

Abstract
Aims
Enoxaparin dosing is currently based on total body weight. It is not known how to dose adjust for patients who are overweight or obese. This population pharmacokinetic pharmacodynamic (PKPD) study was undertaken to determine a suitable dosing strategy for such patients.
Methods
Patients admitted to the Royal Brisbane Hospital and prescribed enoxaparin as part of their normal care were eligible for inclusion into the study. Approximately three blood samples were taken per patient to determine anti-Xa concentrations. The occurrence of bruising was also recorded. A population pharmacokinetic–pharmacodynamic analysis using NONMEM was undertaken. Simulations were performed using MATLAB.
Results
Ninety-six patients were recruited in a prospective study. One-third of patients had a body mass index < 24.9 kg m−2, one-third from 25 to 29.9 kg m−2, and one-third> 30 kg m−2. A two-compartment linear model with additive error was fitted to the data. A covariate analysis showed clearance was best described by lean body weight and the central volume compartment by total body weight. The probability of bruising using a logistic regression model was best described by Cmax and age. Simulations suggest that patients over 50 years of age whose total body weight is> 90 kg, or under 50 years of age whose total body weight is> 120 kg are likely to have a smoother concentration–time profile and less bruising if a dose of 100 IU kg−1 (1 mg kg−1) based on lean body weight is administered every 8 h.
Conclusions
Dose adjustments of enoxaparin in obese patients are likely to reduce the prevalence of bruising, although prospective validation of this is required.
Keywords: enoxaparin, obesity, NONMEM, pharmacokinetics, pharmacodynamics, modelling
Introduction
Obesity is a world-wide health problem that is highly correlated with morbidity and mortality from cardiovascular disease [1], diabetes [2], osteoarthritis [3] and depression [4]. The prevalence of obesity is increasing in adults [5], as well as children [6], with less physical activity, more sedentary occupations and a greater use of automation to complete simple tasks considered as possible contributing factors [5]. It follows that many patients requiring medical intervention with enoxaparin for cardiovascular disease or thromboembolic disorders are obese. However, to date, little dosing information has been presented in the literature for this subpopulation. Accurate dosing of low molecular weight heparins (LMWHs) is of significant clinical importance, to ensure efficacy and minimize the risk of adverse bruising or more severe bleeding events [7]. To date, we are aware of one other study of enoxaparin in obese individuals [8]. This study was carried out in healthy volunteers given a dose of 150 IU kg−1, and the authors suggested no dose adjustment was required for obese individuals. The weight descriptor used to dose enoxaparin was not provided. Because of these factors, it has not been possible to extrapolate these findings to the typical patient requiring management with enoxaparin. We therefore aimed to identify a suitable dosing regimen for enoxaparin in obese patients, using a population pharmacokinetic–pharmacodynamic modelling approach.
Methods
Patients admitted to the Royal Brisbane Hospital, a tertiary referral hospital, and prescribed enoxaparin as part of normal clinical care for the treatment of acute coronary syndrome (ACS), deep vein thrombosis (DVT) or prophylaxis of these conditions were eligible for entry into the study. The patients were typical of those normally receiving enoxaparin, as these conditions are routinely managed as in-patients in the study institution. The investigators did not influence dose in any way, with patients receiving 100 IU kg−1 (1 mg kg−1) of enoxaparin twice daily for the treatment of ACS or DVT. Those receiving prophylactic doses were prescribed 4000 IU (40 mg) once daily. Patients considered for inclusion were required to have normal hepatic enzyme concentrations defined by values of liver enzymes within twice the normal range (defined by the pathology department at the study institution). Patients were also required to have normal values of bilirubin, albumin, and an estimated glomerular filtration rate ≥ 72 ml min−1 according to the method of Cockcroft and Gault [9], but where lean body weight was used instead of total body weight [10]. Seventy-two ml min−1 was chosen, as it is similar to the cut-off point for normal renal function used in studies for other renally cleared drugs [11]. Any patient with an intrinsic coagulation disorder (defined as pretreatment abnormalities in their international normalized ratios (INR)> 1.2 or activated partial thromboplastin time (APTT)> 85 s), or recent childbirth was excluded from the study. Ethical approval was obtained from the Royal Brisbane Hospital Research Ethics Committee.
