

Abstract
Aims
To characterize the in vitro and in vivo inhibitory effect of stiripentol, a new anticonvulsant, on the metabolism of carbamazepine and saquinavir, which are substrates of CYP3A4.
Methods
Human liver microsomes and cDNA-expressed CYP enzymes were used for the in vitro experiments. Pharmacokinetic data from epileptic children and healthy adults were used for the carbamazepine and saquinavir in vivo studies, respectively.
Results
Carbamazepine biotransformation to its 10,11-epoxide by human liver microsomes (Vmax = 10.3 nmol min−1 nmol−1 P450, apparent Km = 362 µm), cDNA-expressed CYP3A4 (Vmax = 1.17 nmol min−1 nmol−1 P450, apparent Km = 119 µm) and CYP2C8 (Vmax = 0.669 nmol min−1 nmol−1 P450, apparent Km = 757 µm) was inhibited by stiripentol (IC50 14, 5.1, 37 µM and apparent Ki 3.7, 2.5, 35 µm, respectively). Saquinavir biotransformation to its major metabolite M7 by human liver microsomes (Vmax = 5.7 nmol min−1 nmol−1 P450, apparent Km = 0.79 µm) was inhibited by stiripentol (IC50 163 µM, apparent Ki 86 µm). In epileptic children treated with carbamazepine and stiripentol, the plasma concentration ratio of carbamazepine epoxide/carbamazepine was decreased by 65%. The in vivo apparent Ki for stiripentol ranged from 10.5 to 41.4 µm. The pharmacokinetics of saquinavir was not modified by stiripentol in healthy adults. The 95% confidence intervals for the difference for Cmax and AUC of saquinavir between the placebo and stiripentol phase were (−39.8, 39.8) and (−33.2, 112), respectively.
Conclusions
These results showed that stiripentol was a weak inhibitor of saquinavir metabolism both in vitro and in vivo. In contrast, stiripentol is a potent inhibitor of carbamazepine 10,11-epoxide formation in vitro and in vivo in epileptic patients.
Keywords: drug metabolism, drug interaction, carbamazepine, antiepileptic agents
Introduction
Metabolic drug interactions are most often the cause of adverse events. However, in some instances they are used for therapeutic purposes. For example, ritonavir, a protease inhibitor, and a potent inhibitor of cytochrome P450 3A4 (CYP3A4)-mediated drug metabolic biotransformations, is administered together with other protease inhibitors such as saquinavir, indinavir or amprenavir to decrease their first-pass effect and to increase the systemic exposure [1]. As a ‘booster agent’ for saquinavir, ritonavir is administered at low dosage. However, under these conditions HIV-resistant strains might develop. Therefore, the use of CYP3A4 inhibitors with no antiretroviral activity and fewer side-effects may be advantageous in this setting. Stiripentol, an anticonvulsant drug under clinical investigation, is an allylic alcohol unrelated structurally to any other antiepileptic drug. Stiripentol has been shown to be an in vitro and in vivo inhibitor of CYP1A2 and 3A4 [2–6], and thus may be useful as an inhibitor of saquinavir metabolism.
The aim of the present study was to characterize the inhibitory effect of stiripentol on the in vitro and in vivo metabolism of carbamazepine (CBZ) and saquinavir in man.
Methods
In vitro studies
Chemicals and reagents
Stiripentol was provided by Laboratoires Biocodex (Montrouge, France). Carbamazepine and carbamazepine 10,11-epoxide were obtained from Laboratoires Novartis Pharma (Rueil-Malmaison, France). Saquinavir and ritonavir were gifts from Produits Roche (Neuilly sur Seine, France) and Abbott Laboratories (Abbott Park, IL, USA), respectively. Glucose-6-phosphate (G6P), glucose-6-phosphate dehydrogenase (G6PD), nicotinamide adenine dinucleotide phosphate (NADP) were purchased from Sigma-Aldrich Chimie S.a.r.l. (St. Quentin Fallavier, France). All other reagents were of the highest purity available.
Human liver samples and preparation of microsomes
Whole human livers (HLM 1–6) were obtained from adult organ donors. Human liver samples were collected according to the recommendations of the Ethics Committee of Institut National de la Santé et de la Recherche Médicale (INSERM, Paris, France). Microsomes were prepared from liver homogenates by differential centrifugation [7] and stored until further use (−80 °C). Total microsomal cytochrome P450 contents were determined by the method of Greim [8]. Microsomal protein concentrations were determined by the BCA® (Pierce) assay based on the method of Smith PK, Krohn RI, Hermanson GT et al. Measurement of protein using bicinchoninic acid. Anal Biochem 1985; 150: 76–85.
