

Abstract
Aims
The F1S and A genetic variants of α1-acid glycoprotein (AAG) change under various physiological and pathological conditions. They also vary in their drug binding abilities. We have studied the stereoselective binding ability of each of the AAG variants using enantiomers of disopyramide (DP) and warfarin (WR).
Methods
The AAG variants were separated by hydroxyapatite chromatography. Binding of drug enantiomers to the AAG variants was studied by the Hummel–Dreyer method. The characteristics of the binding activities were examined by Scatchard plot analysis. The first five amino-terminal amino acids (residues 112–116) of the cyanogen bromide (CNBr) fragment (residues 112–181) of each of the separated AAG fractions were elucidated by Edman degradation.
Results
Commercial AAG was separated into two main fractions. Residues 112–116 of fraction 2 were identical to the amino acid sequences predicted from the AAG A gene, LAFDV, and encode the F1S variant. In fraction 3, the deduced amino acid sequence of the AAG B gene, FGSYL, was established, and encodes the A variant. The binding affinities of both DP enantiomers in fraction 3 were significantly higher than those in fraction 2. The differences between dissociation constants (Kd) in fractions 2 and 3 were 5.2-fold for (S)-DP (P < 0.05) and 3.7-fold for (R)-DP (P < 0.001). The dissociation constant of (S)-DP (0.39 ± 0.08 µm) was lower than that of (R)-DP (0.53 ± 0.10 µm) in fraction 3 [95% confidence interval (CI) −0.282, −0.010; P < 0.05], although the binding activities of the DP enantiomers were almost the same in fraction 2. By contrast WR enantiomers had a higher binding affinity in fraction 2 than in fraction 3, the differences in dissociation constants between fractions 2 and 3 being 12.6-fold for (S)-WR (P < 0.001) and 8.3-fold for (R)-WR (P < 0.001). The dissociation constant of (S)-WR (0.28 ± 0.10 µm) was significantly lower than that of (R)-WR (0.48 ± 0.08 µm) in fraction 2 (95% CI −0.369, −0.028; P < 0.05), but there were no significant differences between the binding activities of WR enantiomers in fraction 3.
Conclusions
DP and WR enantiomers bind preferentially to fraction 3 and fraction 2, respectively. Fractions 2 and 3 are encoded by the AAG A and the AAG B genes, respectively.
Keywords: α1-acid glycoprotein, disopyramide, stereoselective binding, variant, warfarin
Introduction
α1-Acid glycoprotein (AAG) is an acute-phase reactant that increases under various physiological and pathological conditions. AAG is a major binding protein for basic drugs and binds a variety of other ligands, including acidic drugs such as warfarin (WR) [1].
There are AAG genetic variants that differ in several amino acid substitutions. The polymorphism is generated by the presence of two different genes, the AAG A and B (B/B′) genes. The amino acid sequences deduced from the AAG A gene differ in 22 positions from those deduced from the AAG B gene [2]. These variants can be observed when the desialylated form of AAG is analysed by isoelectrofocusing. The three main AAG variants are designated F1, S and A, depending on their electrophoretic migration [3]. F1 and S variants are encoded by the AAG A gene, and the A variant is encoded by the AAG B gene [4]. Several attempts have been made to elucidate the structure–function relationships, such as drug binding differences, between the genetic variants, i.e. the A variant and a mixture of the F1 and S variants (F1S variant) [5–8]. There are few reports, however, of the effect of the AAG variants on the binding activities of drug enantiomers. Many drugs have chiral centres and are available commercially as racemic mixtures. Such drugs may show stereoselective pharmacokinetics and pharmacodynamics [9, 10]. Many of these stereospecific characteristics may be due to stereoselective protein binding in plasma and tissues. Although the binding activities of many racemic drugs to AAG are known to be stereoselective, the mechanism of chiral recognition by AAG has not been elucidated.
In the present study, we have investigated the stereoselective binding abilities of the enantiomers of disopyramide (DP) and WR, which bind selectively to the A and F1S variants, respectively [6, 7], to AAG fractions separated by modified hydroxyapatite chromatography. Ikenaka et al. reported that AAG was separated into four fragments (I–IV) by fragmentation of AAG with cyanogen bromide (CNBr) [13]. The newly formed first six amino-terminal amino acids (residues 112–117) of the CNBr fragment (residues 112–181) are the alternative amino acids [2]. In order to determine the relationship between the fractions separated in this study and the A and F1S variants, we also examined the first five amino-terminal amino acids of CNBr fragment II of each of the separated fractions. The aim of this study therefore was to elucidate the stereoselective binding ability of each of the separated AAG fractions.
