

Abstract
Aims
Voriconazole, a new triazole antifungal agent, is metabolized mainly by cytochrome P450s CYP2C19 and CYP2C9, and also by CYP3A4. The aim of this open-label, placebo-controlled, randomized, three-way crossover study was to determine the effects of cimetidine and ranitidine on the steady-state pharmacokinetics of voriconazole.
Methods
Twelve healthy male subjects received oral voriconazole 200 mg twice daily plus cimetidine 400 mg twice daily, voriconazole 200 mg twice daily plus ranitidine 150 mg twice daily, and voriconazole 200 mg twice daily plus placebo twice daily. Treatment periods were separated by at least 7 days.
Results
When cimetidine was administered with voriconazole, the maximum plasma voriconazole concentration (Cmax) and the area under the plasma concentration–time curve of voriconazole (AUCτ) was increased by 18.3% [90% confidence interval (CI) 6.0, 32.0] and 22.5% (90% CI 13.3, 32.5), respectively. Concomitant ranitidine had no significant effect on voriconazole Cmax or AUCτ. Time of Cmax (tmax) elimination half-life (t1/2) or terminal phase rate constant (kel) for voriconazole were similar in all three treatment groups. Most adverse events were mild and transitory; two subjects were withdrawn due to adverse events.
Conclusions
Coadministration of the histamine H2-receptor antagonists cimetidine or ranitidine does not affect the steady-state pharmacokinetics of voriconazole in a clinically relevant manner.
Keywords: cimetidine, drug interaction, pharmacokinetics, ranitidine, voriconazole
Introduction
Voriconazole is a new triazole antifungal agent, developed as oral and intravenous formulations, with potent activity against a broad spectrum of clinically significant pathogens, including Aspergillus and Candida species [1–3], and emerging fungal pathogens, such as Scedosporium and Fusarium species [4, 5].
The pharmacokinetics of voriconazole have been investigated following single and multiple (7–30 days) doses in both healthy volunteers and patients [6–8]. In vitro and in vivo studies indicate that voriconazole is extensively metabolized by the cytochrome P450 system, mainly by the polymorphically expressed CYP2C19 isoenzyme, by CYP2C9, and to a lesser extent by CYP3A4 [9].
Cimetidine and ranitidine are histamine H2-receptor antagonists, and are widely used in the treatment of gastrointestinal disorders caused by oversecretion of gastric acid. These agents may affect drug absorption by increasing gastric pH, or they may impair hepatic or renal drug clearance by one of several mechanisms including altered cytochrome P450 hepatic drug metabolism. As a nonspecific inhibitor of the cytochrome P450 oxidase system cimetidine therefore has the potential to interact with voriconazole [10]. Cimetidine has been shown to interact with a variety of other drugs as a result of cytochrome P450 inhibition, but its interaction with voriconazole is unknown. Compared with cimetidine, ranitidine has a greatly reduced effect on the cytochrome P450 system [11]; however, the increase in gastric pH caused by either cimetidine or ranitidine could affect the absorption of voriconazole.
This study was therefore designed to investigate the effects of both cimetidine and ranitidine on the steady-state pharmacokinetics of voriconazole, and to evaluate the safety and toleration of voriconazole when coadministered with these histamine H2-receptor antagonists.
Methods
Subjects
Healthy male volunteers aged 18–45 years, weighing 60–100 kg with a body mass index of between 18 and 25 according to Quetelet's Index [weight(kg)/height2(m)], were eligible for inclusion in the study.
Subjects were excluded if they had a history of gastric or duodenal ulcers, allergies (especially drug-related hypersensitivity), any evidence of clinically significant disease, or laboratory abnormalities.
The study design was approved by the Clinical Research Ethics Committee, Anatole France Street, Brussels, Belgium, and all subjects gave written informed consent prior to enrolment. Subjects were asked not to consume caffeine or other methylxanthines, alcohol, or undertake unaccustomed exercise during the 48 h before each dose. In addition, they were advised not to take prescribed or over the counter medicines (except paracetamol) for the duration of the study.
Study design
This was an open, randomized, placebo-controlled, three-way crossover study conducted at the Hôpital Erasme, Brussels, Belgium, in which a minimum of 12 subjects were to be enrolled. In the 3 weeks prior to the start of the study, the subjects underwent physical and laboratory screening tests. The physical examination included measurement of body height and weight, resting blood pressure and pulse rate, and a 12-lead electrocardiogram was recorded. For the laboratory tests, 15 ml blood samples were taken for routine haematological and biochemical testing, a urine sample was also taken for urinalysis and drug screen. The test results were reviewed prior to entry into the study, to ensure that all volunteers enrolled into the study fulfilled the entrance criteria.