Human P450 isozymes
Previous studies have established that human CYP3A4 and CYP2C8 isoforms mediate CBZ biotransformation into CBZE [9]. Therefore CBZ metabolism was studied in human cDNA-expressed CYP2C8 (Gentest P252) and CYP3A4 (Gentest P202) (Baculovirus-Insect-Cell-expressed). These microsomes also contain cDNA-expressed human P450 reductase and human cytochrome b5 and were purchased from Gentest (Woburn, MA, USA).
Incubations with human liver microsomes and with cDNA-expressed CYP3A4 and CYP2C8
Acetonitrile was used to dissolve CBZ, saquinavir, stiripentol and ritonavir and was present in incubations containing those compounds at a final concentration (v/v) of 0.5%. Acetonitrile was chosen as it was shown to have the least inhibitory effect of a range of solvents [10, 11].
Carbamazepine metabolism
Human microsomes and CYP3A4 (0.02 nmol P450 ml−1) or CYP2C8 (0.04 nmol P450 ml−1) microsomal suspensions containing 0.5 mg ml−1 MgCl2, 0.5 mg ml−1 G6P, 0.5 UI ml−1 G6PD were diluted with 100 mm phosphate buffer (KH2PO4 100 mm/Na2HPO4, 2H2O 100 mm, 19.6/80.4, v/v) at pH 7.4 in a final volume of 0.5 ml. The incubations were started by addition of 1 mm NADP and continued for 60 min at 37 °C. Incubations were stopped by adding 250 µl of ice-cold acetonitrile, and the reaction tubes were then stored on ice. Incubations without NADPH-generating system served as controls. Incubations for all studies were conducted in duplicate. CBZ biotransformation was linear with respect to incubation time and to protein concentration (up to 0.2 nmol P450 ml−1 in human liver microsomes and up to 0.04 nmol P450 ml−1 in cDNA-expressed CYP3A4 and CYP2C8).
Saquinavir metabolism
Microsomal suspensions (0.04 nmol ml−1) were preincubated for 3 min at 37 °C. The experimental conditions were similar to those described above. Incubations were stopped at 5 min by 250 µl of ice-cold acetonitrile. Saquinavir biotransformation was linear with respect to incubation time and to P450 concentration (up to 0.1 nmol P450 ml−1 in human liver microsomes).
Enzyme kinetics – carbamazepine
Apparent Km and Vmax were determined in microsomes from six human livers and in cDNA-expressed CYP2C8 and CYP3A4. Carbamazepine was incubated for 60 min at eight different concentrations (10, 25, 50, 100, 250, 500, 750 and 1000 µm).
Enzyme kinetics – saquinavir
Apparent Km and Vmax were determined in microsomes from two human livers. Saquinavir was incubated for 5 min at seven different concentrations (0.1, 0.2, 0.4, 0.6, 0.8, 1 and 2 µm).
Time dependence of stiripentol inhibition
In order to characterize the time-dependent inhibition of CBZ metabolism by stiripentol two experiments were conducted. First, CBZ (100 µm) and human liver microsomes (0.02 µm P450) were incubated without or with stiripentol (2 µm). At 5, 10, 15, 20, 30, 45 and 60 min, reaction was stopped as described above. In a second experiment human liver microsomes (0.2 µm P450) and stiripentol (2 µm) were incubated with or without NADPH generating system (described above). At specific times (0, 5, 15, 30 and 60 min), aliquots (50 µl) were withdrawn and diluted 10-fold by addition of an incubation mixture (450 µl) containing CBZ (500 µm), an NADPH generating system and MgCl2 0.5 mg ml−1 in 100 mm phosphate buffer pH 7.4. Incubation for a further 20 min was allowed.
Inhibitory kinetics – carbamazepine
The apparent Ki of stiripentol was determined with various concentrations of carbamazepine (50–1000 µm) and stiripentol (0–10 µm) in human liver microsomes and in cDNA-expressed CYP3A4 and CYP2C8. An IC50 was determined in human liver microsomes by coincubations of 50 µm carbamazepine with increasing concentrations of stiripentol (1–500 µm) or ritonavir (0.001–1 µm) for comparison.
Inhibitory kinetics – saquinavir
The apparent Ki of stiripentol was determined with various concentrations of saquinavir (0.1–2 µm) and stiripentol (0–200 µm) in human liver microsomes. An IC50 was determined in human liver microsomes by coincubations of 1 µm saquinavir with increasing concentrations of stiripentol (10–500 µm) or ritonavir (0.001–1 µm) for comparison.