Patients were identified for inclusion through the accident and emergency department and by clinical pharmacists based on the medical and surgical wards. Patients were selected consecutively and with replacement for possible inclusion in the study until the required patient number for the study was achieved. They were individually weighed and height measured by the investigators in order to compute body mass index (BMI). They were stratified so that one-third had a BMI < 24.9 kg m−2 (normal weight), one-third had a BMI from 25 to 29.9 kg m−2 (overweight) and one-third had a BMI> 30 kg m−2 (obese). Approximately three blood samples per patient were collected into 3.2% sodium citrate tubes that were centrifuged and the plasma separated. Samples were assayed within 2–4 h of collection. Anti-Xa activity was determined using a chromogenic substrate assay [12, 13] and concentrations initially recorded in IU ml−1. Concentrations throughout this paper are however, reported in IU l−1. The IL Test™ Heparin assay was used to determine anti-Xa concentration using an ACL–Futura analyser manufactured by Instrument Laboratories, Viale Monza, Italy. Quality controls as indicated in the IL Test™ Heparin were performed by the Queensland Health Pathology Service. Blood samples were taken as soon as the patient was recruited into the study, which could have been after the first or any subsequent dose of enoxaparin.
The exact administration time of enoxaparin was documented on specific trial stickers placed on the patient's medication chart. Personnel responsible for taking blood samples recorded the exact time of their collection. All staff involved with the trial were individually advised about the importance of accurate documentation.
Population analysis
A standard three-stage population analysis for identification of covariates was used [14, 15]. Post hoc estimates of the parameters were obtained from the baseline model and the potential influence of covariates was evaluated using linear regression for continuous covariates and a t-test for discrete covariates. Those with the highest correlation coefficients or where a statistical difference in parameter estimates between two discrete groups occurred were considered for inclusion into the covariate model. The various size descriptors considered were total body weight (WT), lean body weight (LBW) [16], ideal body weight (IBW) [17, 18], adjusted body weight (ABW) [19], allometric scaling of the previous size descriptors [20], body surface area (BSA) [21], and body mass index (BMI) [22] where:
-
1. LBW(males) = 1.1 × WT − 120 × (WT/HT)2
LBW (females)= 1.07 × WT − 148 × (WT/HT)2
2. IBW = 45.4 + 0.89 × (HT − 152.4) + 4.5 (if male)
3. ABW = IBW + CF × (WT − (IBW)
where CF, the correction factor, was set to 0.4 as suggested for aminoglycosides [19].
4. Allometric scaling = Size descriptor3/4
5. BSA = √{[HT × WT]/3600}
6. BMI = [WT]/[HT2 (m)] WT = kg, HT = cm
The population analysis was undertaken using the first order method (FO) in NONMEM (version 5) [23]. Standard goodness of fit criteria such as assessment of the objective function, parameter estimates and their between-subject variability (BSV) and diagnostic plots were used to assess model suitability.
The likelihood ratio test at the α = 0.05 significance level was used to discriminate between nested structural models which corresponds to a reduction of 3.84 units (χ2, P < 0.05) in the objective function with one parameter difference between models. No model was accepted based purely on a change of 3.84 points in objective function due to inherent statistical inaccuracies of this change using the FO method [24]. In addition, the values of the parameter estimates and their BSV were assessed. If parameter estimates did not seem biologically plausible (e.g. a central volume less than plasma volume or if Vd decreased as body weight increased) or could not be estimated by NONMEM, the model was rejected. This principle was also applied to between-subject variance, where very small values, e.g. < 1 × 10−4 or inappropriately large values, e.g.> 1 × 102 were considered a sign of over-parameterization of the model. In addition, the clinical significance of covariates was assessed; defined as a change in the parameter value of> 20% over the range of the usual values of the covariate in question.
Pharmacodynamic model
Bruising incidence, at any location including injection site, was recorded over the duration of the study. Patients were divided into two groups: those that developed bruising, and those that did not. Patients that exhibited one or more bruises were considered as having a bruise. Demographic differences between the groups were considered for inclusion into a logistic regression model as well as the predicted maximum and minimum anti-Xa concentrations (Cmax and Cmin, respectively) for the last dose for each patient.
