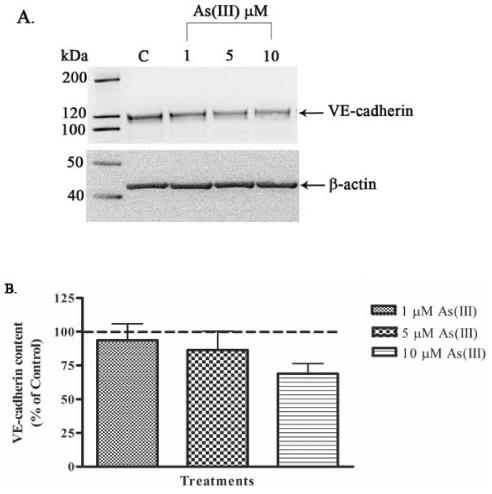
Effect of As(III) treatment on VE-cadherin protein levels. Confluent HAECs were treated with 1, 5 and 10 μM As(III) for 24 h and VE-cadherin protein content was analyzed by Western blotting technique. The VE-cadherin protein content is shown in the (A) upper panel and the lower panel represents the β-actin loading control. The images in (A) are representative of 7 experiments performed. The percent change in VE-cadherin protein relative to the control (untreated) is represented in (B). The control (100%) is indicated by the dotted line in (B). Error bars represent mean ± SE for n=7.
Effect of As(III) exposure on β-catenin localization
The cytoplasmic domain of VE-cadherin is linked with β-catenin, so it was of interest to determine whether As(III) affects β-catenin distribution. Exposure to As(III) for 6 h revealed the most damaging effects on the endothelial monolayer. Hence, the effect of As(III) treatment on β-catenin localization in HAECs was analyzed by immunofluorescence staining at the 6 h time point (Fig. 5). As previously observed with VE-cadherin localization, β-catenin staining showed a corresponding concentration-dependent effect with As(III) treatment. The frequency of gap formation with 10 μM As(III) (Fig. 5D) was greater than 5 μM As(III) (Fig. 5C) and only a few gaps were observed with 1 μM As(III) treatment (Fig. 5B). Non-uniform distribution or loss of β-catenin staining was evident wherever gaps could be observed. Also, 250 nM Gö 6976 treatment inhibited the gap formation induced by 10 μM As(III) (Fig. 5E) and restored the β-catenin staining at cell-cell junctions.
FIG. 5.
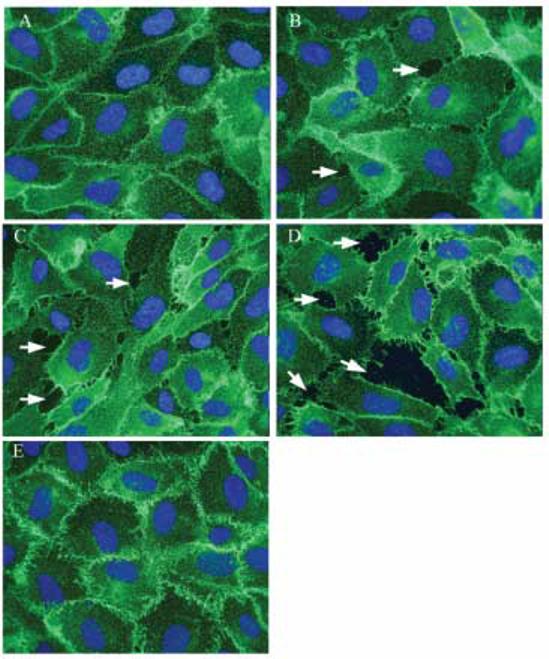
Effect of As(III) on β-catenin localization. Confluent HAECs were either untreated (A) or treated with 1 μM As(III) (B), 5 μM As(III) (C) and 10 μM As(III) (D) for 6 h and β-catenin localization was visualized by immunofluorescence staining (green). Non-uniform staining or loss of β-catenin was observed wherever gaps occurred (indicated by arrows). Pretreatment with 250 nM Gö 6976 for 1 h prior to and during 10 μM As(III) treatment (E) restored the β-catenin localization at cell-cell junctions. The nuclei were stained with Hoechst (blue). Magnification=60X. The images are representative of 3 independent experiments performed.