Subjects were randomly assigned to receive the following three treatments: voriconazole with cimetidine (cimetidine group), voriconazole with ranitidine (ranitidine group), and voriconazole with placebo (placebo group). Each 8-day treatment period was separated by a 7-day washout period. Treatment periods consisted of: voriconazole 200 mg twice daily + cimetidine 400 mg twice daily, ranitidine 150 mg twice daily, or placebo twice daily (days 1–6); a single dose of voriconazole 200 mg + cimetidine 400 mg twice daily, ranitidine 150 mg twice daily, or placebo twice daily (day 7); and single doses of cimetidine 400 mg, ranitidine 150 mg, or placebo (day 8). Subjects were admitted to the unit during the evening prior to initial dosing, and had a physical examination. Urine was collected and tested for drugs of abuse, and a breath alcohol test was conducted. All enrolled subjects remained resident within the unit from the evening of admission until 32 h after administration of the last dose of each treatment period.
For inclusion in the pharmacokinetic analyses, subjects had to complete at least two treatment periods.
Safety analysis
Safety evaluations were made by recording adverse events throughout the study period, and by assessing the subjects' laboratory test data, blood pressure, and pulse rate. Physical examinations were performed prior to discharge (day 8) of each period, and included cardiovascular, respiratory, abdominal, and skin examinations. A follow-up examination (physical and laboratory assessments) 7–10 days after the last dose in the study was also performed. Laboratory assessments (haematology, clinical chemistry, and urinalysis) were performed by Biorim Laboratory (Brussels, Belgium). Objective test findings that resulted in a change in study treatment dosage or discontinuation were recorded as adverse events. For all adverse events, the investigator recorded his/her opinion of the relationship to study treatment. Reasons for withdrawal from the study were recorded.
Pharmacokinetic sampling
Approximately 7 ml of blood were taken prior to the morning doses on days 1–7 and additionally at regular intervals up to 32 h post dose on day 7. Samples were collected into beadless heparinized tubes, centrifuged at 1500 g at 4 °C for 10 min within 1 h of collection, and stored at −20 °C in screw-capped polypropylene tubes prior to analysis.
Assays
Plasma samples were transported on dry ice to Huntingdon Life Sciences (Huntingdon, UK), where voriconazole levels were measured using a previously validated high-performance liquid chromatography assay [12]. Interbatch imprecision and inaccuracy of the assay were in the ranges 3.3–9.0% and −0.1–0.9%, respectively, over a range of 25–2500 ng ml−1. The limit of quantification was 10 ng ml−1, with imprecision and inaccuracy at this level, as determined from back-calculated concentra-tions of the calibration standards, of 0.9% and 0.4%, respectively.
Parameter calculations
The linear trapezoidal method was used to calculate the area under the plasma concentration–time curve over the dosing interval (AUCτ). Visual inspection of the data was used to determine maximum plasma concentration (Cmax), trough plasma concentration (Cmin), and time to first occurrence of Cmax (tmax). The terminal phase rate constant (kel) was estimated by log-linear regression on those data points visually assessed to be on the terminal log-linear phase. Terminal phase plasma half-life, t1/2, was calculated as 0.693/kel.
Statistical analysis
A sample of 12 subjects was considered sufficient to detect a difference of 20% in AUCτ with probability 0.8 when testing (two-sided) at the 5% level. Natural log-transformed AUCτ and Cmax, untransformed kel, and tmax were subjected to an analysis of variance (anova), allowing for variation due to sequence, subject, period, and treatment. All analyses and tabulations were performed using SAS/STAT® (SAS Institute Inc., North Carolina, USA) software [13]. First-order differential carryover was removed from the model as it was not found to be statistically significant.
Results
Subjects
Thirteen subjects were enrolled into the study and randomized to receive treatment. Two subjects were withdrawn from treatment due to adverse events (see below). One subject was replaced, thus 12 subjects completed the three treatment periods. The age of subjects ranged from 21 to 39 years (mean 29 years), their weights ranged from 67 to 82 kg (mean 75 kg), and their heights from 169 to 187 cm (mean 177 cm). None of the subjects received any other treatment (drug or nondrug) prior to entry into the study, and there were no protocol deviations or violations.