Methods
Materials
Human AAG (Cohn fraction VI, lot 118H7613) and racemic-WR were purchased from Sigma (St Louis, MO, USA). The aqueous insoluble WR was converted to the soluble sodium salt by reaction with 50% aqueous solution of 1 mol l−1 sodium hydroxide solution (Wako Pure Chemicals, Osaka, Japan). DP enantiomers were donated by Nippon Roussel K.K., Japan. All other reagents were of the highest grade available commercially.
Resolution of WR enantiomers
Each WR enantiomer was prepared by the resolution of rac-WR on a Chiralcel AD column with a chiral stationary phase. High-performance liquid chromatography (HPLC) was performed using the following conditions for chromatography: apparatus, Hitachi L-7100 (Hitachi, Tokyo, Japan); column, Chiralcel AD, 250 × 4.6 mm internal diameter (i.d.) (Daicel Chemical Industries, Ltd, Tokyo, Japan); mobile phase, n-hexane/2-propanol, 8 : 2 (v/v) containing 0.5% acetic acid; flow rate, 0.5 ml min−1; detection, UV 254 nm; temperature, 40 °C. Their stereochemical purities were ascertained under the same conditions, and the stereochemical purities of (R)-WR and (S)-WR were found to be 97.67% and 97.03%, respectively. Each WR enantiomer was also converted to the sodium salt under the same conditions as those used for rac-WR.
Separation of the AAG fractions
AAG fractions were separated by a modification of a method developed in this laboratory [11, 12]. AAG (approximately 15–20 mg) was dissolved in 0.5 ml of distilled water, and the whole volume was applied into an HPLC system (Hitachi L-7100; Hitachi) equipped with a detector (Hitachi L-7420) set at 280 nm. The separation of the AAG fractions was achieved using a 500 × 30 mm i.d. column packed with hydroxyapatite (Wako Pure Chemicals, Osaka, Japan) with a gradient elution program at a flow rate of 9.5 ml min−1 and at room temperature. A typical chromatogram is shown in Figure 1. Four fractions could be obtained by the gradient elution program. The relative proportions of separated fractions 1–4 were 2.44 ± 0.96%, 72.78 ± 4.36%, 23.03 ± 3.65%, 0.52 ± 0.13% (means ± SD, n = 10), respectively. The sum of the relative proportions of fractions 2 and 3 in the four fractions was larger than 95%. Therefore, only fractions 2 and 3 were analysed in the following study.
Figure 1.
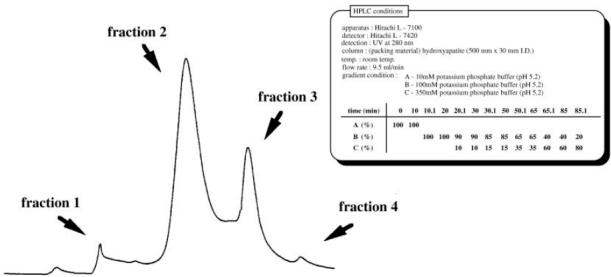
Typical chromatogram of the fractionation of commercial AAG by high-performance liquid chromatography with a hydroxyapatite column (500 × 30 mm i.d.).
Determination of bound drug concentration by the Hummel–Dreyer method
The binding of DP and WR enantiomers to AAG was examined by the Hummel–Dreyer method modified by Pinkerton and Koeplinger [14]. All solutions were prepared in 10 mm citrate phosphate buffer pH 7.0. The measurements were performed on a 50 × 4.0 mm i.d. column and a 100 × 4.0 mm i.d. column packed with 5 µm LiChrosorb DIOL (Merck, Darmstadt, Germany) with a gradient program for DP and WR, respectively. A flow rate of 2.0 ml min−1 was used throughout the binding experiments, which were carried out at 37 °C. Detection was at 254 nm using a UV detector (Hitachi L-7420). The coefficients of variation for replicate assays of (S) (R)-DP (2.5 mg ml−1) and (S) (R)-WR (2.0 mg ml−1) were 18.0% (n = 8), 17.6% (n = 9), 5.8% (n = 6) and 3.8% (n = 4), respectively. The limits of determination of DP and WR enantiomers were 0.1 and 0.05 µg ml−1, respectively.