Effect of As(III) exposure on PY of VE-cadherin and β-catenin
The association of VE-cadherin with β-catenin is critical for the stability of AJ, and PY of either one or both the proteins can alter endothelial integrity. As(III)-induced disruption of the endothelial monolayer was evident starting at the 1 h time point. Therefore, the effect of As(III) exposure on PY of VE-cadherin and β-catenin was analyzed at 1 h by using immunoprecipitation followed by Western blotting. The phosphorylation of β-catenin was determined by probing the membrane with anti-phosphotyrosine monoclonal antibody following electrophoresis (Fig. 6A). The same membrane was stripped and total β-catenin protein content was analyzed (Fig. 6B). The PY levels were determined by normalizing with the total β-catenin protein content (Fig. 6C). A concentration-dependent effect of As(III) exposure on PY of β-catenin was observed. Treatment of cells with 1 μM As(III) showed no change in PY as compared to the control (untreated), whereas an increase in PY was observed with 5 μM As(III), and 10 μM As(III) treatment resulted in a significant increase in PY of β-catenin. The increase in PY induced by 10 μM As(III) was completely inhibited by 250 nM Gö 6976 (Fig. 6C).
FIG. 6.
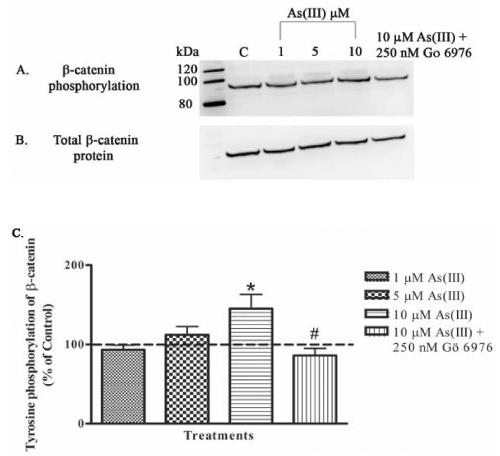
Effect of As(III) on PY of β-catenin. Confluent HAECs were treated with 1, 5, and 10 μM As(III) for 1 h or pretreated with 250 nM Gö 6976 for 1 h prior to and during 10 μM As(III) treatment. The β-catenin was immunoprecipitated, and Western blotting technique was used to analyze the phosphorylation of tyrosine residues (A). The membrane was stripped and reprobed to determine the total β-catenin protein content (B). The images A and B are representative of 4-6 independent experiments performed. The percent change in PY with the various treatments relative to the control (untreated) is represented in (C). The control (100%) is indicated by the dotted line. Error bars represent mean ± SE for n=4-6. * and # indicates significantly different from control and 10 μM As(III), respectively, with p < 0.01.
No PY of VE-cadherin was observed (data not shown). Even a basal level of PY could not be detected. Increasing the concentrations of phosphatase inhibitors in the lysis buffer to confirm that VE-cadherin was not dephosphorylated during harvesting or immunoprecipitation did not yield any positive results. Analysis of the total VE-cadherin protein content established the presence of VE-cadherin and confirmed the adequacy of immunoprecipitation and Western blotting (data not shown).
Effect of As(III) exposure on the formation of stress fibers
The cadherin/β-catenin complex is associated with the actin cytoskeleton, and this association is very important for maintaining junctional stability and structural integrity of the endothelial cell monolayer. As previously mentioned, reorganization of actin microfilaments into stress fibers can adversely affect the monolayer integrity. Hence, the effects of 1, 5 and 10 μM As(III) on reorganization of actin microfilaments were analyzed at 1, 6, 12 and 24 h by immunofluorescence staining using fluorophore-conjugated phalloidin. A concentration and time-dependent effect of As(III) treatment was observed. At the 1 h time point (Fig. 7), As(III) induced a concentration-dependent increase in stress fibers with the maximum stress fibers being observed with 10 μM As(III) treatment (Fig. 7D). Treatment with Gö 6976 abolished the stress fiber formation at all concentrations of As(III) treatment. The images of 250 nM Gö 6976 only treatment (Fig. 7E) and 10 μM As(III) + 250 nM Gö 6976 treatment (Fig. 7F) are shown.
FIG. 7.