One subject was withdrawn (raised liver transaminases) from treatment during the cimetidine period, having completed the placebo and ranitidine periods. This subject was excluded from analysis for cimetidine effects (but included in the other two treatments). One subject who completed only one treatment period was excluded from all pharmacokinetic analyses.
Thus, 11, 12, and 12 subjects were included in the analyses of cimetidine, ranitidine, and placebo effects, respectively. All 13 subjects were included in the safety analyses.
Voriconazole pharmacokinetics
Cimetidine group
Voriconazole Cmin data indicated that most subjects in the cimetidine group achieved steady state by day 6 (Figure 1). The voriconazole Cmax and AUCτ were approximately 18.3% [90% confidence interval (CI) 6.0, 32.0] and 22.5% (90% CI 13.3, 32.5) higher when voriconazole was coadministered with cimetidine, compared with placebo, respectively. There were no differences between voriconazole tmax or kel for cimetidine and placebo treatments. Voriconazole t1/2 was also unaffected (Table 1).
Figure 1.
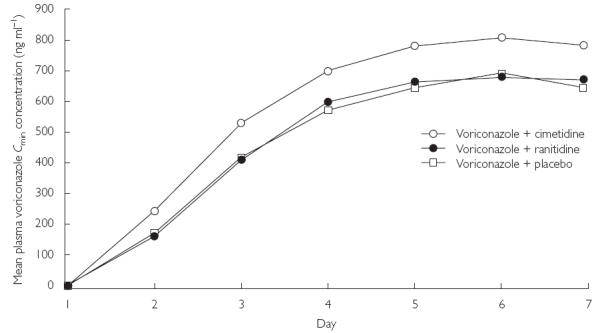
Mean plasma Cmin concentrations of voriconazole.
Table 1.
Summary of mean pharmacokinetic parameters for voriconazole in plasma, following coadministration with placebo, cimetidine, or ranitidine.
| Voriconazole + placebo | Voriconazole + cimetidine | Voriconazole + ranitidine | |||
|---|---|---|---|---|---|
| Parameter | Mean | Mean | Difference/ratio between means*(90% CI) | Mean | Difference/ratio between means*(90% CI) |
| Cmax (ng ml−1)† | 1 906 | 2 254 | 118% | 1 972 | 104% |
| (106, 132) | (93.1, 115) | ||||
| AUCτ (ng·h ml−1)† | 11 131 | 13 634 | 123% | 11 582 | 104% |
| (113, 133) | (96.5, 112) | ||||
| tmax (h)‡ | 1.7 | 1.5 | −0.2 | 1.6 | 0.0 |
| (−0.4, −0.1) | (−0.3, 0.2) | ||||
| kel (h−1)‡ | 0.104 | 0.104 | 0.001 | 0.095 | −0.009 |
| (−0.010, 0.011) | (−0.019, 0.001) | ||||
| t1/2 (h)§ | 6.7 | 6.7 | – | 7.3 | 0.6 |
Ratio (%) between means shown for Cmax and AUCτ, difference between means for tmax, kel, and t1/2.
Geometric mean.
Arithmetic mean.
Harmonic mean.
The decline in mean Cmax up to 32 h post dose was similar in both cimetidine and placebo groups (Figure 2). Throughout the dosing period, mean Cmin values were higher in the cimetidine group compared with the placebo group (Figure 1).
Figure 2.
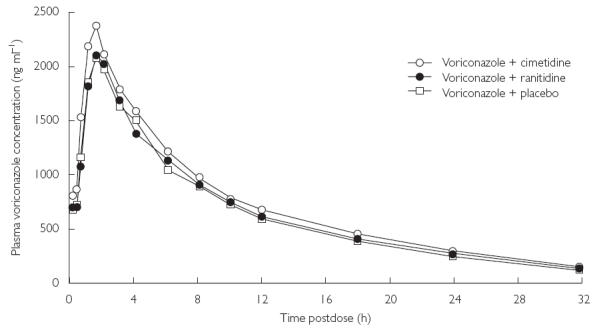
Mean plasma concentrations of voriconazole on day 7.
Ranitidine group
Voriconazole Cmin data indicated that most subjects in the ranitidine treatment group achieved steady state by day 6 (Figure 1). The means of voriconazole Cmax and AUCτ were 3.5% higher (90% CI −6.9, 15.0) and 4.0% higher (90% CI − 3.5, 12.2), respectively, when voriconazole was coadministered with ranitidine, compared with placebo. There were no differences for voriconazole tmax or kel for the comparison between ranitidine and placebo (Table 1).