Dosing simulations
To identify a suitable dosing strategy for enoxaparin in obese patients, a series of stochastic simulations were performed using MATLAB (ver. 6.0.0.88, release 12). The target concentration window to determine an appropriate dosing strategy was initially identified from the literature, although it was anticipated that this could be revised based on data from the pharmacodynamic model. The initial window was set between 500 and 1000 IU l−1 as suggested by the TIMI 11A study [7]. In this study it was noted that the prevalence of side-effects was significantly reduced with no loss in efficacy when 100 IU kg−1 (WT) was administered twice daily rather than 125 IU kg−1 (WT) twice daily [7]. The median trough concentration in the 100 IU kg−1 (WT) arm was 500 IU l−1, compared with 600 IU l−1 in the 125 IU kg−1 (WT) arm. Peak anti-Xa concentrations were reported as 1000 IU l−1 and 1500 IU l−1, respectively.
Results
Ninety-six patients were enrolled in the study, 46 treated for DVT prophylaxis and 50 treated for a variety of clinical indications shown in Table 1. Thirty-two patients had a BMI of < 25, 31 a BMI of 25–29.99 and 33 a BMI> 30 (Table 1). Mean weights (± SD) were 66.2 ± 10.9 kg, 83.3 ± 8.72 kg and 105 ± 17.2 kg in the normal, overweight and obese groups, respectively. Seventy-one males and 25 females were recruited. Age and estimated creatinine clearance did not statistically differ between the groups.
Table 1.
Demographic data.
| Characteristic | Normal (BMI < 25) | Overweight (BMI 25–29.99) | Obese (BMI > 30) | All patients | P |
|---|---|---|---|---|---|
| Number of patients | 32 | 31 | 33 | 96 | |
| Height (cm) | 173 ± 10.5 | 175 ± 7.94 | 173 ± 10.5 | 173 ± 9.62 | NS* |
| Weight (kg) | 66.2 ± 10.9 | 83.3 ± 8.72 | 105 ± 17.2 | 85.0 ± 20.5 | < 0.001* |
| (range) | (41–85) | (67–98) | (76–160) | (41–160) | |
| Body mass index | 22.0 ± 2.53 | 27.0 ± 1.46 | 35.1 ± 2.53 | 28.1 ± 6.27 | < 0.001* |
| (range) | (15.7–24.9) | (25.1–29.1) | (30.8–44.1) | (15.0–44.9) | |
| Age | 52.9 ± 18.0 | 55.1 ± 18.4 | 60.8 ± 18.0 | 56.3 ± 16.9 | NS* |
| Male | 25 (78) | 26 (84) | 20 (61) | 71 (74) | NS** |
| Female | 7 (22) | 5 (16) | 13 (39) | 25 (26) | |
| Indication | |||||
| Deep vein thrombosis | 4 (13) | 3 (10) | 6 (19) | 13 (14) | |
| Pulmonary embolism | 0 (0) | 2 (6) | 3 (9) | 5 (5) | |
| Acute coronary syndrome | 11 (34) | 9 (29) | 12 (36) | 32 (33) | |
| Prophylaxis | 17 (53) | 17 (55) | 12 (36) | 46 (48) | |
| Serum creatinine (µmol l−1) | 68.4 ± 14.5 | 74.0 ± 11.1 | 69.1 ± 14.5 | 70.5 ± 14.0 | NS* |
| Creatinine clearance (ml min−1) | 106 ± 28.2 | 100 ± 35.8 | 93.1 ± 28.2 | 99.6 ± 32.5 | NS* |
Mean ± SD for continuous variables. Number (%) for nominal data.
One-way ANOVA between normal, overweight and obese patient groups.
χ2 Test between normal, overweight and obese patient groups.
Population pharmacokinetics
A two-compartment first order input model with log normal BSV on clearance (CL) and central volume compartment (V2), with additive residual variance was found to be the most suitable baseline structural model. Incorporation of an additive basal anti-Xa component fixed at 20 IU l−1 (as per Schoemaker [25]) or estimated did not improve model fitting further. Final parameter estimates for the baseline model are shown in Table 2, and the weighted residual plot is shown in Figure 1.
Table 2.
Final parameter estimates for baseline model.
| Parameter | Units | Value | SE (CV%) |
|---|---|---|---|
| CL | l h−1 | 0.90 | 7.92 |
| V2 | l | 3.72 | 22.7 |
| Ka | h−1 | 0.181 | 22.7 |
| V3 | l | 12.7 | 48.1 |
| Q | l h−1 | 0.356 | 49.2 |
| ωCL | CV% | 41.7 | 37.9 |
| ωV2 | CV% | 67.6 | 28.7 |
| σ2 | (IU l−1)2 | 6560 | 31.6 |
Parameter estimates expressed in units shown ± SE expressed as % CV.