Analysis of partial amino acid sequences of AAG fractions
Preparation of the CNBr fragments was carried out according to the method of Ikenaka et al.[13]. AAG (approximately 2.0 mg) was dissolved in 20 µl of 70% formic acid. CNBr (Wako Pure Chemicals) (2.5 or 10 mg) was added, and the resulting solution was shaken gently at room temperature for 20 h, diluted with 200 µl of distilled water, and evaporated to dryness in vacuum at 60 °C. The residue was reconstituted with distilled water and subjected to 22% SDS–PAGE. After the electrophoresis, the gels were blotted onto a PVDF membrane (Daiichi Pure Chemicals, Tokyo, Japan) and detected by staining with Coomassie Brilliant Blue R-250 (Figure 2). Peptide bands of approximately 7 kDa of each of the fractions, probably corresponding to CNBr fragment II, were subjected to Edman degradation procedures, and the first five amino-terminal amino acids were elucidated.
Figure 2.
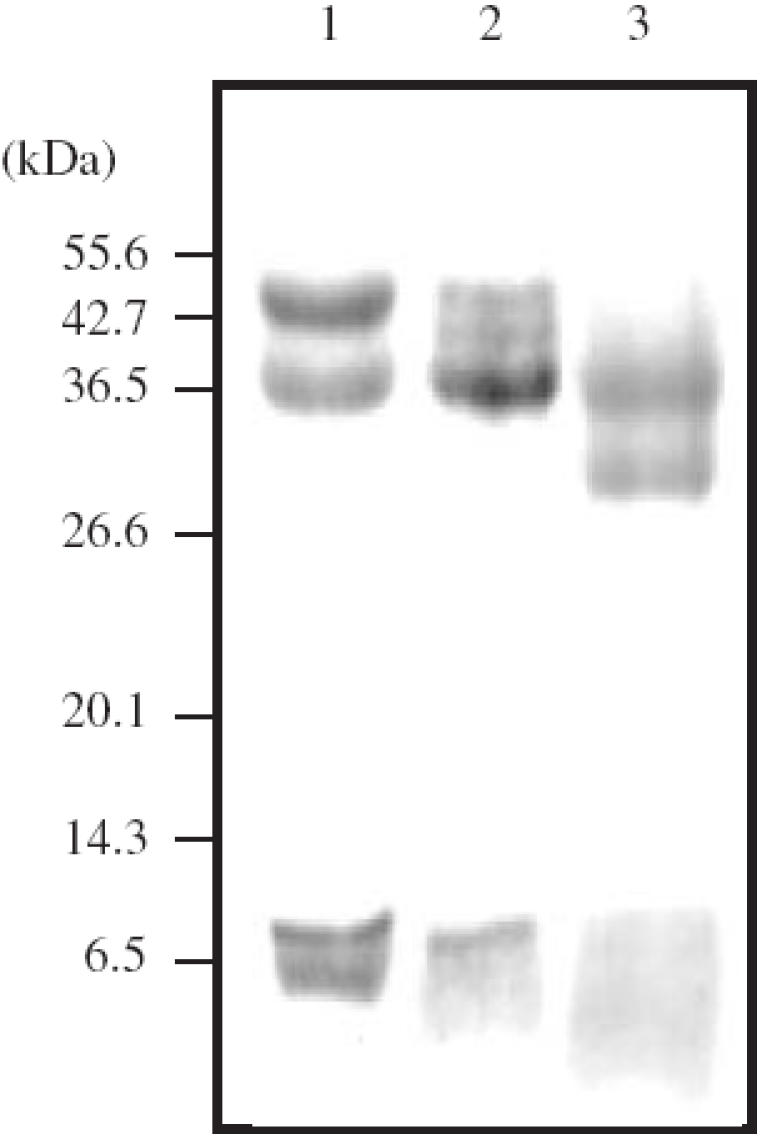
SDS–PAGE (22% acrylamide gel) analysis of the fragmentation of each of the AAG fractions with cyanogen bromide (CNBr). Lane 1, Fraction 2 (2.5 mg of CNBr); lane 2, fraction 3 (2.5 mg of CNBr); lane 3, fraction 3 (10 mg of CNBr).
