

Abstract
Aim
To determine the absolute bioavailability of extravascularly administered dexmedetomidine, a novel a2-adrenoceptor agonist, in healthy subjects.
Methods
Single 2 µg kg−1 doses of dexmedetomidine were given intravenously, intramuscularly, perorally and buccally (where the solution is not swallowed) to 12 healthy male subjects. The drug concentration-time data were analysed using linear one-compartment (buccal and peroral data), or two-compartment modelling (intravenous data), or noncompartmental methods (intramuscular data).
Results
Mean (95% CI) absolute bioavailability after peroral, buccal and intramuscular administration was 16% (12–20%), 82% (73–92%) and 104% (96–112%), respectively.
Conclusion
Dexmedetomidine is well absorbed systemically through the oral mucosa, and therefore buccal dosing may provide an effective, noninvasive route to administer the drug.
Keywords: bioavailability, dexmedetomidine, pharmacokinetics
Introduction
Dexmedetomidine is a potent and specific a2-adrenergic receptor agonist [1], which causes sedation and anaesthesia in animals [2]. In humans, dexmedetomidine has been under clinical evaluation as an adjunct to anaesthesia [3, 4] and, more recently, as an agent for sedation in the intensive care unit [5]. The pharmacokinetics of dexmedetomidine in humans have been studied after intravenous (i.v), intramuscular (i.m) and transdermal administration [6–10]. The mean elimination half-life is 1.5–3 h after i.v and i.m. dosing, and 5.6 h after transdermal dosing. After i.m and transdermal administration, the time to maximum concentration in blood was 1.6–1.7 h and 6 h, and the absolute bioavailability was estimated to be 73% and 88%, respectively. The aim of this study was to determine the absolute bioavailability of dexmedetomidine administered via the peroral (p.o.) and buccal route. An i.m. dose of the drug was used as a reference.
Methods
In this open study, dexmedetomidine hydrochloride (Orion Pharma, Turku, Finland) was given intravenously and extravascularly to 12 healthy male subjects (age 20–27 years; weight 67–85 kg; height 171–191 cm) using a four-period, cross-over design with balanced randomization. The wash-out period between consecutive administrations was at least 8 days. The general health of the subjects was ascertained through medical history and by clinical examinations, including electrocardiography and a thorough laboratory screening. On the study days, subjects abstained from smoking, and fasted from the midnight before and until 3 h after drug administration. During this period, only water was allowed. Alcohol intake was prohibited for 48 h and any medication for 2 weeks prior to the beginning of the study. The protocol was approved by the Ethics Committee of Turku University Central Hospital. Written informed consent was obtained from each subject.
At the beginning of each study session, a single 2 µg kg−1 dose of dexmedetomidine was administered intravenously (over 5 min at a constant rate using an infusion pump), intramuscularly (by injection), perorally (drug solution ingested with 150 ml of water) or buccally (the drug solution retained in the mouth for 5 min without swallowing, then expelled into a tube for the measurement of remaining drug). 10 ml venous blood samples were collected at baseline and at 10 min, 20 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 5 h, 6 h and 7 h after receiving the drug. After i.v. administration, samples were also drawn at 5 and 15 min The blood samples were centrifuged and the sera stored in polyethylene storage tubes at 20 °C. Dexmedetomidine concentrations were measured using a validated [11] gas chromatographic/mass spectrometric method [12]. The lower limit of quantification was 0.02 µg l−1. The qual-ity control samples showed an interassay variability of 5.1–7.1% (CV%) over the concentration range 0.1–2.5 µg l−1. The amount of dexmedetomidine expelled from the mouth was analysed using a simple high-performance liquid chromatography method with UV detection. The coefficient of variation of the method was less than 1%.
The dexmedetomidine concentration-time data after i.v. administration were analysed using a linear two-compartment model with zero-order infusion. The nonlinear least-squares fit to the data was performed using the reciprocal of the predicted concentrations as a weighting factor (PCNonlin program vs 4.0, SCI Software, Lexington, Kentucky, USA). The concentration-time data after p.o. and buccal administration were analysed using a linear one-compartment model with first-order absorption and correction for lag-time (PCNonlin). After i.m. administration, compartment modelling could not be performed, and, thus, the pharmacokinetic parameters were calculated using standard noncompartmental methods (BIOPAK program vs 2.1, SCI Software). The absolute bioavailability (extent of absorption) upon extravascular administrations was calculated using Equation 1:
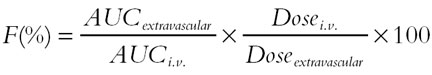 |
where AUC refers to the area under the concentration-time curve. The absolute bioavailability is reported as geometrical mean (95% confidence interval, CI), and other parameters as mean ± s.d.