Figure 1.
Weighted residual plot for baseline first order input additive error model
Sex improved model fitting considerably when included as a covariate, although LBW as a sole covariate on CL was better than sex alone. It should be noted that LBW incorporates sex in its derivation. Incorporation of total body weight as a covariate on V2 further improved model fitting and was included in the final covariate model. The final pharmacokinetic parameters described by this model are presented in Table 3, and the weighted residual plot shown in Figure 2. The residual error of the covariate model decreased marginally compared with the baseline model, and the BSV on CL and V2 reduced by 15% and 14%, respectively. In addition to the statistical improvement in the model, the effect of the covariates was clinically significant with a predicted CL ranging from 0.478 to 1.41 l h−1 and V2 from 2.15 to 8.39 l based on the range of LBW and WT of patients recruited in this study, respectively.
Table 3.
Final parameter estimates for covariate model.
| Parameter | Units | Value | SE (CV%) |
|---|---|---|---|
| CL | l h−1 70 kg−1 (LBW) | 1.03 | 6.80 |
| V2 | l 70 kg−1 (WT) | 3.67 | 24.5 |
| Ka | h−1 | 0.195 | 25.6 |
| V3 | l | 13.1 | 34.4 |
| Q | l h−1 | 0.363 | 33.3 |
| ωCL | CV% | 35.6 | 20 |
| ωV2 | CV% | 58 | 33.3 |
| σ2 | (IU l−1)2 | 6430 | 30 |
Parameter estimates expressed in units shown ± SE expressed as % CV.
Figure 2.
Weighted residual plot for final covariate model
Pharmacodynamic model
Demographic differences that were statistically different between the bruising and non-bruising groups were age, estimated creatinine clearance and the maximum and minimum predicted anti-Xa concentrations, Cmax and Cmin, respectively (Table 4). No difference in WT or BMI was observed, which suggested dosing of 100 IU kg−1 based on WT did not occur in obese patients, and some arbitrary dose adjustment was made by the prescriber.
Table 4.
Differences between bruising and non-bruising patients.
| Bruising | No bruising | P | |
|---|---|---|---|
| Number of patients | 26 | 70 | < 0.0001** |
| Cmax (IU l−1) ± SEM | 890 ± 40.4 | 500 ± 33.3 | < 0.0001* |
| Cmin (IU l−1) ± SEM | 515 ± 38.0 | 178 ± 28.4 | < 0.0001* |
| Total daily dose, IU day−1 ± SD | 154 ± 6.45 | 82 ± 7.22 | < 0.0001* |
| Creatinine clearance, ml min−1 ± SD | 83.5 ± 5.82 | 106 ± 3.78 | < 0.01* |
| Age ± SD | 66.7 ± 2.13 | 52.5 ± 2.05 | < 0.001* |
| Body mass index ± SD | 29.7 ± 1.37 | 27.5 ± 0.71 | 0.14* |
| Weight, kg ± SD | 86.7 ± 4.27 | 84.4 ± 2.4 | 0.632* |
Cmax is the model predicted Cmax. Cmin is the model predicted Cmin. SEM, Standard error of the mean.
χ2 test.
t-Test.
Logistic regression model
The logistic regression model identified Cmax alone improved model fitting compared with Cmin alone or a combination of Cmax and Cmin, with interaction. Further improvements in objective function were seen when age and Cmax were combined. Creatinine clearance did not improve model fitting alone or in combination with age.
The final model was defined by;
where θ1 = − 10.2 (SE = 21.5% CV), θ2 = 1.7 (SE = 21.1% CV) and θ3 = 5.04 (SE = 32.5% CV).
Probability of bruising
Figure 3 shows the empirical probability of developing bruising against the logistic regression model (excluding age). Cmax was categorized into bins sized 200 IU l−1. The number of patients who developed bruising as a fraction of the total number of patients in the bin was plotted against the average concentration of the bin. The probability of bruising was also simulated for patients of variable age (Figure 4). It can be seen that patients of 50 or 70 years of age are more likely to develop bruising compared with those 30 years of age with the same Cmax.
Figure 3.
