

Abstract
Aims
To develop a population pharmacokinetic model for paclitaxel in the presence of a MDR modulator, zosuquidar 3HCl.
Methods
The population approach was used (implemented with NONMEM) to analyse paclitaxel pharmacokinetic data from 43 patients who received a 3-h intravenous infusion of paclitaxel (175 mg m−2 or 225 mg m−2) alone in cycle 2 or concomitantly with the oral administration of zosuquidar 3HCl in cycle 1.
Results
The structural pharmacokinetic model for paclitaxel, accounting for the Cremophor EL™ impact, was a three-compartment model with a nonlinear model for paclitaxel plasma clearance (CL), involving a linear decrease in this parameter during the infusion and a sigmoidal increase with time after the infusion. The final model described the effect of Zosuquidar 3HCl on paclitaxel CL by a categorical relationship. A 25% decrease in paclitaxel CL was observed, corresponding to an 1.3-fold increase in paclitaxel AUC (from 14829 µg l−1 h to 19115 µg l−1 h following paclitaxel 175 mg m−2) when zosuquidar Cmax was greater than 350 µg l−1. This cut-off concentration closely corresponded to the IC50 of a sigmoidal Emax relationship (328 µg l−1). A standard dose of 175 mg m−2 of paclitaxel could be safely combined with doses of zosuquidar 3HCl resulting in plasma concentrations known, from previous studies, to result in maximal P-gp inhibition.
Conclusions
This analysis provides a model which accurately characterized the increase in paclitaxel exposure, which is most likely to be due to P-gp inhibition in the bile canaliculi, in the presence of zosuquidar 3HCl (Cmax > 350 µg l−1) and is predictive of paclitaxel pharmacokinetics following a 3 h infusion. Hence the model could be useful in guiding therapy for paclitaxel alone and also for paclitaxel administered concomitantly with a P-gp inhibitor, and in designing further clinical trials.
Keywords: paclitaxel, P-glycoprotein modulator, population pharmacokinetics, Zosuquidar 3HCl
Introduction
Paclitaxel is indicated for the treatment of breast and ovarian carcinoma as well as in nonsmall cell lung cancer. The dose commonly used in clinical practice varies from 135 mg m−2 to 250 mg m−2 and is administered by intravenous (i.v.) infusions of 1–24 h duration. Owing to its poor solubility, paclitaxel is formulated in a mixture of Cremophor EL™ (CrEL, polyoxyethylated castor oil derivative) and dehydrated ethanol USP (1 : 1, v/v) [1]. Following administration, paclitaxel undergoes extensive metabolism and billiary excretion [2]. The three main metabolites are formed via CYP2C8 and CYP3A4-mediated pathways and are believed to be 10–40-fold less cytotoxic than paclitaxel [1]. The reported efficacy of paclitaxel as a single agent varies substantially, (for example between 6 and 50% in anthracycline-resistant breast cancer) and resistance to therapy is observed in a substantial percentage of cases [3]. In 1999, Dumontet et al.[4] published a review of the mechanisms of action and resistance to antitubulin agents. These authors determined that, in addition to multidrug resistance (involving P-gp), other possible mechanisms of resistance to paclitaxel could occur, (e.g. an alteration in the interaction between the drug and its microtubular target). Paclitaxel is a known substrate of P-gp and it has been shown that basal expression of P-gp plays a role in the resistance of cancer cells to paclitaxel by its action as an efflux pump [5]. The involvement of other multidrug resistance-associated proteins, MRP1 and MRP2, is still unclear but neither is thought to play a role in the loss of efficacy of paclitaxel [5, 6].
Efforts at overcoming multidrug resistance have primarily focused on attempts to inhibit P-gp. Since the discovery that verapamil and cyclosporin A were P-gp inhibitors (Ki about 5–10 µM) [7] and able to reverse P-gp mediated resistance, a considerable amount of research has been performed. Verapamil and cyclosporin A, considered first-generation MDR modulators, could not be developed clinically as such because of tolerability issues but second-generation (e.g. PSC-833; valspodar) [8–10] and third generation (e.g. GF120918 [11], XR9576 [tariquidar][12], VX-710 [biricodar][13, 14], LY335979 [zosuquidar 3HCl][15]) compounds are in clinical development. These third-generation molecules are noncytoxic, bind with high affinity to P-gp (Ki about 20–100 nM) [7], and demonstrate potent in vitro reversible activity against MDR human tumour cell lines. P-gp protects the body from toxicity from xenobiotics or endogenous substances by: (a) preventing their absorption from the intestinal tract; (b) preventing their distribution into specific organs, (e.g. by its action in the blood–brain barrier); and (c) promoting their clearance (e.g. by its action in the bile canaliculi and the kidney). This latter role may explain the results from clinical trials investigating the safety of chemotherapy combined with a P-gp inhibitor such as PSC833 [8–10], GF120919 [11] or VX-710 [13, 14]. These trials showed clinically significant pharmacokinetic interactions [7] characterized by a decrease in the clearance of the anticancer drug and hence increased exposure. The interpretation of subsequent phase II and III clinical trials was complicated since it was not possible to administer the same dose of chemotherapy in the presence and the absence of the MDR modulator.
Zosuquidar 3HCl [16–19] is a novel third-generation potent (Ki = 59 nM) MDR modulator and a specific inhibitor of P-gp but not of MRP1 or MRP2. It was first identified by its ability to restore doxorubicin sensitivity to P-gp-expressing cell lines, thus enhancing the survival time of mice inoculated with P388/ADR cells. It is known that P-gp and cytochrome P450 show substrate commonality and therefore in vitro experiments were performed to study the interaction between LY335979 and cytochrome P450. The results showed that Zosuquidar 3HCl had no affinity for liver enzymes CYP3A, CYP1A, CYP2C9, CYP2D6 at nanomolar concentrations [18]. In the present phase I dose escalation clinical trial, paclitaxel pharmacokinetics were studied in the presence and absence of zosuquidar 3HCl.
It could be hypothesized that the properties of Zosuquidar 3HCl (no interaction with the liver enzymes at nM levels and favorable PK/PD characteristics [15]) should lead to a smaller degree of pharmacokinetic interaction than that observed with PSC-833 and VX-710. This should enable clinically indicated doses of paclitaxel, to be administered allowing more straightforward interpretation of the clinical trial results. The pharmacokinetic data collected were analysed using NONMEM [20] in order to generate a population PK model that could describe and predict paclitaxel PK in the presence and absence of MDR modulator.