Data analysis
The characteristics of the binding of DP and WR enantiomers to the separate fractions and to total (unfractionated) AAG were examined by Scatchard plot analysis at drug concentrations ranging from 2.5 to 10 µg ml−1, eight different concentrations, for DP and 2.0–10 µg ml−1, nine different concentrations, for WR and at protein concentrations from 0.1 to 0.7 mg ml−1. Binding parameters were then obtained by nonlinear regression analysis using ‘MULTI’[15] and the following equation: Cb = N × Pt × Cu/(Kd + Cu) + NSB × Cu, where N and Kd are the number of binding sites and the dissociation constant, respectively, Cb and Cu are the concentrations of bound and unbound drugs, respectively, Pt is the protein concentration (the molecular mass being taken as 40 kDa), and NSB is the nonspecific binding component. Statistical significance (P < 0.05) was determined using Student's t-test.
Results
The binding parameters for the DP and WR enantiomers to the separate fractions and to total (unfractionated) AAG are shown in Tables 1 and 2, respectively. The binding of DP and WR enantiomers to fractions 2 and 3 showed large differences. The dissociation constants (Kd) of both DP enantiomers in fraction 3 were significantly smaller than those in fraction 2, although no significant differences were observed in the number of binding sites (N). The differences between dissociation constants in fractions 2 and 3 were 5.2-fold for (S)-DP [95% confidence interval (CI) 0.141, 3.157; P < 0.05] and 3.7-fold for (R)-DP (95% CI 0.723, 2.149; P < 0.001) (Table 1). There were also differences between the stereoselective binding abilities of DP enantiomers to the separate fractions. In fraction 3 (S)-DP had a smaller dissociation constant than (R)-DP (95% CI −0.282, −0.010; P < 0.05), although no significant difference was observed in the number of binding sites (Table 1). However, no significant differences between the DP enantiomers were observed for either the dissociation constant, or the number of binding sites in fraction 2.
By contrast, completely different results were obtained for the WR enantiomers. Fraction 2 had a higher binding affinity than fraction 3 for both WR enantiomers, although the numbers of binding sites in fractions 2 and 3 were the same. There were differences of 12.6-fold for (S)-WR (95% CI −5.260, −1.228; P < 0.001) and 8.3-fold for (R)-WR (95% CI −4.614, −2.351; P < 0.001) between the dissociation constants in fractions 2 and 3 (Table 1). In fraction 2, the dissociation constant of (S)-WR was lower than that of (R)-WR (95% CI −0.369, −0.028; P < 0.05), although there were no significant differences between the WR enantiomers in either the dissociation constant, or the number of binding sites, in fraction 3. By contrast, only small differences were found between the respective dissociation constants of total (unfractionated) AAG and fraction 3 for each DP enantiomer and fraction 2 for each WR enantiomer (Tables 1 and 2).
SDS–PAGE analysis of the fragmentation of fractions 2 and 3 with 2.5 mg of CNBr showed the presence of four peptide bands of approximately 44, 34, 7 and 5.5 kDa and two peptide bands of approximately 34 and 7 kDa, respectively (Figure 2, lanes 1, 2). SDS–PAGE analysis of fraction 3 also showed the presence of two faint peptide bands of approximately 44 and 41 kDa. Peptide bands smaller than 5 kDa, corresponding to CNBr fragments III and IV, could not be detected. The first five amino-terminal amino acids (residues 112–116) of a peptide band of approximately 7 kDa, probably corresponding to CNBr fragment II (residues 112–181) [13], of each of the fractions was elucidated. Fraction 2 showed the same amino acid sequences as those predicted from the AAG A gene, LAFDV. Fraction 3, however, showed a mixture of deduced amino acid sequences from the AAG A gene and AAG B gene, i.e. a mixture of LAFDV and FGSYL [2]. Furthermore, SDS–PAGE analysis of the fragmentation of fraction 3 with 10 mg of CNBr showed the presence of only two major peptide bands of approximately 34 and 29 kDa, and the 7-kDa peptide could not be detected as a band (Figure 2, lane 3).