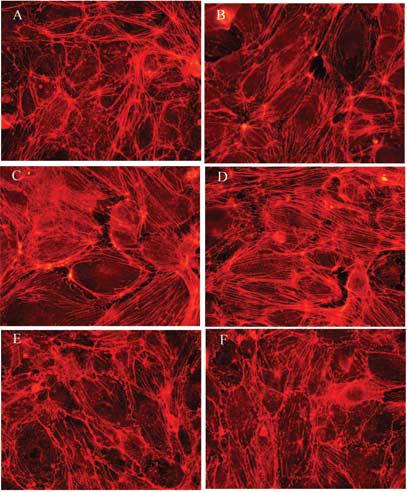
Effect of 1 h As(III) exposure on the formation of stress fibers. Confluent HAEC monolayers were either untreated (A) or treated with As(III); 1 μM (B), 5 μM (C) and 10 μM (D) for 1 h, and the stress fibers induced with As(III) treatment were visualized by immunofluorescence staining with Alexa Fluor 568 conjugated phalloidin (red). Pretreatment with 250 nM Gö 6976 for 1 h prior to and during 10 μM As(III) treatment (F) abolished the As(III) induced stress fibers whereas 250 nM Gö 6976 only treatment (E) did not have an effect on stress fiber formation. Magnification=60X. The images are representative of 4 independent experiments performed.
At the 6 h time point (Fig. 8), the frequency and density of stress fibers induced by As(III) was much higher than that observed at the 1 h time point. Also, a concentration-dependent increase in stress fibers was observed with the maximum stress fibers being induced with 10 μM As(III) treatment (Fig. 8D). Treatment with Gö 6976 reduced the stress fibers at all concentrations of As(III) treatment. The images of 250 nM Gö 6976 only treatment (Fig. 8E) and 10 μM As(III) + 250 nM Gö 6976 treatment (Fig. 8F) are shown. At the 12 and 24 h time points, similar concentration-dependent effects of As(III) treatment were observed (data not shown). Stress fiber formation, like the intercellular gap formation, was maximal at the 6 h time point.
FIG. 8.
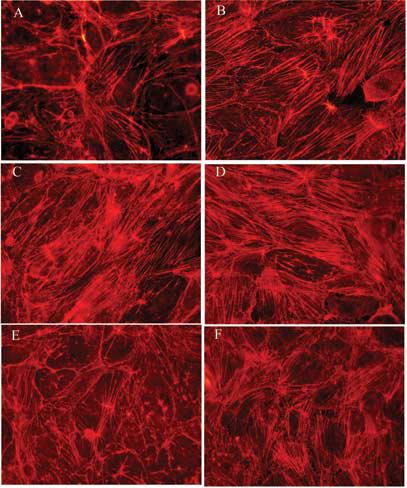
Effect of 6 h As(III) exposure on the formation of stress fibers. Confluent HAEC monolayers were either untreated (A) or treated with As(III); 1 μM (B), 5 μM (C) and 10 μM (D) for 6 h and the stress fibers induced with As(III) treatment were visualized by immunofluorescence staining with Alexa Fluor 568 conjugated phalloidin (red). Pretreatment with 250 nM Gö 6976 for 1 h prior to and during 10 μM As(III) treatment (F) reduced the As(III) induced stress fibers whereas 250 nM Gö 6976 only treatment (E) did not have an effect on stress fiber formation. Magnification=60X. The images are representative of 4 independent experiments performed.
Effect of As(III) on the activation of cPKCs
The translocation of PKC from cytosol to the membrane is an important step in its activation process (Newton, 2001). Therefore, the effect of As(III) on the activation of cPKCs was analyzed by separation of the cytosolic and membrane fractions and determining the protein content in each fraction by Western blotting technique. Preliminary experiments showed that 10 μM As(III) treatment induced translocation of PKCα from the cytosolic fraction to the membrane fraction at 1 h, but not at earlier time points (data not shown). Also, As(III) induced damage to the endothelial monolayer was observed starting at 1 h. Hence, cells were exposed to 1, 5 and 10 μM As(III) and cPKC activation was analyzed at the 1 h time point (Fig. 9). Treatment with 5 and 10 μM As(III) induced a significant increase in PKCα activation as determined by quantifying the protein content in the membrane fraction (Fig. 9A and B). Conversely, a decrease was observed with 1 μM As(III) treatment. In the cytosolic fraction (Fig. 9C and D), a decreasing trend in the PKCα content was observed with increasing As(III) concentration indicative of PKCα translocation from the cytosol to the membrane. An exact proportionate change in the cytosolic fraction was not evident compared to the membrane fraction, but overall, protein levels were much greater in the cytosolic fraction than the membrane fraction. Phorbol esters are very potent activators of PKCs (Newton, 2001) and hence phorbol 12-myristate 13-acetate (PMA) was used as a positive control. Treatment with 1 μM PMA for 1 h induced a robust activation of PKCα (data not shown).
FIG. 9.