HPLC analysis – carbamazepine
Internal standard (3-bromo-N-propylcinnamamide) was added to the microsomal incubation mixture and the samples were extracted once with 5 ml diethyl ether. After evaporation of the organic solvent, the residue was dissolved in the mobile phase (60/40 water/acetonitrile, 100 µl) and 80 µl were injected onto the HPLC system (Thermo Quest, Fremont, CA, USA). Separation was accomplished on an Ultrasphere octyl 5-µm 250 × 4.6-mm column (Beckman, Berkeley, CA, USA) at a flow rate of 1 ml min−1. The eluent was monitored by ultraviolet absorbance at a wavelength of 210 nm. Retention times for CBZE, CBZ and internal standard were 4.5, 7.0 and 16.5 min, respectively. The standard curves were linear from 0.8 to 8 µm and from 8.5 to 85 µm for CBZE and CBZ, respectively. The minimum quantifiable concentrations were 0.8 µm (CBZE) and 8.5 µm (CBZ). The coefficients of variation for interday reproducibility were 6.5, 5.3 and 4.2% for CBZE at 1.2, 3.6 and 4.8 µm, respectively, and 2.2, 1.8 and 2.2% for CBZ at 12.7, 38 and 51 µm, respectively.
HPLC analysis – saquinavir
Saquinavir and its metabolites were assayed using a previously described method [12]. The standard curve was linear from 0.01 to 1 µm of saquinavir. The minimum quantifiable concentration was 0.01 µm. The coefficient of variation for interday precision was 12% for saquinavir. Metabolite concentrations were calculated using saquinavir standard curves, based on the assumption that the extinction coefficients for the metabolites were identical to that of saquinavir [12].
Data analysis
The kinetics of the biotransformations of carbamazepine and saquinavir by liver microsomes or cDNA-expressed CYPs were fitted by a one-enzyme Michaelis-Menten model, a one-enzyme model Hill equation, or a two-enzyme Michaelis-Menten model. Goodness of fit was based on visual examination of the plots and by application of the Akaike's information criterion [13]. Calculated parameters were maximum rate of formation (Vmax) and Michaelis constant (apparent Km), intrinsic clearance (CLint = Vmax/apparent Km), inhibition constant (apparent Ki) and IC50. The type of inhibition was identified from the change in the apparent Km and Vmax and confirmed by the inhibition models (competitive, noncompetitive, mixed, uncompetitive) that fit the data. Calculations were performed using Sigma Plot Software (SPSS Inc., Chicago, IL, USA). IC50s were estimated using GraphPad Prism 3.0 (GraphPad Software, San Diego, CA, USA).
In vivo studies
Carbamazepine-stiripentol interaction
Data used to derive individual patient inhibition constants (apparent Kiin vivo for CYP3A4) came from a previous double-blind placebo-controlled trial, that had evaluated the efficacy of stiripentol in children with refractory partial epilepsy receiving CBZ [5]. During a 1-month baseline period, patients received single-blind add-on placebo. The second period lasted 3 months when they received open add-on stiripentol. At the end of the latter, responders (defined as those experiencing a 50% decrease in seizure rate compared with baseline) were randomized to either stiripentol or placebo for a 2-month double-blind period. Subsequently, all patients received long-term stiripentol in an open fashion. Analysis was performed at the end of the double-blind period. Five venous blood samples were drawn for haematology, biochemistry, and trough plasma concentrations of STP, CBZ and CBZE at steady state. The first sample was taken at week 2 of baseline (S1), the second and third ones, respectively, at weeks 2 (S2) and 10 (S3) of the open period, and the last two, respectively, at weeks 2 (S4) and 7 (S5) of the double-blind period. Stiripentol was determined using a previously described procedure [2]. CBZ and CBZE were determined using the same procedure as described for in vitro studies using 100 µl plasma.
The approach used to calculate in vivo inhibition constants (apparent Kiin vivo) and based on an inhibition model used by others [14, 15, 16] was previously reported by Tran et al. [2]. Inhibition is described by the equation CLf/Clfi = 1 + I/Kiin vivo in which CLf and CLfi are formation clearances of CBZE in the absence and presence of stiripentol, respectively, and I is the plasma concentration of stiripentol. The calculation of apparent Kiin vivo was performed using the Sigma Plot Software (SPSS Inc.). A P-value of 0.05 was considered significant.
Saquinavir-stiripentol interaction
Twelve healthy male volunteers were enrolled in the study after each gave written informed consent. They were 20–29 years old and weighed from 63 to 90 kg. The protocol was approved by the local Ethics Committee (Paris-Cochin, France). A multiple-dose, placebo-controlled, randomized crossover pharmacokinetic study was performed. The study was conducted according to the following schedule. Subjects received stiripentol (Diacomit®) or placebo at a dose of 1000 mg b.i.d. on days 1–7, and on day 8 saquinavir (Fortovase® 400 mg single dose) was coadministered with stiripentol or placebo (1000 mg b.i.d.). Blood samples for determination of plasma concentrations of saquinavir and stiripentol were collected before dosing and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 10, 12, 16, 20 and 24 h after dosing. This first period was followed by a 6-day wash-out period from day 9 to 14 before the second period of the crossover study from day 15 to 23.