Discussion
We previously reported the fractionation of the six AAG glycoforms by chromatography on a hydroxyapatite column (250 × 21.4 mm i.d.), and suggested that the difference between the binding activities of the DP enantiomers was due to differences in association of DP to variants 3–6, while the role of variants 1 and 2 in the binding of DP to AAG was minor [11, 12]. Herve et al. reported that two separate drug-binding sites were present on AAG, the A variant and the F1S variant, and that DP bound specifically to the A variant [7]. In this study therefore we modified the method of separation of AAG so that it was resolved into two main fractions, on a hydroxyapatite column (500 × 30 mm i.d.).We studied the characteristics of the binding of DP enantiomers, which bind selectively to the A variant [7], and WR enantiomers, which bind selectively to the F1S variant [6], the two main fractions of AAG. Fraction 2 exhibited a high binding affinity for WR enantiomers, and fraction 3 strongly bound the DP enantiomers. Only small differences were found between the respective dissociation constants of total (unfractionated) AAG and fraction 3 for each DP enantiomer and fraction 2 for each WR enantiomer (Tables 1 and 2). These results show that DP and WR enantiomers bind selectively to fractions 3 and 2, respectively, and suggest that fraction 2 is the F1S variant and fraction 3 is the A variant.
In order to determine the relationship between the separate fractions and the genetic variants, we studied the amino acid sequences of each of the AAG fractions. SDS–PAGE analysis of the fragmentation products after treatment with 2.5 mg of CNBr showed the presence of peptide bands of 7 kDa, probably corresponding to CNBr fragment II, in both fractions 2 and 3 (Figure 2, lanes 1, 2). The first five amino-terminal amino acids of the 7-kDa peptide of fraction 2 showed amino acid sequences identical to those predicted from the AAG A gene. By contrast, a peptide band of approximately 5.5 kDa of fraction 2 also showed amino acid sequences identical to those predicted from the AAG A gene (Figure 2, lane 1). This band was also detected in the fragmentation of total (unfractionated) AAG (data not shown). The presence of this band can not be explained by the presence of the methionine variant because the product of the AAG A gene does not possess a methionine in position 112–181. No product of the AAG B gene was observed in fraction 2. These results show that fraction 2 is the product of AAG A gene. On the other hand, a peptide of 7 kDa of fraction 3 showed a mixture of the amino acid sequences from the AAG A and B genes. Fragmentation of the product of the AAG B gene with CNBr was expected to lead to the formation of 1 mol of CNBr fragment I and 0.5 mol of CNBr fragments III and IV, because the gene possesses a methionine in position 156 [2]. Therefore, the presence of the product of the AAG B gene in the peptide band of 7 kDa shows that the CNBr cleavage was incomplete, and the presence of a 41-kDa peptide, probably corresponding to CNBr fragments I and III (residues 1–156), also suggests that CNBr cleavage was incomplete. In order to cleave the product of the AAG B gene completely, 10 mg of CNBr was added to 2.0 mg of fraction 3. As a result, the 7- and 41-kDa peptides in fraction 3 could not be detected as a band (Figure 2, lane 3). These results show that fraction 3 is almost identical to the AAG B gene, containing a trace amount of the AAG A gene. The above results suggest that fraction 2 is identical to the F1S variant and that fraction 3 is almost identical to the A variant. On the other hand, the peptide band of approximately 29 kDa appeared to be inaccessible (Figure 2, lane 3). This suggested that the amino-terminal amino acid residue of the 29-kDa peptide might be pyroglutamic acid, as observed in native AAG [16]. Therefore, the peptide band of 29 kDa is thought to be the partially deglycosylated peptide of approximately 34 kDa, corresponding to CNBr fragment I (residues 1–111).
In this study, we also found that there were differences between the stereoselective binding abilities of the separate fractions. In fraction 2, the dissociation constant of (S)-WR was smaller than that of (R)-WR, although no significant differences between DP enantiomers were observed in either the dissociation constant or the number of binding sites. In fraction 3 (S)-DP had a smaller dissociation constant than did (R)-DP, whereas there were no significant differences between the binding parameters of WR enantiomers. These results show that only fraction 3 and fraction 2 had stereoselective binding ability to DP and WR enantiomers, respectively. Herve et al. recently reported that the ligand binding site of the A variant seemed to be of smaller size and of greater ligand specificity. However, they also reported that the ligand binding site of the F1S variant could be a relatively large hydrophobic pocket with no obvious structural requisites for binding [8]. It seems therefore that the difference between the binding behaviours of DP enantiomers to fraction 3, almost identical to the A variant, is probably due to the ligand binding site of smaller size and of greater ligand specificity. By contrast, the ligand binding site of the F1S variant does not have greater ligand specificity. Therefore, it is interesting that fraction 2, almost identical to the F1S variant, had stereoselective binding ability in relation to the WR enantiomers. Several investigators have recently reported stereoselective binding ability of AAG. Hanada et al. reported that enantiomer pairs of DP, propranolol (PL) and verapamil (VP), bound to AAG at the same site but with different affinities [17]. However, the binding parameters of each genetic variant were not determined. It is known that the relative concentrations of the genetic variants of AAG change under various physiological and pathological conditions [18, 19]. Therefore, an understanding of the stereoselective binding ability on each variant could be of interest in clinical pharmacology.