Results
The mean concentration-time curves of dexmedetomidine concentrations in serum after i.v., i.m., buccal and p.o. administration are presented in Figure 1.
Figure 1.
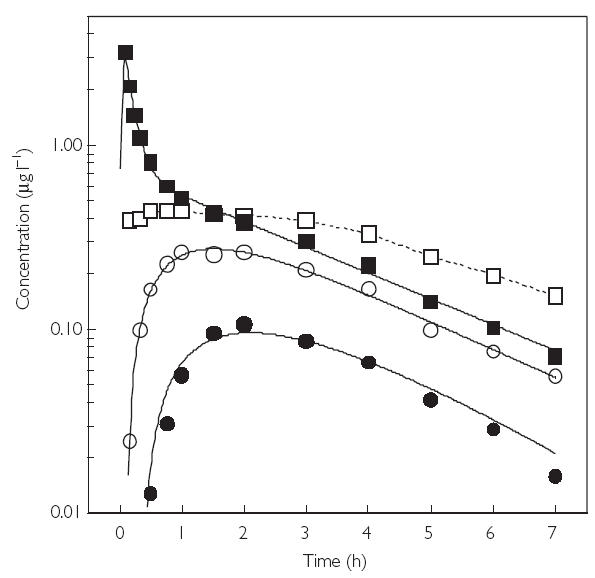
Mean curves of dexmedetomidine concentrations in serum vs time after intravenous (i.v), intramuscular (i.m), buccal and peroral (p.o) administration of a single 2.0 µg kg−1 dose. i.v. (▪); i.m. (□); buccal (○); and p.o. (•).
After i.v. administration, the half-life of the distribution phase was 6.5 ± 3.4 min, and the drug was initially distributed into a volume of 0.43 ± 0.15 l kg−1. The elimination half-life was 2.17 ± 0.42 hours. Total clearance was 0.64 ± 0.14 l h−1 kg−1 (47.4 ± 9.0 l h−1), and the apparent volume of distribution at steady-state was 1.61 ± 0.26 l kg−1 (121 ± 20 l). The AUC was 2.76 ± 0.54 µg h l−1. Interindividual variation in the main pharmacokinetic parameters was relatively low, with standard deviation of less than 20%.
After i.m. administration, AUC was 2.84 ± 0.43 µg h l−1, giving a mean absolute bioavailability of 103.6% (95.5–112.3%). The peak concentration and the time to peak were 0.51 ± 0.08 µg l−1 and 1.7 ± 1.8 h, respectively. The elimination half-life was 2.5 ± 0.6 h.
After p.o. administration, the maximum dexmedetomidine concentration in serum (0.11 ± 0.04 µg l−1) was achieved in 2.2 ± 0.5 h after a lag-time of 0.6 ± 0.3 h. The terminal half-life was estimated to be 1.2 ± 0.3 h which is probably not an accurate estimate, due to the low concentrations of dexmedetomidine in the elimination phase. AUC was 0.45 ± 0.18 µg h l−1, giving a mean absolute bioavailability of 15.6% (12.1–20.1%).
Whereas the i.v., i.m. and p.o. doses were exactly 2.0 µg kg−1, the actual buccal doses varied from 0.49 to 1.75 µg kg−1 (mean ± s.d., 1.12 ± 0.33 µg kg−1), because not all drug was absorbed. After buccal administration, AUC was 1.29 ± 0.50 µg h l−1, and the dose-corrected mean buccal bioavailability was 81.8% (72.6–92.1%). The peak concentration (0.29 ± 0.09 µg l−1) was attained at 1.5 ± 0.2 h after a short lag-time of 0.13 ± 0.04 hours. The apparent elimination half-life was 1.9 ± 0.5 h.
Discussion
Consistent with previous reports [6, 7], dexmedetomidine kinetics after i.v. administration could be described adequately by a two-compartment model. The pharmacokinetic parameters obtained after i.v. infusion are comparable with previously reported data [6, 7, 10].