Plasma saquinavir concentrations were measured by HPLC using a previously published method [17] with slight modifications, namely direct extraction from 100 µl plasma added to desmethylflunitrazepam (Roche) as internal standard with tert-butylmethylether (3 ml) in alkaline medium (NaOH 0.5 N, 100 µl). Retention times for internal standard and saquinavir were 7.7 and 9.5 min, respectively. The standard curve was linear from 0.015 to 7.5 µm of saquinavir. The minimum quantifiable concentration was 0.015 µm. The coefficients of variation for interday precision were 7.2, 4.6 and 5.5% for saquinavir at 0.045, 2.25 and 4.5 µm, respectively. Recovery was 99% for saquinavir.
Kinetics and statistical analysis
Pharmacokinetic and statistical analyses were performed using Triomphe computer software [18]. The pharmacokinetics of saquinavir was characterized with noncompartmental methods. Maximum plasma concentration (Cmax) and time to reach Cmax (tmax) were obtained directly from the observed data. The area under the concentration-time curve profiles (AUC) were determined using the linear trapezoidal rule. Apparent total clearance (CL/F) was determined by the ratio of dose/AUC.
To assess the effects of stiripentol on saquinavir pharmacokinetics, Friedman's test was used to compare tmax, and crossover anova for repeated measurements was performed on logarithmically transformed Cmax and AUC.
Results
In vitro studies
Time-dependence of inhibition by stiripentol
The production of CBZE by human liver microsomes increased linearly as a function of time between 5 and 60 min with or without stiripentol (Figure 1) and was not sensitive to the duration of preincubation time with stiripentol and NADPH (Figure 1). These time-dependent inhibition studies were performed using previously reported methods [19–21] and it can be concluded from the results that stiripentol did not appear to be a mechanism-based inactivator. This was consistent with results previously published by Tran et al. [2].
Figure 1.
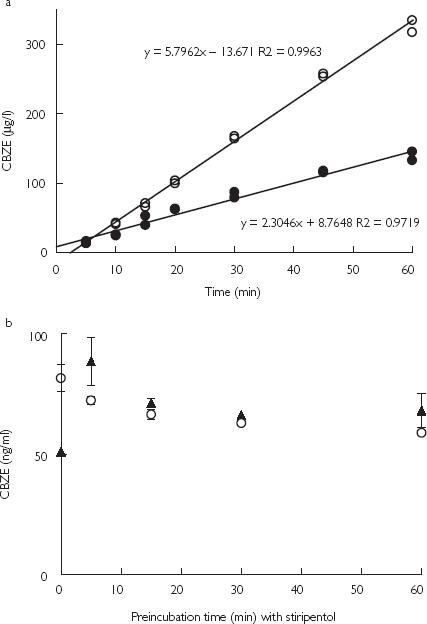
Influence of stiripentol on the time-course of carbamazepine epoxide (CBZE) formation, during incubations of (a) CBZ (100 µm) and human liver microsomes (0.02 µm) with • or without ○ 2 µm stiripentol (STP), (b) CBZ (100 µm) and human liver microsomes (0.2 µm) after a preincubation with 2 µm STP with ▴ or without ○ NADPH. Data points represent the mean of duplicate incubations.
Carbamazepine-stiripentol interaction
Under the experimental conditions used, the formation of CBZE in human liver microsomes was best described by a Hill model (Figure 2a,b) and in cDNA-expressed CYP3A4 and 2C8 (Figure Figure 2d,e) by a Michaelis-Menten model. The apparent Km, Vmax and CLint (calculated as the apparent Km/Vmax ratio) for the formation of CBZE are displayed in Table 1. Formation of CBZE was inhibited by stiripentol and ritonavir in human liver microsomes (Figure 2c) and by stiripentol in cDNA-expressed CYP3A4 and CYP2C8 (Figure 2f). The IC50 and apparent Ki are displayed in Table 1.
Figure 2.