Methods
Patient selection
Forty-three patients (13 males, 30 females) with a histological or cytological diagnosis of metastatic or locally advanced cancer (not amenable to surgery or radiotherapy of curative intent) who had failed conventional therapy, had disease considered refractory to standard chemotherapy regimens, or had disease for which no standard chemotherapy was available, were enrolled into the study. This trial was approved by the relevant ethics committee (ICRF Medical Oncology Unit, Oxford) at the participating medical institutions and sponsored by Eli Lilly. All participants gave written informed consent and the study was conducted in accordance with the ethical principles of the most recent version of the Declaration of Helsinki. Patients were at least 18 years of age, and met other eligibility requirements, which included having received no more than two prior regimens (including adjuvant therapy), having a performance status of 0–2 on the Eastern Cooperative Oncology Group (ECOG) scale and an estimated life expectancy of at least 12 weeks. Prior radiation therapy or chemotherapy must have been completed at least 3 weeks prior to a patient enrolling into the study (6 weeks if the prior treatment was nitrosourea or mitomycin C and 4 weeks if radiation therapy of more than 25% of bone marrow) and patients must have recovered from the acute effects of that therapy. Adequate organ function was required with absolute granulocytes count (AGC) ≥ 1.5 GI l−1 (= 1.5 × 109 l−1), platelets (PTS) ≥ 100 GI l−1, bilirubin (BIL) < 1.5 mg dl−1 (25.5 µmol l−1), alanine transaminase (ALT) and aspartate transaminase (AST) < 2.5 times upper limit of normal. Inclusion criteria also included a serum creatinine (Crserum) ≤ 1.5 mg dl−1 (133 µmol l−1) or a creatinine clearance (CrCL) > 40 ml min−1. A description (demographic and laboratory values) of the patient population enrolled in this study is presented in Table 1.
Table 1.
Information regarding the demographic and biological characteristics (median (range)) of the population enrolled in the study.
| Covariates | Cycle 1 | Cycle 2 |
|---|---|---|
| Number of patients | 43 | 35 |
| Gender | Male 13, female 30 | Male 11, female 24 |
| Smoking status | Smoker 13, nonsmoker 30 | Smoker 12, nonsmoker 23 |
| Age (years) | 59 (29–77) | 61 (29–77) |
| Serum albumin concentration (g l−1) | 41 (26–50) | 40 (26–46) |
| Alkaline phosphatase (U l−1) | 152 (53–1320) | 185 (61–740) |
| Alanine amino transaminase (U l−1) | 17 (7–107) | 23 (8–157) |
| Aspartate amino transaminase (U l−1) | 24 (11–85) | 24 (13–74) |
| Total bilirubin (µmol l−1) | 7 (1–14) | 6 (2–12) |
| Body mass index (kg m−2) | 23.66 (16.65–35.21) | 23.26 (16.55–32.34) |
| Body surface area (m2) | 1.74 (1.38–2.3) | 1.71 (1.7–2.29) |
| Creatinine clearance* (ml min−1) | 75.4 (33.54–148.65) | 79.4 (34.9–225) |
| Height (cm) | 166 (148–197) | 166 (148–197) |
| Phosphate plasma concentration (mmol l−1) | 1.09 (0.59–1.45) | 1.05 (0.55–1.53) |
| Potassium plasma concentration (mEq l−1) | 4.3 (2.7–5.4) | 4.4 (2.9–5.3) |
| Plasma plasma protein (g l−1) | 72 (62–84) | 70 (61–84) |
| Serum creatinine concentration (µmol l−1) | 81 (48–125) | 77 (44–125) |
| Sodium plasma concentration (mEq l−1) | 140.0 (126–145) | 140 (130–154) |
| White blood cells counts (GI l−1) | 8.3 (3.9–41.2) | 6.3 (3.17–27.7) |
| Weight (kg) | 66.7 (45.1–96) | 66.7 (45.1–96) |
based on the Cockcroft & Gault equation X = 1.23 (male) and 1.04 (female).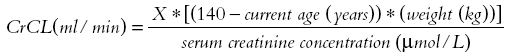
Study design and treatment
Patients were entered in cohorts of a least three and received in cycle 1 a combination of zosuquidar 3HCl (multiple oral administration) and paclitaxel (3 h i.v. infusion), whereas in cycle 2 (21 days later), paclitaxel was administered as a single agent. In cohort 1, zosuquidar 3HCl and paclitaxel doses were 100 mg m−2 and 175 mg m−2, respectively. The dose of both therapeutic agents was escalated through the cohorts as described in Table 2. In addition, the administration schedule was amended in the course of the study based on information from this and other ongoing clinical trials. The paclitaxel dose was adjusted based on the nadir of counts from the preceding cycle [21].
Table 2.
Dose escalation scheme
| Cohort number | Number of patients | Zosuquidar 3HCl dose (n*) | Time between zosuquidar 3HCl doses (h) | Paclitaxel dose (mg m−2) given on day X of the cycle x hour after the nthzosuquidar 3HCl dose |
|---|---|---|---|---|
| 1 | 9 | 100 mg m−2 (10) | 8 | 175 on day 3, 2 h (7th) |
| 2 | 3 | 200 mg m−2 (7) | 12 | 175 on day 3, 2 h (5th) |
| 3 | 4 | 300 mg m−2 (7) | 12 | 175 on day 3, 2 h (5th) |
| 4 | 8 | 300 mg m−2 (3**) | 12 | 175 on day 1, 0.5 h (2nd) |
| 5 | 7 | 250 mg m−2 (3) | 12 | 175 on day 1, 0.5 h (2nd) |
| 6 | 3 | 450 mg (2) | 12 | 225 on day 1, 1 h (1st) |
| 7 | 9 | 500 mg (2+) | 12 | 225++ on day 1, 1 h (1st) |
n, total number of doses of zosuquidar 3HCl administered in cycle1;
only 2 doses for id 153, 250; +only 1 dose for id 200; ++175 mg m−2 for id 165, 166 and 256. From cycle 1 to cycle 2, five patients had their paclitaxel dose reduced (1 from 225 to 175 mg m−2 and 4 from 175 to 135 mg m−2).