Adverse events
The number of subjects experiencing adverse events in each treatment group was similar: nine subjects in the cimetidine group, six in the ranitidine group, and eight in the placebo group. However, the total number of adverse events reported was higher in the cimetidine (n = 16) and placebo (n = 20) groups than in the ranitidine group (n = 9).
All but one of the adverse events were assessed as being of mild or moderate intensity. Nine subjects had visual disturbances considered related to treatment: two in the cimetidine group, two in the ranitidine group, and five in the placebo group. With the exception of two events in the placebo group, which were classified as moderate, all the visual disturbances were classified as mild in intensity and all resolved without medical intervention.
Two subjects were withdrawn from the study due to adverse events considered to be treatment related by the investigator: one due to burning and pruritus of the scrotum which emerged during the placebo period, and one due to elevated aspartate (SGOT) and alanine (SGPT) transaminases which emerged during the cimetidine period. Both adverse events resolved following discontinuation of study drug and neither were considered to be serious by the sponsor. A total of six subjects had urine tests positive for haemoglobin, most of which remained positive for the duration of the study. None of these subjects had urinary tract infections or other adverse events that could explain haemoglobin in the urine.
Discussion
This is the first study investigating the possible effects of cimetidine and ranitidine on voriconazole pharmacokinetics. These two histamine H2-receptor antagonists show no clinically relevant effects on steady-state pharmacokinetics of voriconazole 200 mg twice daily when coadministered either as cimetidine 400 mg twice daily or ranitidine 150 mg twice daily.
When coadministered with cimetidine, voriconazole Cmax and AUCτ were approximately 18.3% and 22.5% higher than when administered with placebo. Values for Cmin corresponded with this higher exposure to voriconazole, and were slightly higher in the cimetidine group than in the placebo group throughout the dosing interval. Values for tmax, kel, and t1/2 were similar in both groups. All treatments were equally well tolerated, with similar numbers and types of adverse events.
Coadministration of voriconazole with ranitidine resulted in voriconazole Cmax and AUCτ increases of 3.5% and 4%, respectively, compared with plasma concentrations found during coadministration with placebo. Other pharmacokinetic parameters were also similar between the two groups. There were fewer adverse events reported in the ranitidine group than in the placebo group or the cimetidine group.
None of the differences in the pharmacokinetic parameters of voriconazole observed in the current study was considered to be clinically relevant. Although cimetidine coadministration increased the group mean values for voriconazole Cmax and AUCτ, compared with placebo coadministration, the increase is considered to be within the normal range of values observed following oral administration of voriconazole. This is supported by the fact that the raised Cmax and AUCτ values associated with cimetidine coadministration in this study are comparable to the values observed in another study in healthy volunteers in which voriconazole was administered alone [14].
Cimetidine is known to have clinically significant interactions with a number of agents (e.g. warfarin, theophylline, phenytoin) that are metabolized by the cytochrome P450 system [11, 15]; histamine H2 antagonists, such as cimetidine, inhibit cytochrome P450 enzymes by reversible binding, thereby decreasing the hepatic clearance of these clinically important drugs. Ranitidine binds to the cytochrome P450 enzymes to a lesser extent than cimetidine [11, 16], thus, the smaller increase in voriconazole Cmax and AUCτ resulting from coadministration with ranitidine compared with cimetidine is expected.
In accordance with previous studies, which showed no significant interactions between cimetidine and the first-generation triazole, fluconazole [17, 18], the current study indicates that the new triazole voriconazole can be coadministered with histamine H2 antagonists without the need for dose adjustment.