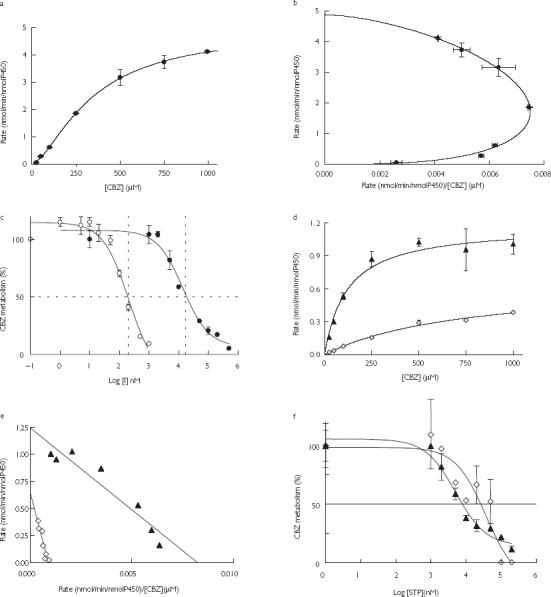
(a,d)The rate of formation of carbamazepine epoxide (CBZE) by human liver microsomes (HLM) and by cDNA-expressed CYP3A4 ▴ and CYP2C8 ◊, respectively, as a function of the concentration of CBZ (0–1000 µm). (b,e) Eadie-Hofstee plot of the data displayed in a and d, respectively CYP3A4 ▴; CYP2C8 ◊. (c) The percentage of CBZ (50 µm) converted to CBZE by human liver microsomes in the presence of increasing concentrations of stiripentol • (STP) (0–500 µm) or ritonavir ○ (RTV) (0–1 µm) (I = inhibitor concentration). (f) The percentage of CBZ (50 µm) converted to CBZE by cDNA-expressed CYP3A4 ▴ and CYP2C8 ◊ in the presence of increasing concentration of STP (0–200 µm). Data points represent the mean of duplicate incubations.
Table 1.
Michaelis-Menten kinetic constants for carbamazepine epoxide (CBZE) formation and the corresponding inhibitory effect of stiripentol and ritonavir, following the incubation of CBZ with human liver microsomes (HLM) [mean ± SD (range) of the six livers], and cDNA-expressed CYP3A4 and 2C8.
| Carbamazepine | HLM( n = 6) | CYP3A4 | CYP2C8 |
|---|---|---|---|
| Apparent Km (µm) | 362 ± 116 (245-609) | 119 | 757 |
| Vmax (nmol min−1 nmol−1 P450) | 10.3 ± 10 (1.6-31.0) | 1.17 | 0.669 |
| CLint (µl min−1 nmol−1) | 24.1 ± 16 (6.5-50.9) | 9.8 | 0.9 |
| IC50stiripentol (µm) | 14.0* | 5.11 | 37.1 |
| IC50ritonavir (µm) | 0.21* | ||
| Apparent Kistiripentol (µm) | 3.7 ± 2.7† | 2.5 | 35 |
| Stiripentol inhibition | Mixed | Competitive | Noncompetitive |
HLM, Human liver microsomes; IC50, 50% inhibitory concentration; apparent Ki, inhibition constant.
Based on a single liver sample.
Mean value from three experiments on the same liver sample.
Inhibition by stiripentol was best described by (i) a mixed inhibition model in human liver microsomes with apparent Ki = 3.7 ± 2.7 µm, (ii) the competitive inhibition model in cDNA-expressed CYP3A4 with apparent Ki = 2.5 µm, and (iii) a noncompetitive inhibition model in cDNA-expressed CYP2C8 with apparent Ki = 35 µm.
Saquinavir-stiripentol interaction
Incubations with human liver microsomes showed that saquinavir was converted to several metabolites, M2 and M7 (major) and M1, M3, M4, M5 and M6 (minor). The formation of M2 and M7 in microsomes from two livers followed Michaelis-Menten kinetics (Figure 3a,b). Apparent Km and Vmax values are displayed in Table 2. Formation of M7 by human liver microsomes was inhibited by stiripentol and ritonavir (Figure 3c). Kinetic analyses suggested that stiripentol was a noncompetitive inhibitor of saquinavir oxidation. IC50 and apparent Ki are displayed in Table 2. Inhibition by stiripentol of saquinavir metabolism was not studied in vitro with cDNA-expressed CYP3A4, because it was only a weak inhibitor in human liver microsomes.
Figure 3.
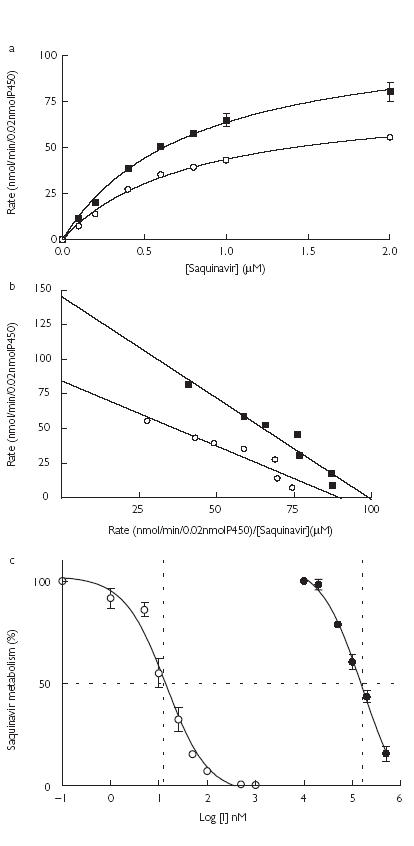
(a) The rate of formation of saquinavir metabolites M2○ and M7▪ by human liver microsomes as a function of the concentration of saquinavir (0–2 µm). (b) Eadie-Hofstee plot of the data represented in a. M2○; M7▪. (c) The percentage of saquinavir (1 µm) converted to its M7 metabolite by human liver microsomes in the presence of increasing concentrations of stiripentol • (STP) (0–500 µm) or ritonavir ○ (RTV) (0–1 µm) (I = inhibitor concentration). Data points represent the mean of duplicate incubations.
