

Abstract
Identification of nitric oxide as the molecule responsible for endothelial dependant vasodilatation has led to an explosion of interest in endothelial function. Oxidative stress has been identified as an important factor in the development of tolerance to organic nitrates. This review examines the evidence supporting this recently developed theory and how mechanisms of nitrate tolerance may link with the wider picture of primary nitric oxide resistance.
Keywords: antioxidants, endothelial dysfunction, nitric oxide, nitrate tolerance, oxidative stress, renin-angiotensin system
Introduction
Nitrates have been used in clinical practice as anti-anginal treatment for over a century [1]. However, the link between nitric oxide and endothelial function has been demonstrated more recently. In 1980, Furchgott et al. published data describing the importance of the endothelial layer in mediating acetylcholine-induced vasodilatation of rabbit aortic rings [2]. The term endothelial derived relaxing factor (EDRF) was used to define this endothelial agent which was easily lost if the endothelium was inadvertently damaged. Soon after this observation, EDRF was identified as nitric oxide (NO) [3] and the pathway responsible for its synthesis from l-arginine was described [4]. With this came the important observation that a closely related analogue of l-arginine, L-NMMA (NG-methyl-L-arginine) could competitively inhibit the pathway. Furchgott's work, along with that of Ignarro and Murad, was recognized with the Nobel Prize for Medicine in 1998.
It is now apparent that NO is important in mediating other cellular signals. Different isoenzymes of nitric oxide synthase (NOS) have been identified in neuronal signalling, and as an inducible form, in mediating inflammation in severe sepsis [4]. NO is also involved in a number of anti-atherogenic processes, such as suppressing platelet aggregation and smooth muscle cell proliferation [4]. Endothelial NOS (eNOS) is the key source of nitric oxide within the vascular endothelium. Constant background release of nitric oxide by eNOS helps maintain physiological vascular tone. However, eNOS agonists such as acetylcholine (Ach) can increase NO levels and thus are able to modulate vascular tone [4].
L-arginine pathway [Figure 1]
Figure 1.
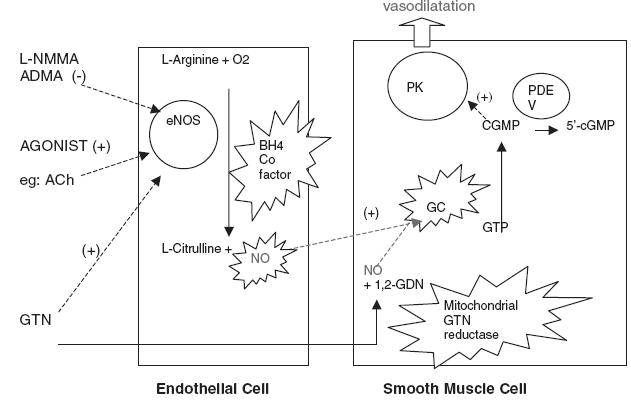
The endothelial nitric oxide pathway. NOS, nitric oxide synthetase; GC, guanylate cyclase; PK, protein kinase; Ach, acetylcholine; NO, nitric oxide; 1,2- GDN – glyceryldinitrate; PDE, phosphodiesterase; 5′-cGMP, cyclic GMP; star burst, target of oxidative stress.
This pathway enables the production of endogenous NO from the substrate l-arginine within the endothelial cell. Situated within the endothelium, NOS converts l-arginine to NO and the by-product l-citrulline. In turn, NO diffuses from the endothelial cell across to the vascular smooth muscle triggering the formation of cyclic GMP via soluble guanylate cyclase (GC) causing smooth muscle relaxation and hence vasodilatation. The pharmacological action of NO, generated by NO donors, short-circuits endothelial NOS by diffusing directly across to the smooth muscle layer. Hence vasodilatation can be endothelium dependent or independent.
NOS can be stimulated by a variety of agonists including acetylcholine, bradykinin, nebivolol [5] and pravastatin [6]. Of these, acetylcholine is the most commonly used experimental agent. Recent evidence based on bovine aortic endothelial cells challenges the generally accepted mechanism of action of pharmacological nitrates by demonstrating that NOS can also be stimulated by nitrate donors (such as GTN) [7]. Thus, GTN may not only act as a nitrate donor but may also be able to stimulate NOS directly [7]. Cross-tolerance to both acetylcholine and GTN can be demonstrated experimentally, however, this effect may only be present at lower doses of acetylcholine [7]. This suggests that the mechanisms of stimulation may differ between different pharmacological substances. In vitro studies suggest that tolerance to GTN may be associated with a modulation of NOS response as reflected by decreased l-arginine uptake in tolerant tissue [7].
Furthermore, the recent discovery of a mitochondrial GTN reductase present in the vascular smooth muscle adds another dimension to the fate of exogenous nitrates [8]. In vitro studies have demonstrated that this enzyme (mitochondrial aldehyde dehydrogenase (mt ALDH)) is able to bioconvert GTN. In vivo work with mt ALDH inhibitors has confirmed that this mechanism results in elevated cyclic guanosine monophosphate (cGMP) and vasorelaxation.
Nitrate tolerance, NO resistance and endothelial dysfunction
Nitrate tolerance can be regarded as an extreme example of nitrate resistance, which exists in a large number of atherogenic conditions such as hypercholesterolaemia, type 2 diabetes, smoking and ischaemic heart disease [9]. The endothelial dysfunction present in these conditions reflects the reduced bioavailability of nitric oxide, which can be measured experimentally [9] and is thought, in part, to result from the increased oxidative stress present in these conditions. Thus, clinically-induced nitrate tolerance associated with nitrate-induced renin-angiotensin activation and oxidant stress may now be regarded as an extension of the primary pathophysiological phenomenon of nitric oxide resistance per se. Hence, the clinical effects of nitrates may be blunted in the first instance by an underlying primary NO resistance to give a ‘primary’ nitrate tolerance while a more dramatic, but mechanistically similar, ‘secondary’ nitrate tolerance develops with continued use [10]. In clinical terms these two nitrate tolerance states are very likely to occur frequently within the same individual. The question of whether secondary nitrate tolerance superimposed on pre-existing nitric oxide resistance results in worsening endothelial function and cardiovascular outcomes remains to be definitively answered. However, evidence supporting this notion exists in the form of a Cox multivariate analysis of two observational coronary secondary prevention studies which showed an increase in mortality in chronic nitrate-treated ischaemic heart disease patients following recovery from an acute cardiac event [11]. Also, randomised controlled trial data exists to show that continuous glyceryl trinitrate (GTN) does worsen endothelial dependant vasodilatation in both ischaemic heart disease patients [12] and healthy volunteers [13].