Blood sampling
Plasma samples were obtained to determine the pharmacokinetics of zosuquidar in cycle 1 and paclitaxel in cycles 1 and 2. Paclitaxel plasma samples were taken at predose and 1, 2, 3 (end of the infusion), 3.25, 3.5, 4, 6, 8, 24 and 48 h after the start of infusion. Zosuquidar plasma samples were taken following the seventh (cohort 1), fifth (cohorts 2 and 3), second (cohorts 4 and 5) or first (cohorts 6 and 7) zosuquidar dose at predose, 1, 2, 3, 4, 6, 8 h for cohort 1–3; predose and 0.5, 1.5, 2.5, 3.5, 5.5, 8.5 h for cohorts 4 and 5; predose and 1, 2, 3, 4, 4.25, 4.5, 5, 7, 10, 12, 24 h for cohorts 6 and 7.
Drug analysis
Zosuquidar concentrations were measured in heparinized plasma samples (following a solid phase extraction with a LMS column) using a validated (20–2000 ng ml−1, with the lower limit of quantification (LOQ) equal to 20 ng ml−1) reverse phase HPLC assay (mobile phase 72 : 28 v/v acetonitrile: 35 mmol L−1 ammonium acetate (pH 4.8); column Zorbax RX-C8, 4.6 × 150 mm, 5 µm (25 °C); flow rate 1.0 ml min−1) using fluorescence detection (excitation and emission wave length, 240 and 415 nm, respectively). Human plasma samples were analysed for paclitaxel using a validated (10–10000 ng ml−1, with LOQ equal to 10 ng ml−1) isocratic reversed-phase HPLC method using UV detection at 227 nm as described previously [22]. Based on the quality control samples, the overall relative standard deviation (an index of precision) was less than 3.5% and 5.8% for zosuquidar and paclitaxel, respectively. The overall relative error (an index of accuracy) was less than 1.5% and 2.0% for zosuquidar and paclitaxel, respectively.
Data analysis
The population pharmacokinetic model for paclitaxel was developed using a population approach implemented in the NONMEM program, version V level 1.1 [20, 23–26] using the first order conditional estimation method with interaction.
A basic structural pharmacokinetic model (defined as describing adequately the mean population and individual tendencies without inclusion of any covariates) for paclitaxel when given on its own was developed using paclitaxel data collected during cycle 2 (in the absence of zosuquidar 3HCl). The nonlinearity observed in paclitaxel plasma pharmacokinetics [27–31] is known to be due to cremophore EL (CrEL) [32–35]. CrEL plasma concentration data were not collected during this study, which prevented the determination of an optimal model. However, based on a consideration of the effect of CrEL on paclitaxel plasma pharmacokinetics [34–36], a basic structural model accounting for the nonlinearity was developed and compared with a simple linear three-compartment pharmacokinetic model.
Hennigsson et al.[36] presented a mechanism-based model for paclitaxel, which demonstrates the linearity of the pharmacokinetics of paclitaxel unbound plasma concentration (Cu) and the inverse relationship between paclitaxel unbound fraction (fu) and CrEL concentration (due to the binding of paclitaxel to CrEL) [35]. In this present study, a true mechanistic model could not be developed because of the absence of paclitaxel total blood and unbound plasma concentrations (Cb and Cu) and CrEL concentrations. In that context, the approach chosen was to build a model taking into account that: (a) paclitaxel plasma CL decreases with time during the infusion when CrEL concentrations were increasing [as paclitaxel binds to CrEL less of the former available for elimination]; and (b) paclitaxel plasma CL increases with time after the end of the infusion when CrEL is eliminated from the body.
The observations were expressed as follows:
| (1) |
where, OBSij is the jth observation (paclitaxel plasma concentration) in the ith individual; θi is the set of PK parameters for the ith individual; Di is the administered dose for the ith individual; tij is the time of collection, after administration, of the jth observation in the ith individual and ∈ij is the residual shift of the observation from the model prediction (random variable assumed to be symmetrically distributed around 0 with variance σ2). Both a proportional and combined additive and proportional residual error model were tested, and a simple proportional error model was found to be appropriate.
For each pharmacokinetic parameter, the possibility of estimating interindividual variability as well as interoccasion variability (within patient variability in PK parameters from cycle 1 to cycle 2) was tested according to the following equation [26].
| (2) |
where, pi is an arbitrary PK parameter of the ith individual; ppop is the mean population estimate; Occ1and Occ2 are fixed to 1 and 0, respectively, if cycle equals 1 and vice versa if cycle equals 2; κ1i, κ2i and ηi are random variables, representing the shift of pi from one occasion to another (interoccasion variability, IOV) and the shift of pi from ppop (inter individual variability; IIV), respectively. These random variables are assumed to be symmetrically distributed around 0 with identical variance for κ1i and κ2i denoted by π2 and with variance-covariance matrix Ω for ηi denoted by diagonal elements (,…, , m being the number of parameters).
Paclitaxel data collected during cycle 1 (in the presence of zosuquidar) were added to the data set and the covariate analysis was carried out in two steps. The impact of zosuquidar on individual paclitaxel PK parameters was assessed firstly through graphical exploratory plots and secondly by testing covariate relationships in NONMEM. The other important covariates and their functional relationship to paclitaxel PK parameters were selected using a stepwise generalized additive model (GAM) based on pi estimates from the basic population model as dependent variables. This GAM analysis was performed outside of NONMEM with the Xpose program, version 2.0 [37]. The covariate relationships selected by the GAM analysis were tested for statistical significance with NONMEM and the following selection criterion was applied. The difference in the minimum value of the objective function between a model with and without a specific covariate relationship was compared with a χ2 distribution in which a difference greater than or equal to 7.88 points was taken as significant at P < 0.005 (for 1 degree of freedom).
Model selection was based on a number of criteria, such as the exploratory analysis of the goodness of fit plots, the estimates and the confidence intervals of the fixed and random parameters, and the minimum value of the objective function. Finally, based on mean and variance parameters from the final model, 1000 Monte-Carlo simulations were carried out in order to generate the population 95% prediction interval.
Noncompartmental analysis (WinNonlin Professional Version 1.5, Pharsight Corporation) of zosuquidar plasma concentration vs time profiles was carried out but is not the objective of this study. As the area under zosuquidar concentration vs time curve during the dose interval (AUCτ) and the observed maximal concentration (Cmax) were correlated, the latter parameter was chosen to study the potential impact of zosuquidar on paclitaxel pharmacokinetics. Previously reported values for zosuquidar pharmacokinetic parameters (following i.v. administration) are a plasma clearance of 90 l h−1 (CV 35.6%), a volume of distribution at steady state of 1105 l (CV 39.6%), distribution and elimination half-lives of 0.7 h (CV 40.1%) and 20 h (58.9%), respectively (geometric mean and coefficient of variation reported (n = 28)) [15].