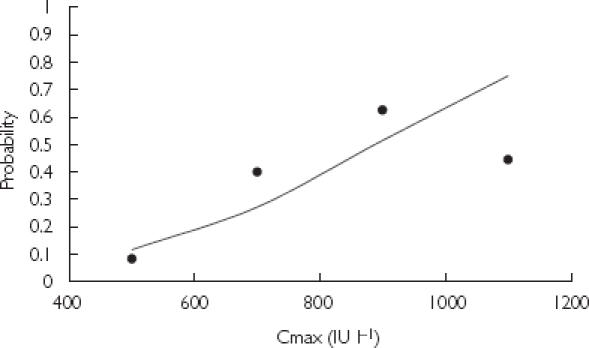
Empirical and predicted probability of bruising based on Cmax. Logistic regression model ( ) and empirical probability of bruising (•).
) and empirical probability of bruising (•).
Figure 4.
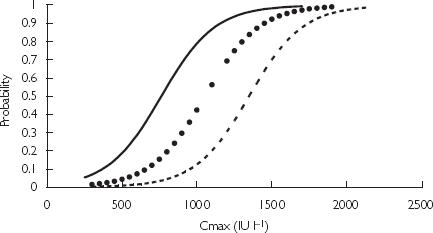
Probability of bruising. Logistic regression model for patients of varying ages; 30 years ( ), 50 years (
), 50 years ( ), 70 years (
), 70 years ( ).
).
Dosing simulations
The Cmax of 1000 IU l−1 identified in Methods predicts a bruising risk of 40% for a 50-year-old patient based on the logistic regression model shown in Figure 4. Since the risk of bruising increases dramatically with age the Cmax was revised to 850 IU l−1 for patients over 50 years of age. This reduces the estimated risk of bruising for a 70-year-old to 50%.
The covariate model was used to simulate anti-Xa concentrations for patients of different weights for the fifth dose. Initial deterministic simulations using the current dosing recommendation of 100 IU kg−1 based on WT every 12 h suggested that the typical patient less than 50 years of age would expect desirable anti-Xa concentrations between 500 and 1000 IU l−1 when total body weight was ≤ 120 kg. For typical patients over 50 years of age, Cmax rises above 850 IU l−1 when total body weight is> 90 kg. Therefore, to identify a suitable dosing strategy for these patients (> 120 kg and < 50 years of age or> 90 kg and> 50 years of age) 5000 steady-state concentration–time profiles were simulated that incorporated variability in CL and V2. Virtual patients were simulated that mimicked the demographic covariates of patients recruited in the original study. The demographics used to simulate these 5000 patients are shown in Table 5. A multivariate lognormal distribution was assumed.
Table 5.
Simulation demographics.
| Males | Females | |
|---|---|---|
| Number | 74% | 26% |
| Weight, kg (%CV) | 86.19 (22.7) | 81.62 (27.8) |
| Height, cm (%CV) | 177.5 (4.28) | 163.3 (4.1) |
| Correlation of weight and height (r) | 0.394 | 0.608 |
r, Correlation coefficient.
The apparent best dosing strategy identified for this patient group (> 120 kg and < 50 years of age or> 90 kg and> 50 years of age) was 100 IU kg−1 based on lean body weight (LBW), administered every 8 h. This is compared with the current dosing strategy of 100 IU kg−1 based on total body weight every 12 h. The criteria for selecting a dosing regimen were based on the percentage of patients that fell within the desired concentration range. The difference between this dosing strategy and current dosing guidelines is graphically represented for patients who weigh> 120 kg in Figure 5a,b shows the 10th, 50th and 90th percentile of the predicted anti-Xa concentration. The 50th percentile is in the range of 700 IU l−1 to 1200 IU l−1 for patients treated with the current dosing guidelines (Figure 5a), and between 700 and 850 IU l−1 for patients managed with the suggested dosing strategy (Figure 5b). Figure 5c,d portrays the percentage of patients> 120 kg whose predicted anti-Xa concentration is> 1000 IU l−1 (solid line),> 850 IU l−1 (dotted line) and < 500 IU l−1 (dashed line) for the current (WT) and new (LBW) dosing strategies, respectively. If current dosing guidelines are used, it is expected that over the dosing interval, 30–70% of patients would expect anti-Xa concentrations> 1000 IU l−1 (Figure 5c), and 45–80%> 850 IU l−1. This can be reduced to 25–40% and 40–60%, respectively, when using the revised dosing strategy (Figure 5d). For patients> 120 kg and> 90 kg, the area under the curve for the 1000 IU l−1 and 850 IU l−1 cut-offs were 2.3 times and 1.9 times greater if the current dosing strategy was used, respectively. The area under the curve for the 500 IU l−1 cut-off was 1.8 times higher for the current WT-based dosing strategy compared with the LBW dosing strategy. The revised dosing strategy was also superior for patients < 120 kg. The number of patients> 1000 IU l−1 and 850 IU l−1 was 1.3 times greater using current WT-based dosing guidelines compared with the LBW dosing strategy, and three times as many patients fell below 500 IU l−1.