References
- 1.Cuenca-Estrella M, Rodriguez-Tudela JL, Mellado E, Martinez-Suarez JV, Monzon A. Comparison of the in vitro activity of voriconazole (UK-109,496), itraconazole and amphotericin B against clinical isolates of Aspergillus fumigatus. J Antimicrob Chemother. 1998;42:531–533. doi: 10.1093/jac/42.4.531. [DOI] [PubMed] [Google Scholar]
- 2.Espinel-Ingroff A. In vitro activity of the new triazole voriconazole (UK-109496) against opportunistic filamentous and dimorphic fungi and common and emerging yeast pathogens. J Clin Microbiol. 1998;36:198–202. doi: 10.1128/jcm.36.1.198-202.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandrasekar PH, Manavathu E. Voriconazole: a second-generation triazole. Drugs Today. 2001;37:135–148. doi: 10.1358/dot.2001.37.2.614849. [DOI] [PubMed] [Google Scholar]
- 4.Torre-Cisneros J, Gonzalez-Ruiz A, Hodges MR, Lutsar I. Program and Abstracts of the 38th Annual Meeting of the Infectious Diseases Society of America. New Orleans, LA: 2000. Voriconazole (VORI) for the treatment of S. apiospermum and S. prolificans infection. Abstract 305, September. [Google Scholar]
- 5.Perfect J, Gonzalez-Ruiz A, Lutsar I. Program and Abstracts of the 38th Annual Meeting of the Infectious Diseases Society of America. New Orleans, LA: 2000. Voriconazole (VORI) for the treatment of resistant and rare fungal pathogens. Abstract 303, September. [Google Scholar]
- 6.Sheehan DJ, Hitchcock CA, Sibley CM. Current and emerging azole antifungal agents. Clin Microbiol Rev. 1999;12:40–79. doi: 10.1128/cmr.12.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koltin Y, Hitchcock CA. Progress in the search for new triazole antifungal agents. Curr Opin Chem Biol. 1997;1:176–182. doi: 10.1016/s1367-5931(97)80007-5. [DOI] [PubMed] [Google Scholar]
- 8.Patterson BE, Coates PE. Program and Abstracts of the 35th Interscience Conference on Antimicrobial Agents and Chemotherapy. San Francisco: 1995. UK 109,496, a novel, wide-spectrum triazole derivative for the treatment of fungal infections: Pharmacokinetics in man. Abstract F78, September. [Google Scholar]
- 9.Patterson BE, Roffey S, Jezequel SG, Jones B. Program and Abstracts of the 35th Interscience Conference on Antimicrobial Agents and Chemotherapy. San Francisco: 1995. UK 109,496, a novel, wide-spectrum triazole derivative for the treatment of fungal infections: disposition in man. Abstract F79, September. [Google Scholar]
- 10.Reynolds JC. The clinical importance of drug interactions with antiulcer therapy. J Clin Gastroenterol. 1990;12(Suppl. 2):S54–S63. doi: 10.1097/00004836-199000000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Rendic S, Kajfez F, Ruf H-H. Characterization of cimetidine, ranitidine, and related structures interaction with cytochrome P-450. Drug Metab Dispos. 1983;11:137–142. [PubMed] [Google Scholar]
- 12.Stopher DA, Gage R. Determination of a new antifungal agent, voriconazole, by multidimensional high-performance liquid chromatography with direct plasma injection onto a size-exclusion column. J Chromatogr B Biomed Sci Appl. 1997;691:441–448. doi: 10.1016/s0378-4347(96)00408-2. [DOI] [PubMed] [Google Scholar]
- 13.SAS/STAT User's Guide. 4. Cary, NC: SAS Institute Inc; 1989. Version 6. [Google Scholar]
- 14.Purkins L, Wood N, Greenhalgh K, Allen MJ, Oliver SD. Voriconazole, a novel wide-spectrum triazole: oral pharmacokinetics and safety. Br J Clin Pharmacol. 2003;56(Suppl. 1):10–16. doi: 10.1046/j.1365-2125.2003.01993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin JH, Cocchetto DM, Yeh KC, Duggan DE. Comparative effects of H2-receptor antagonists on drug interaction in rats. Drug Metab Dispos. 1986;14:649–653. [PubMed] [Google Scholar]
- 16.Martinez C, Albet A, Agundez JA, et al. Comparative in vitro and in vivo inhibition of cytochrome P450 CYP1A2, CYP2D6, and CYP3A by H2-receptor antagonists. Clin Pharmacol Ther. 1999;65:369–376. doi: 10.1016/S0009-9236(99)70129-3. [DOI] [PubMed] [Google Scholar]
- 17.Washton H. Review of fluconazole: a new triazole antifungal agent. Diagn Microbiol Infect Dis. 1989;12(Suppl):229S–233S. doi: 10.1016/0732-8893(89)90141-7. [DOI] [PubMed] [Google Scholar]
- 18.Lazar JD, Wilner KD. Drug interactions with fluconazole. Rev Infect Dis. 1990;12(Suppl. 3):S327–S333. doi: 10.1093/clinids/12.supplement_3.s327. [DOI] [PubMed] [Google Scholar]