Results
A basic structural PK model for paclitaxel, which mimicked the impact of CrEL on paclitaxel PK by describing a paclitaxel plasma CL changing with time (model C below), was defined and compared with model A and B.
A was a linear three compartment pharmacokinetic model.
B was a three compartment pharmacokinetic model with Michaelis-Menten elimination.
C was a three compartment pharmacokinetic model with a nonlinear model for paclitaxel plasma clearance (CL) (equations 3, 4 and 5).
During the infusion:
| (3) |
Postinfusion:
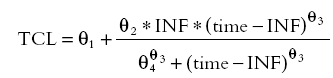 |
(4) |
where INF and time are the length of the infusion and the time from the start of the infusion, respectively, and TCL is the population clearance.
Individual CL values were defined as follows.
| (5) |
where η1 describes the departure of the clearance of individual from the population.
Model C provided a better description of the data when compared with model A and B (Figure 1). Owing to the sparse data available during the infusion phase, a linear decrease of CL over time was found to be the most appropriate relationship (compared with Emax or sigmoidal Emax relationships). In addition, a linear increase of CL over time during the postinfusion period was tested but the sigmoidal Emax model gave a superior fit to the postinfusion data. Furthermore, this latter postinfusion-time-dependent function is more relevant because of the Michaelis–Menten nature of CrEL elimination [38].
Figure 1.
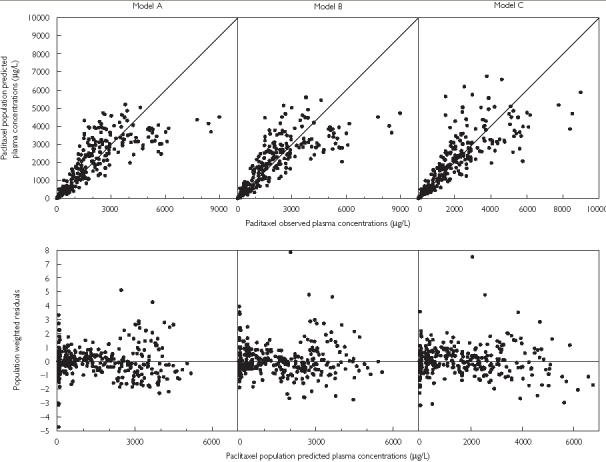
Goodness-of-fit plots for the linear three compartment model (model A – left), the three compartment with Michaelis–Menten elimination model (model B – middle) and the three compartment with the time- varying CL model (model C – right)
Similarly to CL, other disposition parameters could be expected to vary with time due to the binding of paclitaxel to CrEL but no such relationship was found to be significant.
In addition, in order to fully explore paclitaxel nonlinear plasma pharmacokinetics, a proportional linear decreasing relationship between paclitaxel CL and paclitaxel dose was added to model C. This new model did not lead to a better fit than model C.
Model C was then applied to the paclitaxel PK data both in the presence and absence of zosuquidar (cycle 1 and cycle 2, respectively). IIV was estimated on four parameters [paclitaxel total and intercompartmental clearance (CL and Q2), paclitaxel central and peripheral volume of distribution (V1 and V 2)] and IOV was estimated on CL and V1. The results for this basic model are presented Table 3.
Table 3.
Paclitaxel pharmacokinetic parameters from the basic and covariate (categorical and continous) population pharmacokinetic models
| Categorical relationship | |||
|---|---|---|---|
| Basic model | (final modsel) | Continuous relationship | |
| OBJF | 8727.003 | 8703.350 | 8680.895 |
| Parameters (value ± SE) | |||
| CL changing with time* | |||
| Slope (θ2) (l h−2) | 9.35 ± 7.32 | 10.0 ± 7.92 | 9.66 ± 6.95 |
| Min CL (θ1) (l h−1) | 7.64 ± 12.2 | 8.48 ± 12.9 | 8.59 ± 29.9 |
| t50 (θ4) (h) | 8.76 ± 16.4 | 9.36 ± 28.2 | 7.27 ± 102 |
| γ1 (θ3) | 2.94 ± 23.5 | 2.68 ± 30.2 | 2.12 ± 95.3 |
| Effect of zosuquidar on paclitaxel CL | |||
| Decrease with LY Cmax > 350 (µg l−1) (%) | – | 25.2 ± 12.4 | – |
| Emax (l h−1) | – | – | 5.49 ± 43.5 |
| LY Cmax50 (µg l−1) | – | – | 328 ± 15.4 |
| γ2 | – | – | 9.18 ± 129 |
| V1 (l) | 7.93 ± 14.0 | 7.95 ± 13.8 | 8.38 ± 13.0 |
| V2 (l) | 198 ± 7.78 | 196 ± 7.81 | 194 ± 16.0 |
| Q2 (l h−1) | 11.1 ± 7.37 | 10.8 ± 9.35 | 11.2 ± 11.3 |
| Q3 (l h−1) | 6.57 ± 15.8 | 6.76 ± 16.4 | 6.35 ± 39.4 |
| V3(l) | 7.00 ± 15.4 | 7.51 ± 18.9 | 10.2 ± 164 |
| ω CL (%) | 27.2 ± 33.2 | 25.9 ± 29.7 | 24.8 ± 34.3 |
| ω CL-Q2 (%) | 32.6 ± 23.4 | 30.5 ± 23.2 | 29.6 ± 24.7 |
| ω Q2 (%) | 44.5 ± 28.0 | 43.7 ± 26.1 | 43.5 ± 37.0 |
| ω CL-V2 (%) | 29.3 ± 30.7 | 26.1 ± 38.4 | 24.2 ± 39.5 |
| ω Q2-V2 (%) | 40.7 ± 29.9 | 39.6 ± 29.2 | 37.5 ± 35.1 |
| ωV2 (%) | 43.7 ± 26.6 | 42.8 ± 26.0 | 40.9 ± 31.4 |
| ωV1 (%) | 38.5 ± 58.6 | 40.0 ± 52.4 | 41.7 ± 58.6 |
| ω IOV CL (%) | 20.9 ± 33.2 | 15.2 ± 53.9 | 16.1 ± 56.2 |
| ω IOV V1 (%) | 57.5 ± 39.6 | 54.5 ± 42.8 | 46.6 ± 47.9 |
| Residual variance (%) | 22.7 ± 7.75 | 22.9 ± 7.77 | 22.5 ± 8.00 |
During the infusion *postinfusion 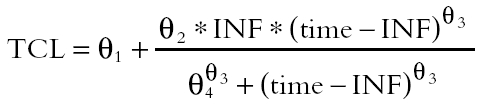 with INF the length of the infusion and time the time from the start of the infusion.
with INF the length of the infusion and time the time from the start of the infusion.