In the present work, dexmedetomidine appeared to show complete bioavailability after i.m. administration, compared to a previously reported value of 73% [9]. However, the time to maximum concentration and the elimination half-life after i.m. dosing are in line with the earlier results [8].
The major finding in the current study was that dexmedetomidine seemed to be well absorbed systemically through oral mucosa, the buccal bioavailability being as high as 82% and the maximum concentration in serum being reached in 1.5 h. In contrast, the bioavailability after p.o. administration was poor (16%), probably due to extensive first-pass effect of dexmedetomidine. Based on these observations, we suggest that buccal dosing of dexmedetomidine provides a clinically relevant, noninvasive alternative to the invasive i.v. or i.m. route of administration.
Acknowledgments
This study was supported by Orion Corporation, Orion Pharma, Finland.
References
- 1.Aantaa R, Kallio A, Virtanen R. Dexmedetomidine, a novel alpha 2-adrenergic agonist. A review of its pharmacodynamic characteristics. Drugs Future. 1993;18:49–56. [Google Scholar]
- 2.Kuusela E, Raekallio M, Anttila M, Falck I, Molsa S, Vainio O. Clinical effects and pharmacokinetics of medetomidine and its enantiomers in dogs. J Vet Pharmacol Ther. 2000;23:15–20. doi: 10.1046/j.1365-2885.2000.00245.x. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Abraham R, Ogorek D, Weinbroum AA. Dexmedetomidine: a promising agent for anesthesia and perioperative care. Isr Med Assoc J. 2000;2:793–796. [PubMed] [Google Scholar]
- 4.Scheinin H, Jaakola ML, Sjövall S, et al. Intramuscular dexmedetomidine as premedication for general anesthesia. A comparative multicenter study. Anesthesiology. 1993;78:1065–1075. doi: 10.1097/00000542-199306000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Angelini G, Ketzler JT, Coursin DB. Use of propofol and other nonbenzodiazepine sedatives in the intensive care unit. Crit Care Clin. 2001;17:863–880. doi: 10.1016/s0749-0704(05)70184-6. [DOI] [PubMed] [Google Scholar]
- 6.De Wolf AM, Fragen RJ, Avram MJ, Fitzgerald PC, Rahimi-Danesh F. The pharmacokinetics of dexmedetomidine in volunteers with severe renal impairment. Anesth Analg. 2001;93:1205–1209. doi: 10.1097/00000539-200111000-00031. [DOI] [PubMed] [Google Scholar]
- 7.Dutta S, Lal R, Karol MD, Cohen T, Ebert T. Influence of cardiac output on dexmedetomidine pharmacokinetics. J Pharm Sci. 2000;89:519–527. doi: 10.1002/(SICI)1520-6017(200004)89:4<519::AID-JPS9>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 8.Scheinin H, Karhuvaara S, Olkkola KT, et al. Pharmacodynamics and pharmacokinetics of intramuscular dexmedetomidine. Clin Pharmacol Ther. 1992;52:537–546. doi: 10.1038/clpt.1992.182. [DOI] [PubMed] [Google Scholar]
- 9.Dyck JB, Maze M, Haack C, Vuorilehto L, Shafer SL. The pharmacokinetics and hemodynamic effects of intravenous and intramuscular dexmedetomidine hydrochloride in adult human volunteers. Anesthesiology. 1993;78:813–820. doi: 10.1097/00000542-199305000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Kivistö KT, Kallio A, Neuvonen PJ. Pharmacokinetics and pharmacodynamics of transdermal dexmedetomidine. Eur J Clin Pharmacol. 1994;46:345–349. doi: 10.1007/BF00194403. [DOI] [PubMed] [Google Scholar]
- 11.Shah VP, Midha KK, Dighe S, et al. Analytical methods validation: Bioavailability, bioequivalence, and pharmacokinetic studies. Conference report. Pharm Res. 1992;9:588–592. doi: 10.1007/BF03189968. [DOI] [PubMed] [Google Scholar]
- 12.Vuorilehto L, Salonen JS, Anttila M. Picogram level determination of medetomidine in dog serum by capillary gas chromatography with negative ion chemical ionization mass spectrometry. J Chromatogr. 1989;497:282–287. doi: 10.1016/0378-4347(89)80030-1. [DOI] [PubMed] [Google Scholar]