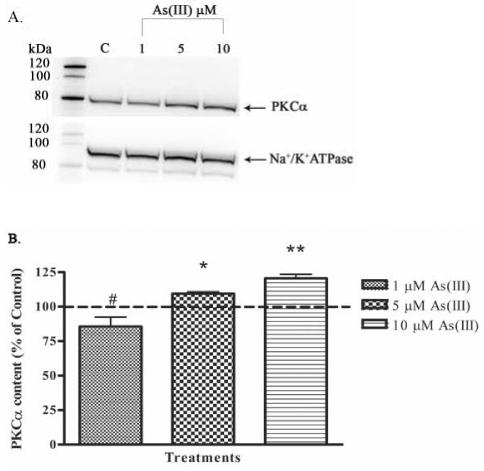
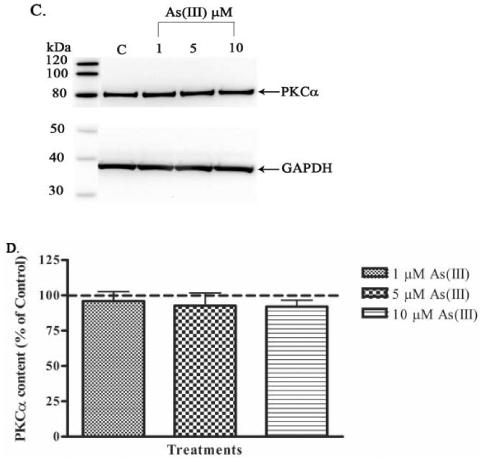
Effect of As(III) on the activation of PKCα. Confluent HAECs were treated with 1, 5, and 10 μM As(III) for 1 h. The lysates were subjected to ultracentrifugation to separate the membrane and cytosolic fractions and the PKCα levels in both fractions were determined by Western blotting. The PKCα content in the membrane fraction is shown in (A) upper panel and the lower panel represents the Na+/K+ ATPase loading control for the membrane fraction. The percent change in PKCα content in the membrane fraction relative to the control (untreated) is represented in (B). The PKCα content in the cytosolic fraction is shown in (C) upper panel and the lower panel represents the GAPDH loading control for the cytosolic fraction. The percent change in PKCα content in the cytosolic fraction relative to the control is represented in (D). The images in (A) and (C) are representative of 3-6 independent experiments performed. The control (100%) is indicated by the dotted line in (B) and (D). Error bars represent mean ± SE for n=3-6. #, * and ** indicates significantly different from control with p < 0.01, 0.05 and 0.001, respectively.
Effect of As(III) on endothelial permeability
Exposure to As(III) caused detrimental effects on the endothelial monolayer and induced reorganization of actin microfilaments into stress fibers. Therefore, we hypothesized that As(III) would also increase permeability. The effect of 1, 5 and 10 μM As(III) treatment on endothelial permeability was analyzed at the 6 h time point (Fig. 10). Confluent monolayers of HAECs grown on Transwell inserts were treated with the various As(III) concentrations and the permeability of the monolayer for FITC-dextran was determined by measuring the fluorescence intensity of the medium in the plate well. A 5% increase in permeability was observed with 1 and 5 μM As(III) treatment, whereas 10 μM As(III) induced approximately a 19% increase. Treatment with 250 nM Gö 6976 inhibited the increase in permeability induced by 10 μM As(III).
FIG. 10.
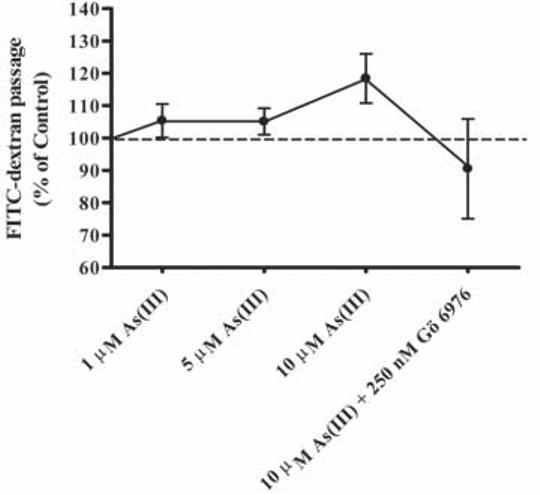
Effect of As(III) on endothelial permeability. Confluent HAECs were treated with 1, 5, and 10 μM As(III) for 6 h or pretreated with 250 nM Gö 6976 for 1 h prior to and during 10 μM As(III) treatment. Monolayer permeability was measured using FITC-dextran. The control (100%) is indicated by the dotted line. Error bars represent mean ± SE for n=4-5.