Paclitaxel AUC increased with increasing zosuquidar Cmax (Figure 2a). Figure 2b illustrates that some individuals showed very similar paclitaxel AUCs in the absence and presence of zosuquidar. The individuals departing from the line of identity are those who had the highest zosuquidar Cmax (> 350 µg l−1).
Figure 2.
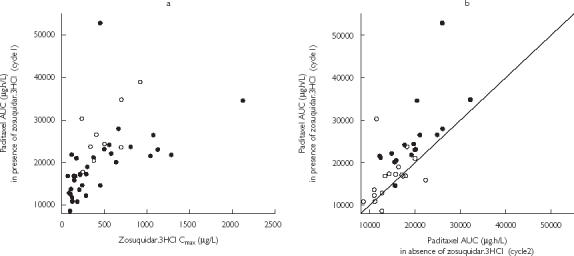
Scatter plots of (a) paclitaxel AUC vs zosuquidar Cmax (•, paclitaxel 175 mg/m2; ○, paclitaxel 225 mg/m2) and (b) paclitaxel AUC in the presence of zosuquidar vs paclitaxel AUC in the absence of zosuquidar (○, zosuquidar.3HCl Cmax < 350 µg/L; •, zosuquidar.3HCl Cmax > 350 µg/L; —, line of identity).
The effect of Zosuquidar 3HCl on paclitaxel AUC reflects the decrease in paclitaxel CL as a consequence of P-gp inhibition. Therefore it was relevant to model this PK interaction on paclitaxel CL rather than on exposure. Both continuous (sigmoidal Emax decrease) and categorical relationships (percentage decrease above a certain threshold) were tested in NONMEM to describe the expected decrease in paclitaxel CL in the presence of zosuquidar. Both these models were found to be significantly better (P < 0.005) than the basic model and to yield very similar results (Table 3). The value of the cut off for the categorical model (zosuquidar Cmax equal to 350 µg l−1) was defined by sensitivity analysis and closely corresponded to the estimated IC50 of the sigmoidal Emax model (zosuquidar Cmax equal to 328 µg l−1). Figure 3 illustrates the improvement in the distribution of the random effects of paclitaxel CL from the basic model C compared with the categorical model accounting for the influence of zosuquidar on paclitaxel plasma CL.
Figure 3.
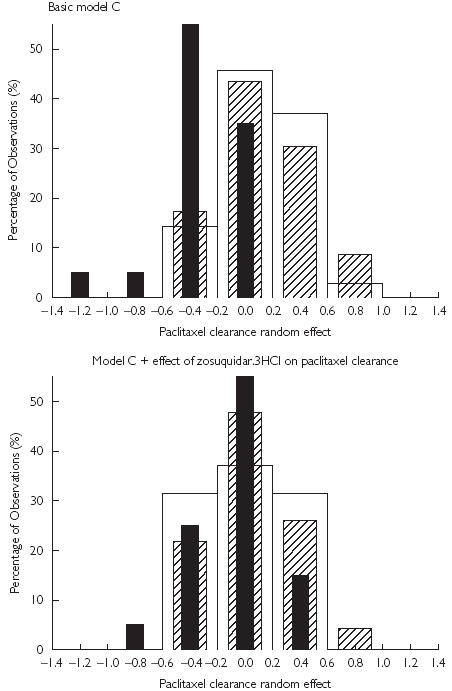
Distribution of paclitaxel clearance random effect estimated from the basic model C (top panel) and the model accounting for the influence of zosuquidar on paclitaxel clearance (lower panel). The difference in the thickness of the bars is for illustrative purposes only. In absence of zosuquidar.3HCl (□); in presence of zosuquidar.3HCl (Cmax ≤ 350 µg/L) ( ); and in presence of zosuquidar.3HCl (Cmax > 350 µg/L) (▪).
); and in presence of zosuquidar.3HCl (Cmax > 350 µg/L) (▪).
The GAM analysis, run on the posterior-individual estimates of paclitaxel CL (one value for each occasion) selected the following other covariates, creatinine clearance (CrCL), body mass index (BMI) and serum bilirubin concentration (BILI). In addition, bilirubin serum concentration and gender were selected as potential covariates on paclitaxel peripheral volume of distribution (V2). When these relationships were tested in NONMEM, only a linear relationship between serum bilirubin and paclitaxel CL was found to significantly improve the model (P < 0.005). However, this relationship was driven by data from two patients and therefore not thought to be robust enough for retention in the final model. Although the continuous model (sigmoidal Emax decrease), describing the effect of zosuquidar on paclitaxel CL, is physiologically more relevant than the categorical model, the latter was retained as the final model.
The corresponding paclitaxel population pharmacokinetic parameters and the observed-predicted concentrations vs time plots are presented in Table 3 and Figure 4, respectively. A 25% decrease in paclitaxel plasma CL in the presence of zosuquidar (Cmax > 350 µg l−1) was estimated, corresponding to an 1.3 fold increase in paclitaxel AUC (from 14829 µg l−1 h to 19115 µg l−1 h following paclitaxel 175 mg m−2). The inclusion of the effect of zosuquidar led to a significant decrease in interoccasion variability in paclitaxel CL (from 20.9% to 15.2%). Other PK parameters, peripheral volumes of distribution (V2 and V3) and intercompartmental clearances (Q2 and Q3) were not significantly altered in the presence of zosuquidar with no cycle differences and no differences between the absence/or presence of zosuquidar (Cmax < 350 µg l−1) and the presence of zosuquidar (Cmax > 350 µg l−1)). All parameters were estimated with acceptable precision and the values are consistent with those reported in the literature [1]. Paclitaxel central and peripheral volumes of distribution (V1, V2, V3) and intercompartmental clearances (Q2 and Q3), were 7.95, 196, 7.51 l and 10.8, 6.76 l h−1, respectively, in both the absence and presence of zosuquidar.
Figure 4.