Table 2.
Michaelis-Menten kinetic constants for M2 and M7 saquinavir metabolite formation and the corresponding inhibitory effect of stiripentol and ritonavir, following the incubation of saquinavir with two different human liver microsomal preparations.
| HLM1 | HLM2 | |||
|---|---|---|---|---|
| Saquinavir | M2 | M7 | M2 | M7 |
| Apparent Km (µm) | 0.78 | 0.79 | 1.23 | 1.58 |
| Vmax (nmol min−1 nmol−1 P450) | 3.86 | 5.70 | 6.8 | 9.50 |
| CLint (µl min−1 nmol−1) | 4949 | 7215 | 5528 | 6013 |
| IC50stiripentol (µm) | – | 163 | – | – |
| IC50ritonavir (µM) | – | 0.0135 | – | – |
| Apparent Kistiripentol (µm) | – | 86 | – | – |
| Stiripentol inhibition | Non-competitive | |||
HLM, Human liver microsomes; IC50, 50% inhibitory concentration; apparent Ki, inhibition constant.
In vivo studies
Carbamazepine-stiripentol interaction
In epileptic children (n = 17) treated with carbamazepine and stiripentol [5], the ratio of the carbamazepine epoxide/carbamazepine trough plasma concentration was decreased by 65.0 ± 7.5% (Table 3). Apparent Kiin vivo values are presented in Table 3 and Figure 4. In four (numbers 14–17) out of the 17 patients, the correlation of the curve (CLf/CLfi = 1 + I/Kiin vivo, see Methods) was not significant and a poor apparent Kiin vivo determination was assumed in these patients (Table 3). These values were not included in the calculation of mean and standard deviation. The mean apparent Kiin vivo 29.6 ± 9.5 µm (10.5–41.4 µm, n = 13) [95% confidence interval (CI) 23.9, 35.3] was in the range of stiripentol plasma concentrations in epileptic patients (10–60 µm) [3, 5] and in that of the mean apparent Kiin vitro 3.7 ± 2.7 µm (1.5–7.5 µm, n = 3) (95% CI −3.1, 10.5). The 95% CI of stiripentol Kiin vitro is large and includes a negative value because of the high variability of the three values estimated (1.5, 2 and 7.5 µm).
Table 3.
Individual calculated Kiin vivo values for the inhibition of carbamazepine (CBZ) metabolism by stiripentol in epileptic children.
| Patient number | Sample number | Age (year) | CBZ (µM) | CBZE (µM) | Stiripentol (µM) | CBZE/ CBZ | Ki (µM) | Correlation r2 | P |
|---|---|---|---|---|---|---|---|---|---|
| 1 | S1 | 12.6 | 26.7 | 4.91 | 0.0 | 0.184 | 41.4 | 0.855 | 0.003 |
| S2 | 46.6 | 3.37 | 29.9 | 0.072 | |||||
| S3 | 60.1 | 3.69 | 73.8 | 0.061 | |||||
| S4 | 65.6 | 4.40 | 62.3 | 0.067 | |||||
| S5 | 65.6 | 4.40 | 65.3 | 0.067 | |||||
| S6 | 58.4 | 3.92 | 55.9 | 0.067 | |||||
| S7 | 49.5 | 4.28 | 32.0 | 0.086 | |||||
| 2 | S1 | 8.35 | 27.1 | 4.84 | 0.0 | 0.179 | 29.0 | 0.745 | 0.027 |
| S2 | 43.2 | 2.89 | 13.7 | 0.067 | |||||
| S3 | 54.2 | 2.73 | 67.9 | 0.050 | |||||
| S4 | 54.6 | 2.89 | 52.1 | 0.053 | |||||