Discussion
The results from this study show that PKCα is a key mediator in a proposed mechanism for As(III)-induced atherosclerosis. The data showed that PKCα plays a crucial role in localization of the AJ proteins, VE-cadherin and β-catenin, and in regulating endothelial gap formation, stress fiber formation and vascular permeability. As(III) treatment induced intercellular gaps thereby disrupting the endothelial monolayer as evidenced by loss or non-uniform staining of VE-cadherin and β-catenin where the gaps appeared. Reduced VE-cadherin protein content was also detected; a finding that may in part be responsible for the observed intercellular gaps. An increased plasma concentration of VE-cadherin has been associated with coronary atherosclerosis (Soeki et al., 2004), and in vitro studies have shown that hydrogen peroxide induces internalization of VE-cadherin and gap formation (Kevil et al., 1998). Hence, the decreased VE-cadherin may be due to internalization and degradation or release into the cell culture medium. β-catenin was found to be phosphorylated with 5 and 10 μM As(III) treatments, but no PY of VE-cadherin was detected. There are several non-receptor tyrosine kinases such as Fyn, Fer, cMet, Abl and Src that have been shown to associate with and modulate the phosphorylation state of β-catenin and its association with cadherin (Lilien and Balsamo, 2005) during various developmental and physiological processes. In particular, phosphorylation of β-catenin at tyrosine 654 by Src has been shown to cause a fivefold reduction in its affinity for E-cadherin (Roura et al., 1999), which belongs to the same class of type I classical cadherins as VE-cadherin (Ivanov et al., 2001). The phosphorylation and dephosphorylation state of AJ proteins plays a critical role in the formation and/or maintenance of stable cell-cell adhesions (Lilien and Balsamo, 2005, Schnittler, 1998). Therefore, PY of β-catenin could potentially reduce its affinity for association with VE-cadherin and cause dissociation or loosening of the AJ.
Arsenic has been shown to increase calcium-dependent PKC activity in Chinese hamster ovary cells at high doses after 4 h treatment (Liu and Huang, 1997). Similarly, transient activation of PKCα was observed with 20 μM arsenic treatment in a mouse epidermal cell line (Chen et al., 2000). Our results showed that 5 and 10 μM As(III) treatment induced significant activation of PKCα at the 1 h time point. The downstream effects of As(III)-induced PKCα activation were confirmed by using the cPKC inhibitor, Gö 6976. Inhibition of PKCα prevented gap formation and restored VE-cadherin and β-catenin at the cell-cell junctions. PY of β-catenin induced by As(III) was also reduced to basal levels. The effects of activation of PKCα on endothelial integrity have been well documented (reviewed in Yuan, 2003). Activation of PKCα by thrombin has been shown to modulate endothelial integrity by induction of stress fibers and increased endothelial permeability (Sandoval et al., 2001). Also, PY of β-catenin has been observed that is PKC dependent (Cohen et al., 1999).
Formation of stress fibers induced by As(III) was also abolished or reduced with Gö 6976 treatment. Studies have demonstrated the relation between PKCα activation, stress fiber formation and cell contraction. It has been found that PKCα induces the activation of myosin by serine/threonine phosphorylation of myosin light chain which results in actin-myosin interaction and subsequent stress fiber formation and cell contraction (Tiruppathi et al., 2003). Therefore, PKC inhibition plausibly inhibited the contractile force exerted by the stress fibers. Confluent endothelial cells consist of a band of actin filaments concentrated along the cell borders known as a dense peripheral band (DPB) that is also present in vivo (Gabbiani et al., 1983, Schnittler, 1998). The cadherin-catenin complex associates with the actin filaments of DPB, which is important for junctional stability. Our results showed that actin filaments were present in untreated cells along the cell borders and were mostly absent in central regions of the cell. Treatment with As(III) caused the loss of DPB and an increase in stress fibers traversing the cells. The effect of As(III) was observed to be concentration and time-dependent with the maximum stress fibers being formed with 10 μM As(III) at the 6 h time point. Actin reorganization associated with the disruption of endothelial barrier is characterized by increased stress fiber density and reduction or loss of DPB (Lum and Roebuck, 2001). Stress fibers have the molecular machinery to generate contractile force and have been associated with gap formation and increased endothelial permeability (Pellegrino et al., 2004, Yuan, 2003).