Figure 5.
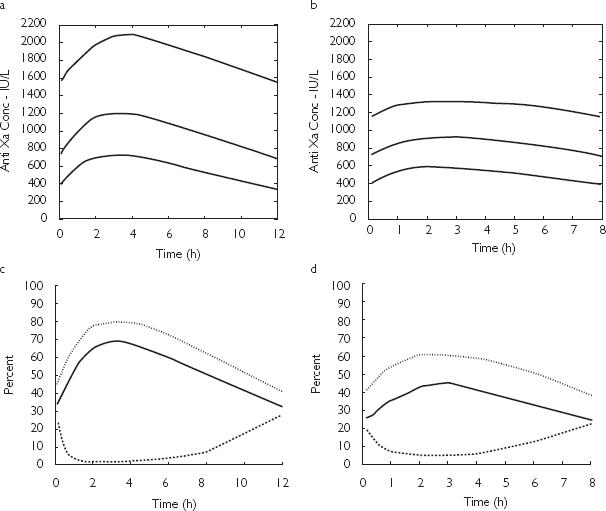
Simulated anti-Xa concentrations and percent of patients falling outside of desired therapeutic range for patients > 120 kg. (a,b) The upper, middle and lower lines represent the 90th, 50th and 10th percentiles, respectively. (c,d) The dashed line represents the percentage of patients whose anti-Xa concentration falls below 500 IU l−1. The solid and dotted lines represent the percentage of patients whose anti-Xa concentration rise above 1000 IU l−1 and 850 IU l−1, respectively. (a) Predicted enoxaparin concentrations (anti-Xa Conc) following a dose of 100 IU kg−1 based on total weight given twice daily. (b) Predicted enoxaparin concentrations (anti-Xa Conc) following a dose of 100 IU kg−1 based on lean body weight given three times daily. (c) Percentage of simulated patients > 1000 IU l−1, >850 IU l−1 or < 500 IU l−1 following a dose of 100 IU kg−1 based on total weight given twice daily. (d) Percentage of simulated patients > 1000 IU l−1, >850 IU l−1 or < 500 IU l−1 following a dose of 100 IU kg−1 based on lean body weight given three times daily.
Discussion
This study has identified that patient characteristics, specifically weight and age, may influence the pharmacokinetics and pharmacodynamics of enoxaparin, since clearance is dependent upon LBW, the central volume of distribution is dependent on WT, and the probability of bruising is dependent on predicted Cmax and age. This is similar to Yee and Duffull [26], who suggested that CL and V for dalteparin were both moderately correlated with WT or ABW, and Sanderink et al. who showed a moderate correlation with CL and WT [8]. Both of these methods used a standard two-stage method rather than a full population approach.
This is the first study to recommend and quantify dose adjustments of enoxaparin for overweight and obese patients. We suggest that the majority of patients would be expected to achieve anti-Xa concentrations between 500 and 850 IU l−1 over the entire dose interval at steady state if either of the following dosing strategies were adopted: (i) all patients are dosed 100 IU kg−1 (1 mg kg−1) based on LBW every 8 h; (ii) patients < 90 kg (> 50 years) or 120 kg (< 50 years) are dosed based on current recommendations of 100 IU kg−1 (1 mg kg−1) total body weight every 12 h, and patients over these nominal values are dosed based on schedule (i).
Schedule (ii) has advantages in that it most closely reflects current practice – with a notional cut-off value added for safety purposes. Schedule (i) is, however, preferable, since all patients can be dosed based on the same criteria, thereby decreasing the risk of adverse medication errors.