Nitrate tolerance
In early accounts of nitrate therapy it was known that prolonged exposure to nitrates resulted in ‘nitrate tolerance’, a diminution of the antianginal and vasodilator response to nitrate treatment. This phenomenon has restricted the clinical use and effectiveness of nitrates to the current day. Consequently, there has been much interest in attempting to understand the mechanisms involved. Tolerance to nitrates given constantly or in regular divided doses occurs quickly, i.e. within days [14]. Nitrate tolerance develops despite an elevation in the drug plasma concentration [15] reflecting a decrease in vascular sensitivity to previously therapeutic levels. This can be prevented or reduced by inclusion of a nitrate free period in the dosing schedule. Much debate has centred on which schedule is best for each nitrate compound. Generally, twice-daily (oral) doses at 8am and 12 noon, or a daily patch with 12- h patch-free period seem effective. A tendency towards nitrate tolerance (as measured by exercise capacity) develops with steadier 24 h plasma concentration profiles [16].
Nitrate-tolerant individuals are more susceptible to enhanced vasoconstriction whenever the plasma nitrate concentration is allowed to fall – the ‘rebound phenomenon’. This is reflected by increased sensitivity to a number of circulating vasoconstrictor substances such as catecholamines and angiotensin II (AII) [17]. Clinically the rebound phenomenon may be more important than is currently recognized. Evidence suggests that even intermittent nitrate patch therapy results in increased vasoconstrictor sensitivity during the patch-off period [18]. Observational studies of explosives industries workers confirm an increased relative risk of sudden cardiac death during off-duty periods [19]. Chronic beta-blocker administration for stable angina protects against ‘rebound vasoconstriction’ in patients treated with intermittent nitrate patches [20]. The mechanism through which beta-blockers exert their protective effect could include a dampening down of the effects of increased vasoconstrictor levels found in nitrate tolerance [21]. This effect could be mediated by not only beta adrenoceptor blockade but also direct inhibition of renin release from the juxtaglomerular apparatus, thus preventing the subsequent cascade of AII release, protein kinase C activation and endothelin 1 mediated sensitization of the vascular smooth muscle to circulating vasoconstrictors [22].
Proposed mechanisms for nitrate tolerance [Figure 2]
Figure 2.
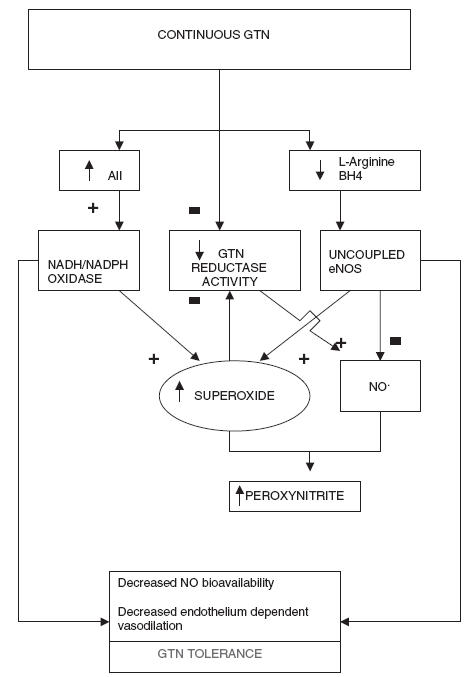
Suggested mechanisms for nitrate tolerance.AII, Angiotensin II; BH4, tetrahydrobiopterin; eNOS, endothelial nitric oxide synthase
Each stage of the nitric oxide pathway has been examined with regard to its importance in nitrate tolerance. Even before the discovery that exogenous and endogenous nitrates shared a common final pathway it was known that tolerance to one organic nitrate frequently implied ‘cross tolerance’ to other nitrogenous and non-nitrogenous substances [23, 24]. In 1973 Needleman [25] documented the importance of the redox status of thiol groups in determining nitrate tolerance. For many years thiol deficiency was thought to be the core mechanism behind the development of tolerance. Later studies disagreed with this notion, failing to demonstrate any clinical benefit from supplementation with N-acetylcysteine, a thiol donor [26]. Finally, measurement of arterial & venous thiol levels in nitrate tolerant/nontolerant rats individuals showed that thiol groups were not deplete in nitrate tolerant tissues [27]. In the 1990s attention focused on neurohormonal activation. Parker et al. noted an increase in circulating catecholamines, stimulation of the renin-angiotensin system and associated increase in body weight and lower haematocrit in tolerant subjects reflecting an increase in intravascular volume [28]. This physiological response to vasodilatation has been termed ‘pseudotolerance’. The counter-regulatory hormonal response has been noted to peak at 24 h but disappears at three days despite a persistent impairment in vascular response [17]. Another suggested mechanism for tolerance is increased breakdown of cGMP by an increase in phosphodiesterase activity. An inhibitor of phosphodiesterase 1 (a1 subclass) has been effective at limiting tolerance in vitro [29]. However, recent attention has been focused on a novel mechanism that involves increased free radical activity.