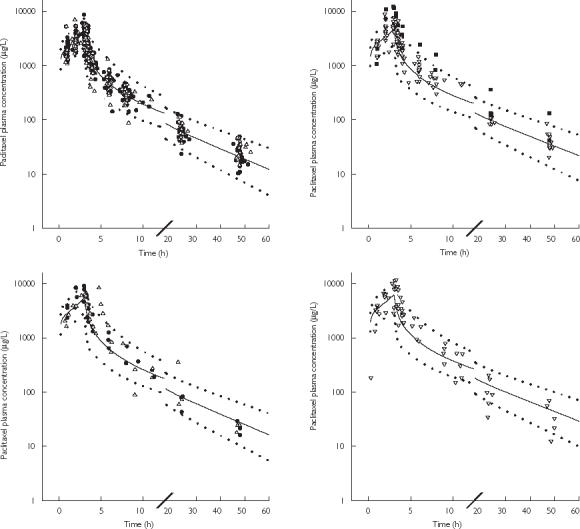
Paclitaxel observed and predicted (mean,  95% prediction interval) plasma concentration vs time profile following a 3 h i.v. administration of 175 mg m−2 (upper panels) and 225 mg m−2 (lower panels) of paclitaxel in the absence (•) or presence of zosuquidar (Cmax≤ 350 µg l−1) (▵) (left panels) and the presence of zosuquidar (Cmax > 350 µg l−1) (▿) (right panels). ▪ highlights patients 253 and 251 with high bilirubin serum concentrations. — represents median prediction.
95% prediction interval) plasma concentration vs time profile following a 3 h i.v. administration of 175 mg m−2 (upper panels) and 225 mg m−2 (lower panels) of paclitaxel in the absence (•) or presence of zosuquidar (Cmax≤ 350 µg l−1) (▵) (left panels) and the presence of zosuquidar (Cmax > 350 µg l−1) (▿) (right panels). ▪ highlights patients 253 and 251 with high bilirubin serum concentrations. — represents median prediction.
The median paclitaxel plasma concentration vs time profiles in the absence and presence of zosuquidar (obtained from 1000 Monte–Carlo simulations) are presented in Figure 5a and b. This illustrates the increase in the time paclitaxel remains above a concentration of 0.1 µM (equivalent to 84 µg l−1) in the presence of zosuquidar (Cmax > 350 µg l−1) from 18 h to 27 h and 23 h to 34 h, following the administration of paclitaxel doses of 175 mg m−2 and 225 mg m−2, respectively. This corresponds to an increase in paclitaxel AUC(0.1µM) (area under paclitaxel plasma concentration vs time curve above 0.1 µmol l−1) of 31% (from 13052 to 17134 µg l−1 h following paclitaxel 175 mg m−2) in the presence of zosuquidar (Cmax > 350 µg l−1). The median paclitaxel CL vs time course during and after a 3 h infusion is presented in Figure 6 showing the effect of zosuquidar (Cmax > 350 µg l−1) remaining constant over time (25% decrease).
Figure 5.
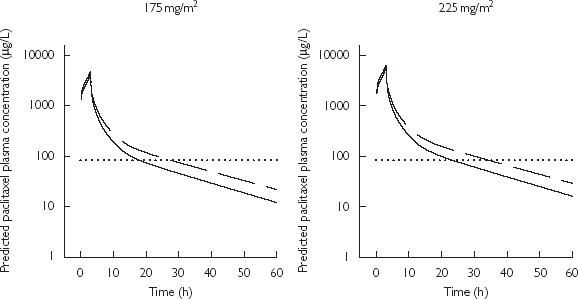
Median paclitaxel simulated plasma concentrations vs time profile following a 3 h i.v. administration of 175 mg m−2 (left panel) and 225 mg m−2 (right panel) in the absence or presence of zosuquidar (Cmax≤ 350 µg l−1) (solid line) and the presence of zosuquidar (Cmax > 350 µg l−1) (dashed line); Dotted line highlights paclitaxel concentrations of 0.1 µM (84 µg l−1)
Figure 6.
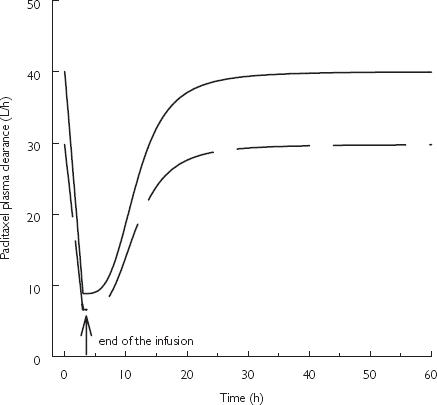
Median paclitaxel plasma clearance vs time profile following a 3 h i.v. administration of paclitaxel in the absence or presence of zosuquidar (Cmax≤ 350 µg l−1) (solid line) and the presence of zosuquidar (Cmax > 350 µg l−1) (dashed line)
Discussion
The basic structural model presented in this study, though empirical in nature, is based on the underlying effect of CrEL on paclitaxel plasma CL. It mimics the influence of CrEL on the paclitaxel plasma unbound fraction by describing paclitaxel plasma CL changing with time. Van Zuylen et al.[34] reported that following 175 and 225 mg m−2 3 h i.v. infusion of paclitaxel, CrEL pharmacokinetics was linear. Therefore, given that only a 3-h infusion regimen of a small range of paclitaxel doses was administered in this study, dose nonproportionality in paclitaxel PK due to CrEL was not expected. Total paclitaxel plasma pharmacokinetics were adequately described by the addition of the time dependency function in paclitaxel CL, accounting for the interaction between CrEL and paclitaxel, without requiring an additional dose-dependent function. Hence, the present model may be considered appropriate. It is predictive for paclitaxel plasma phamacokinetics following a 3 h infusion of 175–225 mg m−2 and provides the structural model on which the influence of zosuquidar can be studied.
Although the continuous model (sigmoidal Emax decrease), describing the effect of zosuquidar on paclitaxel CL, is likely to be more physiologically relevant than the categorical model, the latter was retained. Both models yielded similar results, but the continuous model was certainly overparameterized as some PK parameters were not precisely estimated.
The estimated maximal 25.2% reduction in paclitaxel plasma CL in the presence of zosuquidar at a Cmax > 350 µg l−1 is consistent with the percentage of the paclitaxel dose recovered unchanged in the bile after administration of tritium-labelled drug [2]. Hence, the influence of zosuquidar on paclitaxel CL is most likely to result from P-gp inhibition in the bile canaliculi. Zosuquidar Cmax is believed to be a better predictor of the potential pharmacokinetic interaction with paclitaxel compared with the area under the zosuquidar plasma vs time curve because of the direct nature of the relationship between the degree of P-gp inhibition and zosuquidar concentration [15]. Time above a threshold concentration or an AUC above the same threshold concentration may be a better predictor. However, the limited data from this study precludes performing such an analysis but this will be the focus of future studies.