Vascular permeability changes are associated with loss of endothelial integrity. Chen et al., (2004) and Tsai et al., (2005) showed that As(III) alters endothelial permeability in rodents. Using FITC-dextran, we found a 5% increase in endothelial permeability with 1 and 5 μM As(III) and approximately a 19% increase with 10 μM As(III) treatment that was inhibited by Gö 6976 treatment. These changes were not as great as expected based on the gap formation observed and actin stress fiber formation. However, Chen et al., (2004) and Tsai et al., (2005) have clearly demonstrated that even a small increase in permeability could be pathologically important. Taken together, these results suggest that As(III) induces PY of β-catenin, which modulates AJ assembly resulting in non-uniform distribution or loss of VE-cadherin as well as β-catenin at the cell-cell junctions and promotes the formation of stress fibers which culminates in intercellular gap formation and increased permeability.
While the results presented here show PKCα dependency for the effects of As(III) on endotheial cell-cell adhesion, monolayer integrity and vascular permeability, they do not suggest that As(III) effects on endothelium are restricted to the PKCα pathway. Other signal transduction pathways intersect with PKCα signaling. The PKC enzymes belong to the family of serine/threonine kinases, and it has been suggested that crosstalk exists between the tyrosine and serine/threonine kinase signaling cascades (Bauman and Scott, 2002, Chang et al., 2001). Involvement of tyrosine kinases introduces the possibility that vascular endothelial growth factor (VEGF), a key modulator of AJ stability that signals through a tyrosine kinase pathway, may be involved. VEGF has been implicated in arsenic-related toxicity through activation of PKCδ (Soucy et al., 2004). In addition, the VEGF receptor tyrosine kinases have been shown to signal with PKCα activation and downstream AKT signaling (Gliki et al., 2002). Moreover, VEGF signals through a Ras-MEK-ERK pathway independent of PKCα suggesting that not all As(III)-related effects may be PKCα dependent. Stress kinases such as p38 and c-Jun amino-terminal kinase (JNK) could also potentially be involved in the observed effects of As(III). Barchowsky et al., (1999) showed that high levels of As(III) can induce cellular stress and activate stress kinases including p38. On the other hand, lower concentrations have been shown to induce DNA synthesis and reactive oxygen species (Barchowsky et al., 1996) without activating stress kinases (Barchowsky et al., 1999). The role of these alternative pathways in endothelial arsenic toxicity is the subject for future investigations.
In summary, we have determined a potential mechanism for arsenic-induced atherosclerosis. This study demonstrates that As(III)-induced activation of PKCα causes PY of β-catenin and formation of stress fibers. PY of β-catenin may cause weakening of the AJ, and this effect, in association with the contractile force generated by stress fibers, results in gap formation and increased endothelial permeability. Proteins at the AJ are largely responsible for maintaining the barrier and normally allow passage of selected molecules. Injury to the endothelium can promote accumulation of oxidized low density lipoproteins (oxLDLs) and monocytes/macrophages in the intima of blood vessels (Lusis, 2000), ultimately leading to formation of foam cells. The intercellular gaps and increased permeability caused by arsenic could potentially accelerate/enhance influx of oxLDLs and monocytes/macrophages across the endothelium and contribute significantly to the progression of atherosclerosis.
Acknowledgments
This study was supported by a grant from the National Institutes of Health (P20 RR017670). We thank Lou Herritt and Dr. Celine Beamer for assistance with fluorescence microscopy.
Footnotes
Conflict of Interest Statement
There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler. Thromb. Vasc. Biol. 1999;19:2286–2297. doi: 10.1161/01.atv.19.10.2286. [DOI] [PubMed] [Google Scholar]
- Barchowsky A, Dudek EJ, Treadwell MD, Wetterhahn KE. Arsenic induces oxidant stress and NF-κB activation in cultured aortic endothelial cells. Free Radic. Biol. Med. 1996;21(6):783–790. doi: 10.1016/0891-5849(96)00174-8. [DOI] [PubMed] [Google Scholar]