The link with oxidative stress
An important development in the quest for the key to tolerance was made in 1995 by Munzel et al. [30] who proposed that nitrate tolerance induced in vivo was associated with an endothelial dependent production of superoxide anion. This notion was supported by work demonstrating that tolerance (and the production of superoxide) could be attenuated by the addition of the enzyme superoxide dismutase (SOD). Interestingly, as far back as 1986 Gryglewski [31] had noted a link between superoxide anion and EDRF instability and the protective effect of SOD. Further supporting evidence for this mechanism includes the reduced production of superoxide anion associated with the inclusion of a nitrate free period in vivo [18]. A randomised controlled trial (RCT) of patients undergoing elective coronary artery bypass grafting, randomised to preoperative intravenous GTN (vs no intravenous GTN), confirms increased superoxide generation in internal mammary artery samples in the nitrate tolerant group [32]. This revelation has led many to attempt to prevent tolerance through the use of antioxidants. Whilst not all work is confirmatory [33], many have had success with a variety of antioxidants. Superoxide anion reacts with NO to form peroxynitrite that is less effective at stimulating eNOS and also has a shorter half-life [34]. Peroxynitrite can cause nitration of tyrosine residues resulting in the formation of 3-nitrotyrosine (a useful marker of activity) and is elevated in nitrate tolerance [35].
Potential sources of superoxide anion
Exactly which enzymes are responsible for superoxide anion generation in the context of nitrate tolerance remains undecided. β-nicotinamide adenine dinucleotide phosphate-dependant membrane-associated oxidase is a major source of superoxide anion and has been shown to be activated by AII infusions in vivo and also in vascular smooth muscle cell cultures [36] [37]. Concomitant administration of losartan and AII in rats has been shown to normalize superoxide production [36]. Supporting evidence of NADPH oxidase involvement in the development of tolerance comes from work looking at the protective effect of hydralazine on nitrate tolerance and subsequent reduction of superoxide anion production via an NADH oxidase pathway in rabbits [38]. However, a clinical RCT of heart failure patients and healthy controls has failed to demonstrate any positive effect of hydralazine (50 mg tds) on nitrate patch-generated tolerance [39]. In situations with decreased l-arginine substrate, endothelial NOS is known to become uncoupled from its production of NO and switch to the production of the superoxide anion itself [40]. Although l-arginine is rarely deplete in clinical practice, there is evidence that L-arginine supplementation helps to partially prevent tolerance in vitro [7]. GTN tolerant endothelium also displays reduced L arginine uptake in vitro [7]. It has been suggested that an apparent deficiency of substrate may result if eNOS is in some way compartmentalized within the endothelial cell [41]. An endogenous NOS inhibitor exists in the form of asymmetric dimethylarginine (ADMA) which, some groups have postulated, may be able to modulate endothelial NOS responses [42]. ADMA, a competitive antagonist of eNOS, which is equipotent with L-NMMA [42], has also been shown to increase oxidative stress in human umbilical endothelial cells [43]. As with other methylarginines l-arginine and L-NMMA, ADMA enters cells through the y+ transporter and thus can compete for uptake [42]. Importantly, excess L- arginine is required to reverse inhibition by L-NMMA or ADMA [44]. In view of the possible compartmentalization of eNOS in caveolae within the endothelial cell [44] and the identification of the y+ transporter on these caveolae [41] it is possible that the l-arginine/ADMA ratio is capable of influencing eNOS activity. Whether this mechanism is important in iatrogenic nitrate tolerance or just primary nitric oxide resistance remains to be shown. Outside the context of nitrate tolerance supplemental l-arginine has been shown clinically to restore endothelium dependent relaxation in patients with heart failure [45].
Certain mechanisms have been postulated for the observed phenomenon of eNOS ‘uncoupling’. Some interest surrounds the interrelation of enzymes NADPH oxidase, protein kinase C (PKC) and eNOS. Long-term nitroglycerin therapy is also known to activate PKC [22]. PKC in turn has been shown to increase superoxide anion levels in vitro [46]. eNOS is known to be a phosphorylation target for PKC [47]. This mechanism is associated with NO induced formation of superoxide [48]. Hence the uncoupling of eNOS necessary for the production of superoxide anion seems to be dependant on PKC activation (via PKC activated eNOS phosphorylation) [49]. Possible candidates for PKC activators include AII [36] and NO itself.
An alternative mechanism involves tetrahydropbiopterin (BH4). Endothelial NOS requires BH4 as a cofactor to enable the transfer of electrons to l-arginine and thus form NO. NOS uncoupling can occur not only in the presence of reduced l-arginine but also reduced BH4[50]. Likewise, BH4 supplementation can reverse this effect in vivo [51]. Furthermore, supplementation with folic acid (which is involved in BH4 regeneration [52]) has been shown in a clinical, double blind RCT (n = 18) to prevent nitrate tolerance in healthy subjects [53]. This effect appears to be independent of the integral antioxidant effect of folic acid [53].
The renin angiotensin system (RAS) and nitrate tolerance
The RAS is inexorably linked with nitrate tolerance. RAS activation seems to be involved in both pseudotolerance, the initial baroreflex response which occurs with all vasodilatory agents and the later true tolerance represented by resistance to vasodilatation. Many but not all groups have found angiotensin converting enzyme (ACE) inhibitors and AII (AII) receptor blockers to have a beneficial effect on nitrate tolerance and this protective effect seems to be enhanced with higher doses. Animal work has demonstrated a beneficial effect on tolerance and prevention of rebound vasoconstriction with high-dose (1.0 mg kg−1) enalapril ex vivo [21]. Similarly, Katz et al. in a randomised, controlled clinical study with 34 healthy volunteers, demonstrated that concomitant enalapril 10 mg bd or captopril 25 mg tds completely prevented tolerance after a 7-day study period [54]. Another clinical RCT of 26 patients, with a history of chest pain, randomised to seven days captopril 50 mg day−1 or placebo demonstrated that tolerance to a 48-h infusion of GTN could be prevented in the captopril group [55]. These effects have been confirmed in a clinical randomised controlled trial of 60 patients on IV nitrate therapy due to unstable angina. In this study addition of captopril 25 mg tds or losartan 50 mg od not only prevented/reduced the effect of nitrate tolerance but also significantly reduced the frequency of recurrent angina and invasive intervention in the treated group vs the control group who received IV nitrates alone [56]. Conversely, two studies using a slightly lower dose of captopril (mean 60 mg day−1 for 24 h prior to GTN) in patients with severe congestive cardiac failure (n = 21), and benazapril in healthy volunteers (n = 20), respectively, have failed to demonstrate a positive effect [57, 58]. AII receptor blockade has been studied in animal models using Losartan and has been shown to be protective against the development of tolerance [59]. This study also demonstrated an exaggeration of superoxide production (two-fold increase) in tolerant individuals, which was normalized in the losartan treated group [59]. A nonselective ET-1 inhibitor (bosentan) had a less potent but similar effect [59]. Combination therapy of nitrates and ace inhibitors has also proved useful in clinical practice in the prevention of ventricular remodelling in heart failure patients [60].