| S5 | 55.9 | 2.77 | 40.5 | 0.050 | |||||
| S6 | 73.2 | 3.88 | 42.7 | 0.053 | |||||
| 3 | S1 | 9.91 | 27.9 | 5.39 | 0.0 | 0.193 | 28.2 | 0.776 | 0.021 |
| S2 | 64.7 | 3.92 | 50.4 | 0.061 | |||||
| S3 | 43.6 | 2.54 | 44.8 | 0.058 | |||||
| S4 | 36.4 | 2.14 | 41.0 | 0.059 | |||||
| S5 | 44.4 | 2.58 | 54.6 | 0.058 | |||||
| S6 | 39.8 | 2.54 | 66.2 | 0.064 | |||||
| 4 | S1 | 7.63 | 30.0 | 6.58 | 0.0 | 0.219 | 28.0 | 0.925 | 0.009 |
| S2 | 33.4 | 2.93 | 46.1 | 0.088 | |||||
| S3 | 54.6 | 3.33 | 71.7 | 0.061 | |||||
| S4 | 56.3 | 3.49 | 64.9 | 0.062 | |||||
| S5 | 55.0 | 4.20 | 35.4 | 0.076 | |||||
| 5 | S1 | 11.8 | 26.7 | 4.44 | 0.0 | 0.167 | 40.2 | 0.859 | 0.008 |
| S2 | 38.9 | 2.89 | 22.6 | 0.074 | |||||
| S3 | 44.9 | 2.30 | 92.2 | 0.051 | |||||
| S4 | 41.5 | 2.14 | 58.9 | 0.052 | |||||
| S5 | 41.0 | 2.02 | 71.3 | 0.049 | |||||
| S6 | 33.0 | 2.62 | 25.2 | 0.079 | |||||
| 6 | S1 | 7.48 | 44.0 | 8.84 | 0.0 | 0.201 | 40.6 | 0.688 | 0.041 |
| S2 | 37.7 | 3.37 | 22.6 | 0.089 | |||||
| S3 | 63.1 | 3.21 | 51.6 | 0.051 | |||||
| S4 | 53.3 | 2.97 | 93.0 | 0.056 | |||||
| S5 | 58.8 | 3.17 | 99.4 | 0.054 | |||||
| S6 | 47.4 | 2.70 | 51.6 | 0.057 | |||||
| 7 | S1 | 6.47 | 27.9 | 7.97 | 0.0 | 0.285 | 16.6 | 0.810 | 0.006 |
| S2 | 47.0 | 4.52 | 32.4 | 0.096 | |||||
| S3 | 51.2 | 3.29 | 58.0 | 0.064 | |||||
| S4 | 70.7 | 4.72 | 59.3 | 0.067 | |||||
| S5 | 70.7 | 3.80 | 49.5 | 0.054 | |||||
| S6 | 82.1 | 6.90 | 49.5 | 0.084 | |||||
| S7 | 66.4 | 6.34 | 35.9 | 0.095 | |||||
| 8 | S1 | 14.8 | 40.2 | 3.53 | 0.0 | 0.094 | 41.3 | 0.975 | 0.002 |
| S2 | 51.2 | 1.74 | 56.8 | 0.036 | |||||
| S3 | 55.9 | 1.90 | 69.6 | 0.036 | |||||
| S4 | 54.6 | 1.66 | 76.0 | 0.033 | |||||
| S5 | 52.9 | 2.02 | 55.1 | 0.041 | |||||
| 9 | S1 | 8.99 | 32.6 | 6.34 | 0.0 | 0.208 | 29.1 | 0.685 | 0.022 |
| S2 | 38.5 | 4.40 | 26.0 | 0.122 | |||||
| S3 | 38.9 | 2.81 | 40.5 | 0.077 | |||||
| S4 | 35.1 | 3.25 | 50.4 | 0.099 | |||||
| S5 | 55.4 | 5.11 | 41.8 | 0.098 | |||||
| S6 | 38.5 | 2.85 | 42.7 | 0.079 | |||||
| S7 | 44.4 | 2.70 | 47.4 | 0.065 | |||||
| 10 | S1 | 6.75 | 34.3 | 7.25 | 0.0 | 0.226 | 10.5 | 0.846 | 0.009 |
| S2 | 30.0 | 3.33 | 0.0 | 0.118 | |||||
| S3 | 45.7 | 3.13 | 16.2 | 0.073 | |||||
| S4 | 44.0 | 3.01 | 17.9 | 0.073 | |||||
| S5 | 55.0 | 4.00 | 13.7 | 0.078 | |||||
| S6 | 37.7 | 3.25 | 6.8 | 0.092 | |||||
| 11 | S1 | 15 | 25.4 | 3.77 | 0.0 | 0.158 | 27.1 | 0.802 | 0.016 |
| S2 | 37.2 | 1.63 | 37.6 | 0.047 | |||||
| S3 | 51.2 | 2.22 | 61.0 | 0.046 | |||||
| S4 | 54.6 | 2.58 | 54.6 | 0.050 | |||||
| S5 | 52.1 | 2.58 | 51.6 | 0.053 | |||||
| S6 | 49.5 | 2.54 | 32.9 | 0.055 | |||||
| 12 | S1 | 7.38 | 27.5 | 5.19 | 0.0 | 0.202 | 28.3 | 0.735 | 0.029 |
| S2 | 41.5 | 3.21 | 9.0 | 0.083 | |||||