The goal of the current work was to achieve comparable concentration–time profiles of enoxaparin in obese patients to the concentration–time profile of patients typically enrolled in the large randomized clinical trials. Based on this analysis, the above dosing recommendations should achieve this. The decrease in the upper end of Cmax to 850 IU l−1 for older patients was recommended based on the occurrence of minor bruising, where this was considered as a biomarker for major bleeding. Although there are no data confirming the predictive nature of minor bruising as a marker for major bleeding, it would seem to be a prudent expectation.
While this work provides some insight into dosing of enoxaparin in the obese, limitations of the study design should be considered. It is recognized that LBW itself has mathematical inconsistencies that could lead to inadequate dosing in morbidly obese subjects [27], and this covariate is more difficult to calculate in the clinical environment. However, the dosing strategy recommended here was based on a patient population where the height and weight ratio did not cause estimates of LBW to decline rapidly [27]. Also, compliance by nursing staff using a thrice daily regimen might be worse than a twice daily regimen, although no evidence of this could be found in the literature. It should be noted that if one dose were missed on a thrice daily regimen, the risk of Cmin falling below that required for efficacy would be less than if a dose were missed on a twice daily regimen. Therefore the thrice daily regimen is actually more forgiving than a twice daily regimen. Also, it should be noted that the bruising rate for patients recruited in this study was higher than that found in other studies, and the dosing strategy recommended is in part based on a pharmacodynamic model to predict the risk of bruising. Finally, it is recognized that data accuracy is dependent upon individuals involved in its collection. Undoubtedly an element of variability is associated with these processes; however, accurate estimates of pharmacokinetic parameters based on asymptotic standard errors were obtained.
In summary, we have identified that lean body weight is an important covariate when dosing enoxaparin, and suggest dose adjustments are required for patients that are overweight or obese, and particularly the elderly. The dosing regimen suggested warrants further investigation.
Acknowledgments
We wish to acknowledge Aventis, the Pharmaceutical Society of Australia and the University of Queensland who provided financial support to complete this study. We also wish to acknowledge the pharmacy, phlebotomy, haematology and nursing staff at the Royal Brisbane Hospital for their various roles during the study period.
References
- 1.Lurbe E, Alvarez V, Redon J. Obesity, body fat distribution, and ambulatory blood pressure in children and adolescents. J Clin Hypertens. 2001;3:362–367. doi: 10.1111/j.1524-6175.2001.00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson JW, Konz EC. Obesity and disease management: effects of weight loss on comorbid conditions. Obes Res. 2001;9:326S–334S. doi: 10.1038/oby.2001.138. [DOI] [PubMed] [Google Scholar]
- 3.Toda Y, Toda T, Takemura S, Wada T, Morimoto T, Ogawa R. Change in body fat, but not body weight or metabolic correlates of obesity, is related to symptomatic relief of obese patients with knee osteoarthritis after a weight control program. J Rheumatol. 1998;25:2181–2186. [PubMed] [Google Scholar]
- 4.Troisi A, Scucchi S, San Martino L, Montera P, D’Amore A, Moles A. Age specificity of the relationship between serum cholesterol and mood in obese women. Physiol Behav. 2001;72:409–413. doi: 10.1016/s0031-9384(00)00422-4. [DOI] [PubMed] [Google Scholar]
- 5.National Health and Medical Research Council. Acting on Australia's weight—a strategic plan for the prevention of overweight and obesity. Australian Government Publishing Services; 1997. [28th September 2001]. Available at http://www.aihw.gov.au/publications/health/hsvd.html. [Google Scholar]
- 6.Magarey AM, Daniels LA, Boulton TJC. Prevalence of overweight and obesity in Australian children and adolescents: reassessment of 1985 and 1995 data against new standard international definitions. Med J Aust. 2001;174:561–554. doi: 10.5694/j.1326-5377.2001.tb143435.x. [DOI] [PubMed] [Google Scholar]