Nitrate tolerance is associated with increased levels of AII [14]. There is evidence linking AII with enhanced ET-1 transcription [61] which may partly explain the enhanced vasoconstrictor response found in nitrate tolerance. It has been suggested that enhanced ET-1 activity acts as an ‘autocrine priming stimulus’ for hypersensitivity to circulating vasoconstrictors such as catecholamines [17]. AII may also represent a link between nitrate tolerance and superoxide formation. AII is also known to stimulate NADH/NADPH oxidase in smooth muscle cell cultures [37] making ace inhibitors and AII blockers potential ‘antioxidants’ themselves.
Antioxidant therapy and the prevention of nitrate tolerance
Following the development of the superoxide theory in nitrate tolerance, evidence of the possible beneficial effects of antioxidant therapy is now emerging.
Classification of antioxidants can be divided simply into two: enzymatic and nonenzymatic (e.g. superoxide dismutase and vitamin E, respectively) and further subdivided by other properties such as solubility. These properties reflect the biological distribution of each antioxidant and hence its likely potency in a given situation. For example, vitamin E is profoundly lipophillic and hence widely distributed throughout the phospholipid bilayer. Antioxidants act to quench free radicals formed by the result of incomplete reduction of oxygen. Free radicals (including reactive oxygen intermediates, ROIs) hold an odd number of electrons that can set in motion a powerful chain of electron transfer reactions. This is the manner in which free radicals cause their toxic effects. As a result, a number of protective mechanisms exist to quench such activity. The variety and quantity of these defences reflect the relative damage unchecked reactions can incur. Recent interest in transcription factors such as nuclear factor-κB (NF-κB) has demonstrated that some redox reactions can activate transcription factors leading to up-regulated expression of various inflammatory cytokines and cell adhesion molecules such as ICAM-1 [62]. Consequently, NF-κB blockade has been shown to prevent vascular lesion formation in rats [63].
The link between elevated oxidative stress and endothelial dysfunction has been well documented in disease states such as atherosclerosis, dyslipidaemia and type 2 diabetes, for review see Liplinski [64]. The increased oxidative stress measurable in these conditions is thought to reflect varying degrees of inflammation, lipoprotein oxidation and nonenzymatic protein glycation [65].
The observation that increased vitamin E consumption appears to be linked to a reduced incidence of cardiovascular events [66] has prompted a number of studies to investigate the benefits of vitamin E supplementation. Rather disappointingly, larger studies have failed to demonstrate any significant reduction in cardiovascular mortality. An exception is a Chinese study of almost 30 000 patients who were randomised to vitamin E, beta-carotene and selenium or placebo [67]. This study found a 9% decrease in all cause deaths over a 5.2 year follow up. However, the underlying nutritional status of this group is unlikely to match that of the general population in the Western hemisphere and makes the relatively small benefit even more disappointing. The HOPE study of over 9000 high-risk patients failed to demonstrate any significant difference in cardiovascular events between those given vitamin E or placebo over the average 4.5 year follow-up period [68]. Most recently, the Heart Protection Study of 20 500 UK patients with diabetes or vascular disease who were randomised to a vitamin combination (vitamins E, C and beta-carotene) or placebo failed to find significant differences in the incidence of vascular/nonvascular death, nonfatal MI, stroke or cancer despite measurable increases in plasma concentrations of each vitamin over the 5 year study period [69]. The discrepancy between earlier observational and more recent randomised controlled trials is likely to be related to the additional lifestyle differences (particularly dietary consumption of additional antioxidant supplements) in those groups observed to be at lower risk.
The relatively short follow-up period of many antioxidant studies has been thought to be a possible reason for these negative results. However, 12-year follow-up of the Physicians health study has shown no benefit in cardiovascular mortality [41]. Comparisons between patient subgroups with, and without, evidence of cardiovascular disease has also failed to show protective benefit [69] countering arguments that antioxidants would be beneficial in disease prevention rather than progression. Similarly, subgroup analysis of diabetes patients, a group known to have increased markers of oxidative stress, has failed to show an effect [69]. Clinical trials on antioxidant supplementation in nitrate tolerant cardiovascular patients is, however, rather more encouraging. Randomised controlled trials of ischaemic heart disease patients have shown vitamin E (n = 48) and vitamin C (n = 48), respectively, to have a protective effect on the development of nitrate patch tolerance [70, 71]. Similar work on vitamin C supplementation in 20 congestive cardiac failure patients (New York Heart Association grades II-IV) has also demonstrated prevention of an attenuated haemodynamic effect during continuous intravenous GTN administration [72]. Interestingly, this work also revealed a significant reduction in measurable vitamin E levels in the placebo treated group compared to the vitamin C treated group. This suggests that antioxidants work together to reduce oxidative stress so that one antioxidant may regenerate another [72]. The rate of reaction between nitric oxide and superoxide to produce peroxynitrite is notoriously rapid [73]. Rate constants of secondary antioxidants such as vitamins C and E are much lower and this difference is probably overcome by high concentrations of both vitamins.