- Barchowsky A, Roussel RR, Klei LR, James PE, Ganju N, Smith KR, Dudek EJ. Low levels of arsenic trioxide stimulate proliferative signals in primary vascular cells without activating stress effector pathways. Tox. Appl. Pharmacol. 1999;159:65–75. doi: 10.1006/taap.1999.8723. [DOI] [PubMed] [Google Scholar]
- Bauman AL, Scott JD. Kinase- and phosphatase-anchoring proteins: harnessing the dynamic duo. Nat. Cell Biol. 2002;4:E203–E206. doi: 10.1038/ncb0802-e203. [DOI] [PubMed] [Google Scholar]
- Bunderson M, Brooks DM, Walker DL, Rosenfeld ME, Coffin DJ, Beall HD. Arsenic exposure exacerbates atherosclerotic plaque formation and increases nitrotyrosine and leukotriene biosynthesis. Toxicol. Appl. Pharmacol. 2004;201:32–39. doi: 10.1016/j.taap.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Bunderson M, Coffin JD, Beall HD. Arsenic induces peroxynitrite generation and cyclooxygenase-2 protein expression in aortic endothelial cells: possible role in atherosclerosis. Toxicol. Appl. Pharmacol. 2002;184:11–18. [PubMed] [Google Scholar]
- Byers HR, White GE, Fujiwara K. Organization and function of stress fibers in cells in vitro and in situ. Cell Muscle Motil. 1984;5:83–137. doi: 10.1007/978-1-4684-4592-3_2. [DOI] [PubMed] [Google Scholar]
- Chang BY, Chiang M, Cartwright CA. The interaction of Src and RACK1 is enhanced by activation of protein kinase C and tyrosine phosphorylation of RACK1. J. Biol. Chem. 2001;276(23):20346–20356. doi: 10.1074/jbc.M101375200. [DOI] [PubMed] [Google Scholar]
- Chen N, Ma W, Huang C, Ding M, Dong Z. Activation of PKC is required for arsenite-induced signal transduction. J. Environ. Pathol. Toxicol. Oncol. 2000;19(3):297–305. [PubMed] [Google Scholar]
- Chen S-C, Tsai M-H, Wang H-J, Yu H-S, Chang LW. Vascular permeability alterations induced by arsenic. Hum Exp Toxicol. 2004;23:1–7. doi: 10.1191/0960327104ht407oa. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Carbajal JM, Schaeffer RC., JR VEGF stimulates tyrosine phosphorylation of β-catenin and small-pore endothelial barrier dysfunction. Am. J. Physiol. 1999;277:H2038–H2049. doi: 10.1152/ajpheart.1999.277.5.H2038. [DOI] [PubMed] [Google Scholar]
- Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J.Cell Sci. 1998;111:1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- Gabbiani G, Gabbiani F, Lombardi D, Schwartz SM. Organization of actin cytoskeleton in normal and regenerating arterial endothelial cells. Proc Natl. Acad. Sci. USA. 1983;80:2361–2364. doi: 10.1073/pnas.80.8.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliki G, Wheeler-Jones C, Zachary I. Vascular endothelial growth factor induces protein kinase C (PKC)-dependent Akt/PKB activation and phosphatidylinositol 3'-kinase-mediated PKCδ phosphorylation: Role of PKC in angiogenesis. Cell Biol. Int. 2002;26(9):751–759. doi: 10.1016/s1065-6995(02)90926-1. [DOI] [PubMed] [Google Scholar]
- Hordijk PL, Anthony E, Mul FPJ, Rientsma R, Oomen LCJM, Roos D. Vascular-endothelial-cadherin modulates endothelial monolayer permeability. J.Cell Sci. 1999;112:1915–1923. doi: 10.1242/jcs.112.12.1915. [DOI] [PubMed] [Google Scholar]
- Ivanov DB, Philippova MP, Tkachuk VA. Structure and functions of classical cadherins. Biochemistry. 2001;66(10):1174–1186. doi: 10.1023/a:1012445316415. [DOI] [PubMed] [Google Scholar]
- Kevil CG, Ohno N, Gute DC, Okayama N, Robinson SA, Chaney E, Alexander JS. Role of cadherin internalization in hydrogen peroxide-mediated endothelial permeability. Free Radic. Biol. Med. 1998;24(6):1015–1022. doi: 10.1016/s0891-5849(97)00433-4. [DOI] [PubMed] [Google Scholar]
- Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of β-catenin. Curr. Opin. Cell Biol. 2005;17:459–465. doi: 10.1016/j.ceb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Liu Y, Huang H. Involvement of calcium-dependent protein kinase C in arsenite-induced genotoxicity in Chinese hamster ovary cells. J. Cell. Biochem. 1997;64:423–433. [PubMed] [Google Scholar]
- Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am. J. Physiol. Cell Physiol. 2001;280:C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marmé D, Schächtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J. Biol. Chem. 1993;268(13):9194–9197. [PubMed] [Google Scholar]