A limitation of any nonrandomized drug interaction study is the sequential nature of the administration. Toxicities in particular, may be additive over repeated cycles of administration or may be of sufficient severity to necessitate a dose reduction in the next cycle. This clearly may be a confounding factor in the interpretation of the data. Only in a randomized setting can the true influence of combination therapy be evaluated.
Results from clinical trials, with other MDR modulators (PSC833 [8, 9] and VX-710 [14]) showed: (a) a greater than 50% decrease in paclitaxel CL with a maximum tolerated dose (MTD) of 50–70 mg m−2 when administered with PSC833 (valspodar) compared with the usual 175 mg m−2; and (b) a paclitaxel MTD of about 60–80 mg m−2 when administered with VX-710 (biricodar) compared with the usual 175 mg m−2. The influence of both PSC833 and VX-710 on paclitaxel CL is greater than the present finding with zosuquidar. Owing to the interaction of PSC833 and VX-710 with cytochrome P450, these compounds are likely to inhibit paclitaxel metabolism in the liver as well as to decrease its biliary excretion through P-gp inhibition in bile canaliculi. Hence it is understandable that a greater pharmacokinetic interaction is observed with these MDR modulators compared with zosuquidar, which is believed to interact only with P-gp.
The decrease in paclitaxel CL observed in the presence of zosuquidar led to a increase in the time that the plasma paclitaxel concentration remains above 0.1 mol L−1, which is known to be a marker of both toxicity (neutropenia) and efficacy [1]. Although the effect of zosuquidar on the toxicity of a clinically used dose of paclitaxel (175 mg m−2) was minimal, the finding is important in planning further clinical trials to compare the toxicity–efficacy of paclitaxel in the presence and absence of zosuquidar. In essence, a paclitaxel dose of 225 mg m−2 in the absence of zosuquidar and a dose of 175 mg m−2 in the presence of zosuquidar should lead to a similar time that the paclitaxel plasma concentration stays above 0.1 µmol L−1 and to a similar AUC(0.1 µmol L−1). Hence these doses will probably be necessary to delineate the pharmacodynamic effect of P-gp inhibition on disease from the effect that could simply arise from the pharmacokinetic interaction.
References
- 1.Grochow LB, Ames MM. A clinician's guide to chemotherapy pharmacokinetics and pharmacodynamics. 1. Baltimore: Williams & Wilkins Publishers; 1998. pp. 93–122. [Google Scholar]
- 2.Walle T, Walle UK, Kumar GN, Bhalla KN. Taxol metaoblism and disposition in cancer patients. Drug Metab Dispos. 1995;23:506–512. [PubMed] [Google Scholar]
- 3.Pivot X, Asmar L, Hortobagyi GN. The efficacy of chemotherapy with docetaxel and paclitaxel in anthracycline-resistant breast cancer. Int J Oncol. 1999;15:381–386. [PubMed] [Google Scholar]
- 4.Dumontet C, Sikic BI. Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport and cell death. J Clin Oncol. 1999;17:1061–1070. doi: 10.1200/JCO.1999.17.3.1061. [DOI] [PubMed] [Google Scholar]
- 5.Allen JD, Brinkhuis RF, van Deemter L, Wijnholds J, Schinkel AH. Extensive contribution of the multidrug transporters P-glycoprotein and MRP1 to basal drug resistance. Cancer Res. 2000;60:5761–5766. [PubMed] [Google Scholar]
- 6.Sekine I, Saijo N. Polymorphisms of metabolizing enzymes and transporter proteins involved in the clearance of anticancer agents. Ann Oncol. 2001;12:1515–1525. doi: 10.1023/a:1013164013375. [DOI] [PubMed] [Google Scholar]
- 7.Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer; mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11:265–283. doi: 10.1016/s0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 8.Advani R, Fisher GA, Lum BL, et al. A phase I trial of doxorubicin, paclitaxel, and valspodar (PSC833), a modulator of multidrug resistance. Clin Cancer Res. 2001;7:1221–1229. [PubMed] [Google Scholar]
- 9.Chico I, Kang MH, Bergan R, et al. Phase I study of infusional paclitaxel in combination with the P-glycoprotein antagonist PSC833. J Clin Oncol. 2001;19:832–842. doi: 10.1200/JCO.2001.19.3.832. [DOI] [PubMed] [Google Scholar]
- 10.Giaccome G, Linn SC, Welink J, et al. A dose-finding and pharmacokinetic study of reversal of multidrug resistance with SDZ PSC 833 in combination with doxorubicin in patients with solid tumors. Clin Cancer Res. 1997;3:2005–2015. [PubMed] [Google Scholar]
- 11.Sparreboom A, Planting AST, Jewell RC, et al. Clinical pharmacokinetics of doxorubicin in combination with GF120918, a potent inhibitor of MDR1 P-glycoprotein. Anti-Cancer Drugs. 1999;10:719–728. doi: 10.1097/00001813-199909000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Ferry D, Price L, Atsmon J, et al. A phase IIA pharmacokinetic and pharmacodynamic study of the P-glycoprotein inhibitor, XR9576, in patient treated with doxorubicin chemotherapy. Proc AACR. 2001;42:950–5160. [Google Scholar]
- 13.Peck RA, Hewett J, Harding MW, et al. Phase I and pharmacokinetic study of the novel MDR1 and MRP1 inhibitor biricodar administered alone and in combination with doxorubicin. J Clin Oncol. 2001;19:3130–3141. doi: 10.1200/JCO.2001.19.12.3130. [DOI] [PubMed] [Google Scholar]
- 14.Rowinsky EK, Smith L, Wang YM, et al. Phase I and pharmacokinetic study of paclitaxel in combination with biricodar, a novel agent that reverses multidrug resistance conferred by over expression of both MDR and MRP. J Clin Oncol. 1998;16:2964–2976. doi: 10.1200/JCO.1998.16.9.2964. [DOI] [PubMed] [Google Scholar]
- 15.De Alwis DP, Pouliquen I, Burgess M, et al. A phase I dose escalating study of LY335979, a novel Pgp modulator – administered intravenously in combination with doxorubicin. Proceedings ASCO. 2001;20(part 1):284–272a. [Google Scholar]