Results of clinical trials on cardiovascular patients are reflected in work on healthy volunteers: Bassenge et al. demonstrated that following three days of transdermal GTN administration with concurrent high dose vitamin C, vascular tolerance could be ‘eliminated’ in 9 healthy volunteers [74]. A wide variety of pharmacological compounds exhibit antioxidant properties including carvedilol a cardio-selective beta-blocker. A randomised controlled trial of 40 heart failure patients treated with either carvedilol, metoprolol, doxazosin or placebo (n = 40) [75] has demonstrated that carvedilol, a beta-blocker with antioxidant properties, also has protective qualities against the development of nitrate tolerance. A further randomised controlled study of 24 patients with untreated hypertension confirmed the protective benefits of carvedilol vs arotinolol (which does not display antioxidant properties) or placebo [76]. It remains to be seen whether the positive effects of antioxidant use obtained in these trials concur with any future large-scale trials of longer duration.
Conclusion
The evidence supports a free radical mechanism as a key factor in the development of nitrate tolerance, which is predominantly an endothelium dependant process. Specific therapy for this multifactorial phenomenon will, however, probably rely on blocking a combination of pathways [Figure 3], not least of which is the renin angiotensin system [77]. Future work on elucidating these mechanisms should enlighten clinical practice with treatment strategies to reduce or even prevent nitrate tolerance (for a mini review see [78] and [79]). The link between the nitric oxide pathway and endothelial dysfunction may provide profound insights into the cornerstones of cardiovascular disease including insulin resistance and atherosclerosis.
Figure 3.
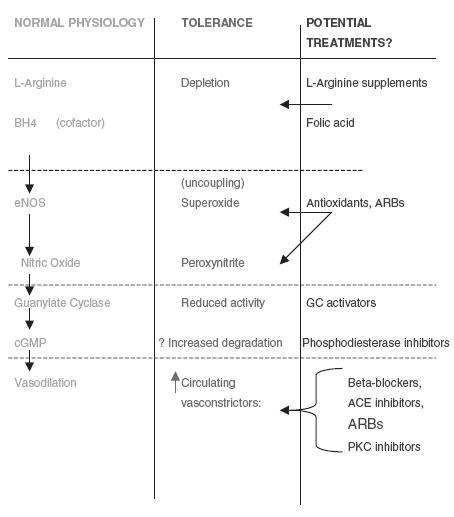
Potential therapy for nitrate tolerance. ENOS, endothelial nitric oxide; ARBs, angiotensin receptor blockers; cGMP, cyclic guanosine mono-phosphate; PKC, Protein kinase C; BH4, tetrahydrobiopterin; GC, Guanylate Cyclase.
References
- 1.Marsh N, Marsh A. A short history of nitroglycerine and nitric oxide in pharmacology and physiology. Clin Exp Pharmacol Physiol. 2000;27:313–319. doi: 10.1046/j.1440-1681.2000.03240.x. [DOI] [PubMed] [Google Scholar]
- 2.Furchgott R, Zawadzki J. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–3766. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 3.Ignarro L. Biological actions and properties of endothelium derived nitric oxide formed and released from artery and vein. Circ Res. 1989;65:1–21. doi: 10.1161/01.res.65.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Moncada S, Higgs A. The L-Arginine-Nitric Oxide pathway. NEJM. 1993;329(27):2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 5.Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature. J Pharmacol Exp Ther. 1995;274:1067–1071. [PubMed] [Google Scholar]
- 6.Kaesemeyer W, Caldwell RB, Huang JZ, Caldwell RW. Pravastatin sodium activates endothelial nitric oxide synthase independent of its cholesterol lowering properties. J Am Coll Cardiol. 1999;31:234–241. doi: 10.1016/s0735-1097(98)00514-2. [DOI] [PubMed] [Google Scholar]
- 7.Abou-Mohammed G, Kaesemeyer WH, Caldwell RB, Caldwell RW. Role of 1-arginine in the vascular actions and development of tolerance to nitroglycerin. Br J Pharmacol. 2000;130:211–218. doi: 10.1038/sj.bjp.0703293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z, Zhang J, Stamler J. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drexlar H, Hornig B. Endothelial dysfunction in human disease. J Mol Cell Cardiol. 1999;31:51–60. doi: 10.1006/jmcc.1998.0843. [DOI] [PubMed] [Google Scholar]
- 10.McVeigh G. Primary nitrate tolerance in diabetes mellitus. Diabetologia. 1994;37:115–117. doi: 10.1007/BF00428787. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura Y, Moss AJ, Brown MW, Kinoshita M, Kawai MD. Long-term nitrate use may be deleterious in ischaemic heart disease: a study using the databases from two large-scale postinfarction studies. Am Heart J. 1999;138:577–585. doi: 10.1016/s0002-8703(99)70163-8. [DOI] [PubMed] [Google Scholar]
- 12.Caramori PR, Adelman AG, Azevedo ER, Newton GE, Parker AB, Parker JD. Therapy with nitroglycerin increases coronary vasoconstriction in response to acetylcholine. J Am Coll Cardiol. 1998;32:1969–1974. doi: 10.1016/s0735-1097(98)00456-2. [DOI] [PubMed] [Google Scholar]
- 13.Gori T, Mak SS, Kelly S, Parker JD. Evidence supporting abnormalities in nitric oxide synthase function induced by nitroglycerin in humans. J Am Coll Cardiol. 2001;38:1096–1101. doi: 10.1016/s0735-1097(01)01510-8. [DOI] [PubMed] [Google Scholar]
- 14.Munzel T, Heitzer T, Kurz S, et al. Dissociation of coronary vascular tolerance and neurohormonal adjustments during long-term nitroglycerin therapy in patients with stable coronary artery disease. J Am Coll Cardiol. 1996;27:297–303. doi: 10.1016/0735-1097(95)00475-0. [DOI] [PubMed] [Google Scholar]