- Mattila P, Majuri M-L, Tiisala S, Renkonen R. Expression of six protein kinase C isotypes in endothelial cells. Life Sci. 1994;55(16):1253–1260. doi: 10.1016/0024-3205(94)90063-9. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein Kinase C: Structural and Spatial Regulation by Phosphorylation, Cofactors, and Macromolecular interactions. Chem. Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- Pellegrino M, Kazmierczak EF, LeBlanc JC, Cho T, Cao K, Marcovina SM, Boffa MB, Cote GP, Koschinsky ML. The apolipoprotein(a) component of lipoprotein(a) stimulates actin stress fiber formation and loss of cell-cell contact in cultured endothelial cells. J. Biol. Chem. 2004;279(8):6526–6533. doi: 10.1074/jbc.M309705200. [DOI] [PubMed] [Google Scholar]
- Rahman M, Tondel M, Ahmad SA, Chowdhury IA, Faruquee MH, Axelson O. Hypertension and arsenic exposure in Bangladesh. Hypertension. 1999;33(1):74–78. doi: 10.1161/01.hyp.33.1.74. [DOI] [PubMed] [Google Scholar]
- Roura S, Miravet S, Piedra J, Herreros AG, Dunach M. Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J. Biol. Chem. 1999;274(51):36734–36740. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- Sandoval R, Malik AB, Minshall RD, Kouklis P, Ellis CA, Tiruppathi C. Ca2+ signaling and PKCα activate increased endothelial permeability by disassembly of VE-cadherin junctions. J. Physiol. 2001;533(2):433–445. doi: 10.1111/j.1469-7793.2001.0433a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittler HJ. Structural and functional aspects of intercellular junctions in vascular endothelium. Basic Res. Cardiol. 1998;93(3):30–39. doi: 10.1007/s003950050205. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Hulderman T, Harki D, Luster MI. Arsenic exposure accelerates atherogenesis in apolipoprotein E−/− mice. Environ. Health Perspect. 2003;111:1744–1748. doi: 10.1289/ehp.6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonova PP, Luster MI. Arsenic and atherosclerosis. Toxicol. Appl. Pharmacol. 2004;198:444–449. doi: 10.1016/j.taap.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Soeki T, Tamura Y, Shinohara H, Sakabe K, Onose Y, Fukuda N. Elevated concentration of soluble vascular endothelial cadherin is associated with coronary atherosclerosis. Circ J. 2004;68:1–5. doi: 10.1253/circj.68.1. [DOI] [PubMed] [Google Scholar]
- Soucy NV, Klei LR, Mayka DD, Barchowsky A. Signaling pathways for arsenic-stimulated vascular endothelial growth factor-A expression in primary vascular smooth muscle cells. Chem. Res. Toxicol. 2004;17:555–563. doi: 10.1021/tx034193q. [DOI] [PubMed] [Google Scholar]
- Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol. Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- Tiruppathi A, Minshall RD, Paria BC, Vogel SM, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul Pharmacol. 2003;39:173–185. doi: 10.1016/s1537-1891(03)00007-7. [DOI] [PubMed] [Google Scholar]
- Tsai M-H, Chen S-C, Wang H-J, Yu H-S, Chang LW. A mouse model for study of vascular permeability changes induced by arsenic. Toxicol. Mech. Methods. 2005;15:433–437. doi: 10.1080/15376520500195640. [DOI] [PubMed] [Google Scholar]
- Tseng CH, Chong CK, Tseng CP, Hsueh YM, Chiou HY, Tseng CC, Chen CJ. Long-term arsenic exposure and ischemic heart disease in arseniasis-hyperendemic villages in Taiwan. Toxicol. Lett. 2003;137(12):15–21. doi: 10.1016/s0378-4274(02)00377-6. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, van Hinsbergh VMN. Cytoskeletal effects of Rho-like small guanine nucleotide–binding proteins in the vascular system. Arterioscler. Thromb. Vasc. Biol. 2001;21:300–311. doi: 10.1161/01.atv.21.3.300. [DOI] [PubMed] [Google Scholar]
- Wang CH, Jeng JS, Yip PK, Chen CL, Hsu LI, Hsueh YM, Chiou HY, Wu MM, Chen CJ. Biological gradient between long-term arsenic exposure and carotid atherosclerosis. Circulation. 2002;105:1804–1809. doi: 10.1161/01.cir.0000015862.64816.b2. [DOI] [PubMed] [Google Scholar]
- White GE, Gimbrone MA, Jr., Fujiwara K. Factors influencing the expression of stress fibers in vascular endothelial cells in situ. J. Cell Biol. 1983;97:416–424. doi: 10.1083/jcb.97.2.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AJ, Pollard JD, Herman IM. Actin filament stress fibers in vascular endothelial cells in vivo. Science. 1983;219:867–869. doi: 10.1126/science.6681677. [DOI] [PubMed] [Google Scholar]
- Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul. Pharmacol. 2003;39:213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]