- 7.Dose-ranging trial of enoxaparin for unstable angina: results of TIMI 11A. J Am Coll Cardiol. 1997;29:1474–1482. [PubMed] [Google Scholar]
- 8.Sanderlink G, Le Liboux A, Jariwala N, et al. Enoxaparin pharmacokinetics and pharmacodynamics in obese subjects. J Am Coll Cardiol. 2001;37:229A. (Abstract) [Google Scholar]
- 9.Cockroft DW, Gault H. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 10.Pesola GR, Akhavan I, Madu A, Shah NK, Carlon GC. Prediction equation estimates of creatinine clearance in the intensive care unit. Intensive Care Med. 1993;19:39–43. doi: 10.1007/BF01709276. [DOI] [PubMed] [Google Scholar]
- 11.Begg EJ, Barclay ML, Duffull SB. A suggested approach to once-daily aminoglycoside dosing. Br J Clin Pharmacol. 1995;39:605–609. [PMC free article] [PubMed] [Google Scholar]
- 12.Teien AN, Lie M. Evaluation of an amidolytic heparin assay method: increased sensitivity by adding purified antithrombin III. Thromb Haemost. 1977;10:399. doi: 10.1016/0049-3848(77)90150-5. [DOI] [PubMed] [Google Scholar]
- 13.Laposata M, Green D, Van Cott EM, Barrowcliffe TW, Goodnight SH, Sosolik RC. College of American Pathologists conference XXXI on laboratory monitoring of anticoagulant therapy. The clinical use and laboratory monitoring of low-molecular-weight heparin, danaparoid, hirudin and related compounds, and argatroban. Arch Pathol Lab Med. 1998;122:799–807. [PubMed] [Google Scholar]
- 14.Maitre PL, Bührer M, Thomson D, Stanski DR. A three-step approach combining Bayesian regression and NONMEM population analysis: application to midazolam. J Pharmacokinet Biopharm. 1991;19:377–384. doi: 10.1007/BF01061662. [DOI] [PubMed] [Google Scholar]
- 15.Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic-pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm. 1992;20:511–528. doi: 10.1007/BF01061469. [DOI] [PubMed] [Google Scholar]
- 16.Cheymol G. Effects of obesity on pharmacokinetics. Clin Pharmacokinet. 2000;39:215–231. doi: 10.2165/00003088-200039030-00004. [DOI] [PubMed] [Google Scholar]
- 17.Devine B. Case study number 25 gentamicin therapy. DICP. 1974;8:650–655. [Google Scholar]
- 18.New weight standards for men and women. Stat Bul. 1959;40:1–3. [Google Scholar]
- 19.Bauer LA, Edwards WAD, Dellinger EP, Simonowitz DA. Influence of weight on aminoglycoside pharmacokinetics in normal weight and morbidly obese patients. Eur J Clin Pharmacol. 1983;24:643–647. doi: 10.1007/BF00542215. [DOI] [PubMed] [Google Scholar]
- 20.Holford NHG. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30:329–332. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 21.Du Bois D, Du Bois EF. Clinical calorimetry. Tenth paper. A formula to estimate the approximate surface area if height and weight be known. Arch Intern Med. 1916;17:863. [PubMed] [Google Scholar]
- 22.World Health Organization. Report of a WHO Consultation on obesity: preventing and managing the global epidemic. Geneva: World Health Organization; 1998. [PubMed] [Google Scholar]
- 23.Beal SL, Sheiner LB. NONMEM user's guide. San Francisco: University of California at San Francisco; 1992. Part I. [Google Scholar]
- 24.Wahlby U, Jonsson EN, Karlsson MO. Assessment of actual significance levels for covariate effects in NONMEM. J Pharmacokinet Pharmacodyn. 2001;28:231–252. doi: 10.1023/a:1011527125570. [DOI] [PubMed] [Google Scholar]
- 25.Schoemaker RC, Cohen M. Estimating impossible curves using NONMEM. Br J Clin Pharmacol. 1996;42:283–290. doi: 10.1046/j.1365-2125.1996.04231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yee J, Duffull S. The effect of body weight on dalteparin pharmacokinetics. Eur J Clin Pharmacol. 2000;56:293–297. doi: 10.1007/s002280000141. [DOI] [PubMed] [Google Scholar]
- 27.Green B, Duffull S. Caution when using lean body weight as a size descriptor for obese subjects. Clin Pharmacol Ther. 2002;72:743–744. doi: 10.1067/mcp.2002.129306. [DOI] [PubMed] [Google Scholar]
- 28.Urquhart J, De Klerk E. Contending paradigms for the interpretation of data on patient compliance with therapeutic drug regimens. Stat Med. 1998;17:251–267. doi: 10.1002/(sici)1097-0258(19980215)17:3<251::aid-sim762>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]