- 15.Fung H. Pharmacokinetics and pharmacodynamics of organic nitrates. Am J Cardiol. 1987;15:4H–9H. doi: 10.1016/0002-9149(87)90543-1. [DOI] [PubMed] [Google Scholar]
- 16.Packer M, Lee W, Kessler P, Gottlieb S, Medina N, Yushak M. Prevention and reversal of nitrate tolerance in patients with congestive heart failure. N Engl J Med. 1987;317:799–804. doi: 10.1056/NEJM198709243171304. [DOI] [PubMed] [Google Scholar]
- 17.Munzel T, Kurz S, Heitzer T, Harrison D. New insights into mechanisms underlying nitrate tolerance. Am J Cardiol. 1996;77:24C–30C. doi: 10.1016/s0002-9149(96)00185-3. [DOI] [PubMed] [Google Scholar]
- 18.Munzel T, Mollnau H, Hartmann M, et al. Effects of a nitrate-free interval on tolerance, vasoconstrictor sensitivity and vascular superoxide production. J Am Coll Cardiol. 2000;36:628–634. doi: 10.1016/s0735-1097(00)00754-3. [DOI] [PubMed] [Google Scholar]
- 19.Hogstedt C, Andersson K. A cohort study on mortality among dynamite workers. J Occup Med. 1979;21:553–556. [PubMed] [Google Scholar]
- 20.Holdright DR, Katz RJ, Wright CA, et al. Lack of rebound during intermittent transdermal treatment with glyeryl trinitrate in patients with stable angina on background beta blocker. Br Heart J. 1993;69:223–227. doi: 10.1136/hrt.69.3.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munzel T, Bassenge E. Long-term angiotensin-converting enzyme inhibition with high-dose enalapril retards nitrate tolerance in large epicardial arteries and prevents rebound coronary vasoconstriction in vivo. Circulation. 1996;93:2052–2058. doi: 10.1161/01.cir.93.11.2052. [DOI] [PubMed] [Google Scholar]
- 22.Munzel T, Giaid A, Kurz S, Stewart DJ, Harrison DG. Evidence for a role of endothelin 1 and protein kinase C in nitroglycerin tolerance. Proc Natl Acad Sci USA. 1995;92:5244–5248. doi: 10.1073/pnas.92.11.5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thadani U, Manyari D, Parker J, Fung H. Tolerance to the circulatory effects of oral isosorbide dinitrate. Rate of development and cross-tolerance to glyceryl trinitrate. Circulation. 1980;61:526–535. doi: 10.1161/01.cir.61.3.526. [DOI] [PubMed] [Google Scholar]
- 24.Thadani U, Fung HL, Darke AC, Parker JO. Oral isosorbide dinitrate in angina pectoris. comparison of duration of action and dose–response relation during acute and sustained therapy. Am J Cardiol. 1982;49:411–419. doi: 10.1016/0002-9149(82)90518-5. [DOI] [PubMed] [Google Scholar]
- 25.Needleman P, Johnson EJ. Mechanism of tolerance development to organic nitrates. J Pharmacol Exp Ther. 1973;184:709–715. [PubMed] [Google Scholar]
- 26.Paker JO, Farrell B, Lahey KA, Rose BF. Nitrate tolerance. the lack of effect of N-acetylcysteine. Circulation. 1987;76:572–576. doi: 10.1161/01.cir.76.3.572. [DOI] [PubMed] [Google Scholar]
- 27.Boesgaard S, Aldershvile J, Poulsen H, Loft S, Anderson ME, Meister A. Nitrate tolerance in vivo is not associated with depletion of arterial or venous thiol levels. Circ Res. 1994;74:115–120. doi: 10.1161/01.res.74.1.115. [DOI] [PubMed] [Google Scholar]
- 28.Parker J, Farrell B, Fenton T, Cohanim M, Parker J. Counter-regulatory responses to continuous and intermittent therapy with nitroglycerin. Circulation. 1991;84:2336–2345. doi: 10.1161/01.cir.84.6.2336. [DOI] [PubMed] [Google Scholar]
- 29.Kim D, Rybalkin SD, Pi X, et al. Upregulation of phosphodiesterase 1A1 expression is associated with the development of nitrate tolerance. Circulation. 2001;104:2338–2343. doi: 10.1161/hc4401.098432. [DOI] [PubMed] [Google Scholar]
- 30.Munzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. J Clin Invest. 1995;95:187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gryglewski R, Palmer R, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 32.Sage PR, de la Lande IS, Stafford I, et al. Nitroglycerin tolerance in human vessels: Evidence for impaired nitroglycerin bioconversion. Circulation. 2000;102:2810–2815. doi: 10.1161/01.cir.102.23.2810. [DOI] [PubMed] [Google Scholar]
- 33.Milone S. Biochemical, hemodynamic, and vascular evidence concerning the free radical hypothesis of nitrate tolerance. J Cardiovasc Pharmacol. 1999;33:685–690. doi: 10.1097/00005344-199905000-00002. [DOI] [PubMed] [Google Scholar]
- 34.White C, Brock T, Chang L, et al. Superoxide and peroxynitrite in atherosclerosis. Proc Natl Acad Sci USA. 1994;91:1044–1048. doi: 10.1073/pnas.91.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mihm M, Coyle C, Jing L, Bauer J. Vascular peroxynitrite formation during organic nitrate tolerance. J Pharmacol Exp Ther. 1999;291:194–198. [PubMed] [Google Scholar]