- 16.Dantzig AH, Law KL, Cao J, et al. Reversal of multidrug resistance by the P-glycoprotein modulator LY335979, from the bench to the clinic. Curr Med Chem. 2001;8:39–50. doi: 10.2174/0929867013373903. [DOI] [PubMed] [Google Scholar]
- 17.Dantzig AH, Shepard RL, Cao J, et al. Reversal of P-glycoprotein-mediated multidrug resistance by a potent cyclopropyldibenzosuberane modulator, LY335979. Cancer Res. 1996;56:4171–4179. [PubMed] [Google Scholar]
- 18.Dantzig AH, Shepard RL, Law KL, et al. Selectivity of the multidrug resistance modulator, LY335979, for P-glycoprotein and effect on cytochrome P-450 activities. J Pharmacol Exp Ther. 1999;290:854–862. [PubMed] [Google Scholar]
- 19.Green LJ, Marder P, Slapak CA. Modulation by LY335979 of P-glycoprotein function in multidrug-resistant cell lines and human natural killer cells. Biochem Pharmacol. 2001;61:1393–1399. doi: 10.1016/s0006-2952(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 20.Boeckmann AJ, Sheiner LB, Beal SL. NONMEM Users guideNONMEM Project Group. . San Francisco: University of California; 1994. [Google Scholar]
- 21.Kruijtzer CMF, Vasey PA, Harris A, et al. A phase I dose ranging study of the P-glycoprotein inhibitor zosuquidar trihydrochloride ( LY335979) administered in combination with paclitaxel in patients with solid tumors) Clin Cancer Res. in press. [Google Scholar]
- 22.Sparreboom A, de Bruijn P, Nooter K, et al. Determination of paclitaxel in human plasma using single solvent extraction prior to isocratic reversed-phase high-performance liquid chromatography with ultraviolet detection. J Chromatogr B Biomed Sci Appl. 1998;705:159–164. doi: 10.1016/s0378-4347(97)00502-1. [DOI] [PubMed] [Google Scholar]
- 23.Scheiner LB, Steimer JL. Pharmacokinetic/pharmacodynamic modeling in drug development. Annu Rev Pharmacol Toxicol. 2000;40:67–95. doi: 10.1146/annurev.pharmtox.40.1.67. [DOI] [PubMed] [Google Scholar]
- 24.Sun H, Fadiran EO, Jones CD, et al. Population pharmacokinetics – A regulatory perspective. Clin Pharmacokinet. 1999;37:41–51. doi: 10.2165/00003088-199937010-00003. [DOI] [PubMed] [Google Scholar]
- 25.Wade JR, Beal SL, Sambol NC. Interaction between structural, statistical and covariates models in population pharmacokinetic analysis. J Pharmacokinet Biopharm. 1994;22:165–177. doi: 10.1007/BF02353542. [DOI] [PubMed] [Google Scholar]
- 26.Karlsson MO, Sheiner LB. The importance of modeling interoccassion variability in population pharmacokinetic analysis. J Pharmacokinet Biopharm. 1993;21:735.750. doi: 10.1007/BF01113502. [DOI] [PubMed] [Google Scholar]
- 27.Gianni L, Kearns CM, Gianni A, et al. Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationship in humans. J Clin Pharmacol. 1995;13:180–190. doi: 10.1200/JCO.1995.13.1.180. [DOI] [PubMed] [Google Scholar]
- 28.Huizing MT, Giaccone G, van Warmerdam LJC, et al. Pharmacokinetics of paclitaxel and carboplatin in a dose-escalating and dose-sequencing study in patients with non-small-cell lung cancer: an ECC trial. J Clin Oncol. 1997;15:317–329. doi: 10.1200/JCO.1997.15.1.317. [DOI] [PubMed] [Google Scholar]
- 29.Huizing MT, Keung CF, Rosing H, et al. Pharmacokinetics of paclitaxel and metabolites in a randomized comparative study in platinum-pretreated ovarian cancer patients. J Clin Oncol. 1993;11:2127–2135. doi: 10.1200/JCO.1993.11.11.2127. [DOI] [PubMed] [Google Scholar]
- 30.Huizing MT, van Warmerdam LJC, Rosing H, et al. Phase I and pharmacologic study of the combination paclitaxel and carboplatin as first-line chemotherapy in stage III and IV ovarian cancer. J Clin Oncol. 1997;15:1953–1964. doi: 10.1200/JCO.1997.15.5.1953. [DOI] [PubMed] [Google Scholar]
- 31.Huizing MT, Vermorken JB, Rosing H, et al. Pharmacokinetics of paclitaxel and three major metabolites in patients with advanced breast carcinoma refractory to anthracycline therapy treated with a 3-hour paclitaxel infusion: a European Cancer Centre (ECC) trial. Ann Oncol. 1995;6:699–704. doi: 10.1093/oxfordjournals.annonc.a059287. [DOI] [PubMed] [Google Scholar]
- 32.Gelderblom H, Verweij J, Nooter K, et al. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–1598. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 33.Sparreboom A, van Zuylen L, Brouwer E, et al. Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implication. Cancer Res. 1999;59:1454–1457. [PubMed] [Google Scholar]
- 34.Van Zuylen L, Gianni L, Verweij J, et al. Inter-relationships of paclitaxel disposition, infusion duration and cremophor EL kinetics in cancer patients. Anti-Cancer Drugs. 2000;11:331–337. doi: 10.1097/00001813-200006000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Van Zuylen L, Karlsson MO, Verweij J, et al. Pharmacokinetic modeling of paclitaxel encapulation in cremophor EL micelles. Cancer Chemother Pharmacol. 2001;47:309–318. doi: 10.1007/s002800000215. [DOI] [PubMed] [Google Scholar]
- 36.Henningson A, Karlsson MO, Vigano L, et al. Mechanism-based pharmacokinetic model for paclitaxel. J Clin Oncol. 2001;19:4065–4073. doi: 10.1200/JCO.2001.19.20.4065. [DOI] [PubMed] [Google Scholar]
- 37.Jonsson EN, Karlsson MO. Xpose – an S-PLUS based population pharmacokinetic / pharmacodynamic model building aid for NONMEM. Comput Meth Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 38.Van der Bongard HJ, Mathot RA, van Telligen O, Schellens JH, Beijnen JH. A population analysis of the pharmacokinetics of cremophor EL using non-linear mixed effect modelling. Cancer Chemother Pharmacol. 2002;50:16–24. doi: 10.1007/s00280-002-0459-x. [DOI] [PubMed] [Google Scholar]