Furthermore, Haginaka and Matsunaga reported that WR enantiomers are not resolved on a pd-AAG (partially deglycosylated AAG) column. On the basis of this finding, they suggested that a sugar chain (s) of AAG cleaved by N-glycosidase might be involved in the stereoselective binding of the WR enantiomers [20]. Shiono et al. reported that sialic acid groups of AAG were responsible for chiral recognition of the enantiomers of PL [21]. These findings suggest that not only the peptide chain but also the carbohydrate chains play an important role in the stereoselective binding ability of AAG. Conversely, Kuroda et al. have reported that the branching type of glycan chains of AAG do not play a significant role in the chiral recognition between enantiomers of PL and VP [22]. However, these studies did not investigate the stereoselective binding ability of each of the variants but of total (unfractionated) AAG. Further study is therefore required to elucidate the effect of the carbohydrate moiety on the stereoselective binding abilities of AAG and its variants.
Acknowledgments
We thank Mr Yukichi Abe (Centre for Instrumental Analysis, Hokkaido University) for his technical assistance with analysis of amino acid sequences.
References
- 1.Urien S, Albengres E, Zini R, Tillement JP. Evidence for binding of certain acidic drugs to α1-acid glycoprotein. Biochem Pharmacol. 1982;31:3687–3689. doi: 10.1016/0006-2952(82)90597-4. [DOI] [PubMed] [Google Scholar]
- 2.Dente L, Pizza MG, Metspalu A, Cortese R. Structure and expression of the genes coding for human α1-acid glycoprotein. EMBO J. 1987;6:2289–2296. doi: 10.1002/j.1460-2075.1987.tb02503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eap CB, Baumann P. Isoelectric focusing of alpha-1 acid glycoprotein (orosomucoid) in immobilized pH-gradients with 8 m urea: detection of its desialylated variants using an alkaline phosphatase-linked secondary antibody system. Electrophoresis. 1988;9:650–654. doi: 10.1002/elps.1150091005. [DOI] [PubMed] [Google Scholar]
- 4.Tomei L, Eap CB, Baumann P, Dente L. Use of transgenic mice for the characterization of human alpha 1-acid glycoprotein (orosomucoid) variants. Hum Genet. 1989;84:89–91. doi: 10.1007/BF00210681. [DOI] [PubMed] [Google Scholar]
- 5.Eap CB, Cuendet C, Baumann P. Binding of d-methadone, 1-methadone, and dl-methadone to proteins in plasma of healthy volunteers: role of the variants of α1-acid glycoprotein. Clin Pharmacol Ther. 1990;47:338–346. doi: 10.1038/clpt.1990.37. [DOI] [PubMed] [Google Scholar]
- 6.Herve F, Gomas E, Duche JC, Tillement JP. Evidence for differences in the binding of drugs to the two main genetic variants of human α1-acid glycoprotein. Br J Clin Pharmacol. 1993;36:241–249. doi: 10.1111/j.1365-2125.1993.tb04224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herve F, Duche JC, D'Athis P, et al. Binding of disopyramide, methadone, dipyridamole, chlorpromazine, lignocaine and progesterone to the two main genetic variants of human α1-acid glycoprotein: evidence for drug-binding differences between the variants and for the presence of two separate drug-binding sites on α1-acid glycoprotein. Pharmacogenetics. 1996;6:403–415. doi: 10.1097/00008571-199610000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Herve F, Caron G, Duche JC, et al. Ligand specificity of the genetic variants of human α1-acid glycoprotein: generation of a three-dimensional quantitative structure–activity relationship model for drug binding to the A variant. Mol Pharmacol. 1998;54:129–138. doi: 10.1124/mol.54.1.129. [DOI] [PubMed] [Google Scholar]