- 36.Rajagopalan S, Kurz S, Munzel T, Freeman B, Griendling K, Harrison D. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH Oxidase activation. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griendling K, Minieri C, Ollerenshaw J, Alexander R. Angiotensin II stimulates NADH and NADPH oxidase axtivity in cultures vascular smooth muscle cells. Circ Res. 1994;74(6):1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 38.Munzel T, Kurz S, Rajagopalan S, et al. Hydralazine prevents nitroglycerin tolerance by inhibiting activation of a membrane-bound NADH oxidase. J Clin Invest. 1996;98:1465–1470. doi: 10.1172/JCI118935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parker J, Parker A, Farrell B, Parker J. The effect of hydralazine on the development of tolerance to continuous nitroglycerin. J Pharmacol Exp Therapeutics. 1997;280:866–875. [PubMed] [Google Scholar]
- 40.Pritchard K, Grooszek L, Smalley DM, et al. Low-density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res. 1995;77:510–518. doi: 10.1161/01.res.77.3.510. [DOI] [PubMed] [Google Scholar]
- 41.McDonald KK, Zharikov S, Block ER, Kilberg MS. A caveolar complex between the cationic amino acid transporter and endothelial nitric-oxide synthase may explain the ‘arginine paradox’. J Biol Chem. 1997;272:31213–31216. doi: 10.1074/jbc.272.50.31213. [DOI] [PubMed] [Google Scholar]
- 42.Leiper J, Vallance P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovascular Res. 1999;43(3):542–548. doi: 10.1016/s0008-6363(99)00162-5. [DOI] [PubMed] [Google Scholar]
- 43.Boger RH, Bode-Boger SM, Tsao PS, Chan JR, Cooke JP. An endogenous inhibitor of nitric oxide synthase regulates endothelial adhesiveness for monocytes. J Am Coll Cardiol. 2000;36(7):2287–2295. doi: 10.1016/s0735-1097(00)01013-5. [DOI] [PubMed] [Google Scholar]
- 44.MacAllister RJ, Vallance P. Nitric oxide in essential and renal hypertension. J Am Soc Nephrol. 1994;5:1057–1056. doi: 10.1681/ASN.V541057. [DOI] [PubMed] [Google Scholar]
- 45.Drexlar H, Hayes D, Munzel T, Hornig B, Just H, Brunner HR. Endothelial function in chronic congestive cardiac failure. Am J Cardiol. 1992;69:1596–1601. doi: 10.1016/0002-9149(92)90710-g. [DOI] [PubMed] [Google Scholar]
- 46.Ohara Y, Peterson T, Zheng B, Kuo J, Harrison D. Lysophosphatidylcholine increases vascular superoxide anion production via protein kinase C activation. Arterioscler Thromb. 1994;14:1007–1013. doi: 10.1161/01.atv.14.6.1007. [DOI] [PubMed] [Google Scholar]
- 47.Hirata K, Kuroda R, Sakoda T, et al. Inhibition of endothelial nitric oxide synthase activity by protein kinase C. Hypertension. 1995;25:180–185. doi: 10.1161/01.hyp.25.2.180. [DOI] [PubMed] [Google Scholar]
- 48.Sheehy A, Burson M, Black S. Nitric oxide exposure inhibits endothelial NOS activity but not gene expression. Am J Physiol. 1998;274:L833–L841. doi: 10.1152/ajplung.1998.274.5.L833. [DOI] [PubMed] [Google Scholar]
- 49.Munzel T, Li H, Mollnau H, et al. Effects of long-term nitroglycerin treatment on endothelial nitric oxide synthase (NOSIII) gene expression, Nos III-Mediated Superoxide Production, and Vascular NO Bioavailability. CircRes. 2000;86:7–12. doi: 10.1161/01.res.86.1.e7. [DOI] [PubMed] [Google Scholar]
- 50.Vasquez-Vivar J, Kalyanaraman B, Martasek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gruhn N, Aldershvile J, Boesgaard S. Tetrahydrobiopterin improves endothelium-dependant vasodilatation in nitroglycerin-tolerant rats. Eur J Pharmacol. 2001;416:245–249. doi: 10.1016/s0014-2999(01)00879-2. [DOI] [PubMed] [Google Scholar]
- 52.Stroes E, van Faassen E, Yo M, et al. Folic acid reverts dysfunction of endothelial nitric oxide synthase. Circ Res. 2000;86:1129–1134. doi: 10.1161/01.res.86.11.1129. [DOI] [PubMed] [Google Scholar]
- 53.Gori T, Burstein J, Ahmed S, et al. Folic acid prevents nitroglycerin-induced nitric oxide synthetase dysfunction and nitrate tolerance. Circulation. 2001;104:1119–1123. doi: 10.1161/hc3501.095358. [DOI] [PubMed] [Google Scholar]
- 54.Katz R, Levy WS, Buff L, Wasserman AG. Prevention of nitrate tolerance with angiotensin converting enzyme inhibitors. Circulation. 1991;83:1271–1277. doi: 10.1161/01.cir.83.4.1271. [DOI] [PubMed] [Google Scholar]
- 55.Pizzulli L, Hagendorff A, Zirbes M, et al. Influence of captopril on nitroglycerin-mediated vasodilation and development of nitrate tolerance in arterial and venous circulation. Am J Cardiol. 1996;131:342–349. doi: 10.1016/s0002-8703(96)90364-6. [DOI] [PubMed] [Google Scholar]
- 56.Cotter G, Metzkor-Cotter E, Kaluski E, et al. Usefulness of Losartan, Captopril, and Furosemide in preventing nitrate tolerance and improving control of unstable angina pectoris. Am J Cardiol. 1998;82:1024–1029. doi: 10.1016/s0002-9149(98)00548-7. [DOI] [PubMed] [Google Scholar]
- 57.Parker JD, Parker JO. Effect of therapy with an angiotensin-converting enzyme inhibitor on hemodynamic and counterregulatory responses during continuous therapy with nitroglycerin. J Am Coll Cardiol. 1993;21(6):1445–1453. doi: 10.1016/0735-1097(93)90322-r. [DOI] [PubMed] [Google Scholar]
- 58.Dakak N, Makhoul N, Flugelman MY, et al. Failure of captopril to prevent nitrate tolerance in congestive heart failure secondary to coronary artery disease. Am J Cardiol. 1990;66:608–613. doi: 10.1016/0002-9149(90)90489-n. [DOI] [PubMed] [Google Scholar]
- 59.Kurz S, Hink U, Nickenig G, Borthayre A, Harrison D, Munzel T. Evidence for a causal role of the renin-angiotensin system in nitrate tolerance. Circulation. 1999;99:3181–3187. doi: 10.1161/01.cir.99.24.3181. [DOI] [PubMed] [Google Scholar]
- 60.Levine T, Levine A, Keteyian S, Narins B, Lesch M. Reverse remodelling in heart failure with intensification of vasodilator therapy. Clin Cardiol. 1997;20(8):697–702. doi: 10.1002/clc.4960200806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sung C, Aarleth A, Storer B, Ohlstein E. Angiotensin type I receptors mediate smooth muscle proliferation and endothelin biosynthesis in rat vascular smooth muscle. J Pharmacol Exp Ther. 1994;271(1):429–437. [PubMed] [Google Scholar]
- 62.van den Berg R, Haenen GR, van den Berg H, Bast A. Transcription factor NF-kappaB as a potential biomarker for oxidative stress. Br J Nutr. 2001;86(Suppl 1):S121–S127. doi: 10.1079/bjn2001340. [DOI] [PubMed] [Google Scholar]
- 63.Kitamoto S, Egashira K, Kataoka C, et al. Increased activity of nuclear factor-kappaB participates in cardiovascular remodelling induced by chronic inhibition of nitric oxide in rats. Circulation. 2000;102(7):806–812. doi: 10.1161/01.cir.102.7.806. [DOI] [PubMed] [Google Scholar]
- 64.Lipinski B. Pathophysiology of oxidative stress in diabetes mellitus. J Diabetes Complications. 2001;15:203–210. doi: 10.1016/s1056-8727(01)00143-x. [DOI] [PubMed] [Google Scholar]
- 65.Laight DW, Carrier MJ, Anggard EE. Antioxidants, diabetes and endothelial dysfunction. Cardiovascular Res. 2000;47:457–464. doi: 10.1016/s0008-6363(00)00054-7. [DOI] [PubMed] [Google Scholar]
- 66.Kuschi L, Folsom A, Prineas R, Mink P, Wu Y, Bostick RM. Dietary antioxidant vitamins and death from coronary heart disease in postmenopausal women. N Engl J Med. 1996;334:1156–1162. doi: 10.1056/NEJM199605023341803. [DOI] [PubMed] [Google Scholar]
- 67.Blot WJ, Li JY, Taylor PR, et al. Nutritional intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J Natl Cancer Inst. 1993;273:1483–1492. doi: 10.1093/jnci/85.18.1483. [DOI] [PubMed] [Google Scholar]
- 68.Yusuf S, Dagenais G, Pogue J, Bosch J, Davies R, Dagenais G. Vitamin E supplementation and cardiovascular events in high-risk patients: the Heart Outcomes Protection Evaluation Study investigators. N Engl J Med. 2000;342:154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 69.MRC/ BHF. Heart protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):23–33. doi: 10.1016/S0140-6736(02)09328-5. [DOI] [PubMed] [Google Scholar]
- 70.Wantabe H, Kakihana M, Ohtsuka S, Sugishita Y. Randomised, double-blind, placebo controlled study of supplemental vitamin E on attenuation of the development of nitrate tolerance. Circulation. 1997;96:2545–2550. doi: 10.1161/01.cir.96.8.2545. [DOI] [PubMed] [Google Scholar]
- 71.Watanabe H, Kakihana M, Ohtuska S, Sugishita Y. Randomised, double blind, placebo-controlled study of the preventative effect of supplemental oral vitamin C on attenuation of nitrate tolerance. J Am Coll Cardiol. 1998;31:1323–1329. doi: 10.1016/s0735-1097(98)00085-0. [DOI] [PubMed] [Google Scholar]
- 72.Watanabe H, Kakihana M, Ohtuska S, Sugishita Y. Randomised, double-blind, placebo-controlled study of ascorbate on the preventative effect of nitrate tolerance in patients with congestive heart failure. Circulation. 1998;97:886–891. doi: 10.1161/01.cir.97.9.886. [DOI] [PubMed] [Google Scholar]
- 73.Huie R, Padmaja S. The reaction of NO with superoxide. Free Radic Res Commun. 1993;18(4):195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 74.Bassenge E, Fink N, Skatchov M, Fink B. Dietary supplement with vitamin C prevents nitrate tolerance. J Clin Invest. 1998;102:67–71. doi: 10.1172/JCI977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watanabe H, Kakihana M, Ohtuska S, Sugishita Y. Randomised, double-blind, placebo-controlled study of carvedilol on the prevention of nitrate tolerance in patients with chronic heart failure. J Am Coll Cardiol. 1998;32(5):1194–1200. doi: 10.1016/s0735-1097(98)00392-1. [DOI] [PubMed] [Google Scholar]
- 76.Watanabe H, Kakihana M, Ohtuska S, Sugishita Y. Preventative effects of carvedilol on nitrate tolerance – a randomised, double-blind, placebo-controlled comparative study between carvedilol and arotinolol. J Am Coll Cardiol. 1998;32(5):1201–1206. doi: 10.1016/s0735-1097(98)00398-2. [DOI] [PubMed] [Google Scholar]
- 77.Munzel T, Keaney J. Are ACE inhibitors a ‘magic bullet’ against oxidative stress? Circulation. 2001;104:1571–1574. doi: 10.1161/hc3801.095585. [DOI] [PubMed] [Google Scholar]
- 78.Gori T, Parker JD. Nitrate Tolerance: a unifying hypothesis. Circulation. 2002;106:2510–2513. doi: 10.1161/01.cir.0000036743.07406.53. [DOI] [PubMed] [Google Scholar]
- 79.Gori T, Parker JD. The puzzle of nitrate tolerance. Circulation. 2002;106:2404–2408. doi: 10.1161/01.cir.0000036742.52907.91. [DOI] [PubMed] [Google Scholar]