- 9.Charles P, Kathleen MG, Terrencen FB, et al. The cardiac effects of d- and 1-disopyramide in normal subjects: a noninvasive study. Circulation. 1982;66:447–453. doi: 10.1161/01.cir.66.2.447. [DOI] [PubMed] [Google Scholar]
- 10.Lima JJ, Boudoulas H. Stereoselective effects of disopyramide enantiomers in humans. J Cardiovascular Pharmacol. 1987;9:594–600. doi: 10.1097/00005344-198705000-00014. [DOI] [PubMed] [Google Scholar]
- 11.Kishino S, Nomura A, Saitoh M, et al. Single-step isolation method for six glycoforms of human α1-acid glycoprotein by hydroxylapatite chromatography and study of their binding capacities for disopyramide. J Chromatogr B. 1997;703:1–6. doi: 10.1016/s0378-4347(97)00403-9. [DOI] [PubMed] [Google Scholar]
- 12.Kishino S, Itoh S, Nakagawa T, Miyazaki K. Enantioselective binding of disopyramide to α1-acid glycoprotein and its variants. Eur J Clin Pharmacol. 2001;57:583–587. doi: 10.1007/s002280100354. [DOI] [PubMed] [Google Scholar]
- 13.Ikenaka T, Ishiguro M, Emura J, et al. Isolation and partial characterization of the cyanogen bromide fragments of α1-acid glycoprotein and the elucidation of the amino acid sequence of the carboxyl-terminal cyanogen bromide fragment. Biochemistry. 1972;11:3817–3829. doi: 10.1021/bi00770a022. [DOI] [PubMed] [Google Scholar]
- 14.Pinkerton TC, Koeplinger KA. Determination of warfarin–human serum albmin protein binding parameters by an improved Hummel–Dreyer high-performance liquid chromatographic method using internal surface reversed-phase columns. Anal Chem. 1990;62:2114–2122. doi: 10.1021/ac00218a013. [DOI] [PubMed] [Google Scholar]
- 15.Yamaoka K, Tanigawara Y, Nakagawa T, Uno T. A pPharmacokinetic analysis program (multi) for the microcomputer. J Pharmacobio-Dyn. 1981;4:879–885. doi: 10.1248/bpb1978.4.879. [DOI] [PubMed] [Google Scholar]
- 16.Schmid K, Kaufmann H, Isemura S, et al. Structure of α1-acid glycoprotein. The complete amino acid substitutions, and homology with the immunoglobulins. Biochemistry. 1973;12:2711–2724. doi: 10.1021/bi00738a026. [DOI] [PubMed] [Google Scholar]
- 17.Hanada K, Ohta T, Hirai M, Arai M, Ogata H. Enantioselective binding of propranolol, disopyramide, and verapamil to human α1-acid glycoprotein. J Pharm Sci. 2000;89:751–757. doi: 10.1002/(SICI)1520-6017(200006)89:6<751::AID-JPS6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Eap CB, Fischer JF, Baumann P. Variations in relative concentrations of variants of human α1-acid glycoprotein after acute phase conditions. Clinica Chimica Acta. 1991;203:379–386. doi: 10.1016/0009-8981(91)90312-z. [DOI] [PubMed] [Google Scholar]
- 19.Herve F, Duche JC, Jaurand MC. Changes in expression and microheterogeneity of the genetic variants of human α1-acid glycoprotein in malignant mesothelioma. J Chromatogr B. 1998;715:111–123. doi: 10.1016/s0378-4347(98)00085-1. [DOI] [PubMed] [Google Scholar]
- 20.Haginaka J, Matsunaga H. Separation of enantiomers on HPLC chiral stationary phases based on human plasma α1-acid glycoprotein: effect of sugar moiety on chiral recognition ability. Enantiomer. 2000;5:37–45. [PubMed] [Google Scholar]
- 21.Shiono H, Shibukawa A, Kuroda Y, Nakagawa T. Effect of sialic acid residues of human α1-acid glycoprotein on stereoselectivity in basic drug-protein binding. Chirality. 1997;9:291–296. [Google Scholar]
- 22.Kuroda Y, Shibukawa A, Nakagawa T. The role of branching glycan of human α1-acid glycoprotein in enantioselective binding to basic drugs as studied by capillary electrophoresis. Anal Biochem. 1999;268:9–14. doi: 10.1006/abio.1998.3050. [DOI] [PubMed] [Google Scholar]