| S3 | 60.5 | 3.41 | 65.7 | 0.060 | |||||
| S4 | 51.2 | 2.58 | 48.2 | 0.054 | |||||
| S5 | 30.5 | 1.82 | 38.4 | 0.064 | |||||
| S6 | 26.7 | 1.43 | 41.4 | 0.057 | |||||
| 13 | S1 | 10.8 | 28.8 | 4.60 | 0.0 | 0.171 | 24.9 | 0.815 | 0.002 |
| S2 | 32.2 | 2.62 | 7.3 | 0.087 | |||||
| S3 | 50.4 | 2.54 | 41.4 | 0.054 | |||||
| S4 | 51.6 | 2.89 | 29.9 | 0.060 | |||||
| S5 | 65.2 | 3.33 | 41.8 | 0.055 | |||||
| S6 | 69.8 | 4.72 | 40.5 | 0.072 | |||||
| S7 | 44.0 | 2.30 | 42.3 | 0.056 | |||||
| S8 | 46.1 | 2.70 | 33.7 | 0.062 | |||||
| 14 | S1 | 2.65 | 15.2 | 3.05 | 0.0 | 0.200 | 4.5 | 0.769 | 0.051 |
| S2 | 19.9 | 1.94 | 3.0 | 0.098 | |||||
| S3 | 38.9 | 3.77 | 5.1 | 0.097 | |||||
| S4 | 29.2 | 2.77 | 2.6 | 0.095 | |||||
| S5 | 45.3 | 3.80 | 5.1 | 0.084 | |||||
| 15 | S1 | 3.7 | 21.2 | 3.53 | 0.0 | 0.178 | 50.7 | 0.436 | 0.153 |
| S2 | 23.7 | 1.90 | 9.4 | 0.086 | |||||
| S3 | 33.4 | 1.98 | 19.2 | 0.063 | |||||
| S4 | 51.2 | 3.37 | 32.9 | 0.070 | |||||
| S5 | 57.1 | 3.65 | 61.9 | 0.068 | |||||
| S6 | 47.4 | 3.41 | 32.9 | 0.077 | |||||
| 16 | S1 | 8.11 | 29.2 | 4.44 | 0.0 | 0.162 | 46.2 | 0.742 | 0.139 |
| S2 | 33.4 | 3.01 | 11.5 | 0.096 | |||||
| S3 | 45.3 | 3.25 | 30.7 | 0.077 | |||||
| S4 | 47.4 | 3.65 | 44.4 | 0.082 | |||||
| 17 | S1 | 10.8 | 23.7 | 3.77 | 0.0 | 0.159 | 38.7 | 0.734 | 0.064 |
| S2 | 30.0 | 2.18 | 17.9 | 0.073 | |||||
| S3 | 30.9 | 1.70 | 36.3 | 0.055 | |||||
| S4 | 36.8 | 1.98 | 63.6 | 0.054 | |||||
| S5 | 39.8 | 2.50 | 55.9 | 0.063 | |||||
| Mean | (1 to 13) | 9.84 | 29.6 | 0.81 | |||||
| SD | 2.94 | 9.47 | 0.09 |
r represents the coefficient of correlation of the regression line: CLf/Clfi = 1 + I/Kiin vivo in which CLf and CLfi represent formation clearances of CBZE in the absence and presence of stiripentol, respectively, I represents the plasma concentration of stiripentol.
Figure 4.
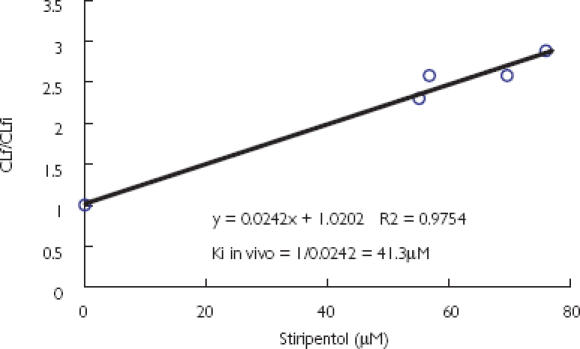
The ratio of carbamazepine epoxide (CBZE) formation clearances without (CLf) and with stiripentol (CLfi) as a function of total plasma concentration of stiripentol in one representative child. The slope is 1/Kiin vivo.
Saquinavir-stiripentol interaction
All 12 subjects completed the study. Treatments were well tolerated. Two subjects presented with adverse effects during the stiripentol phase. One had abdominal pain and another an episode of headache. These effects were moderate and disappeared spontaneously. Treatment was not discontinued.
The plasma concentration of saquinavir during the placebo period was low and variable. Co-administration of stiripentol (1000 mg b.i.d. per 7 days) had no effect on the plasma concentration of saquinavir (Figure 5), or on its pharmacokinetics (Table 4). Neither a period nor order effect was found.
Figure 5.
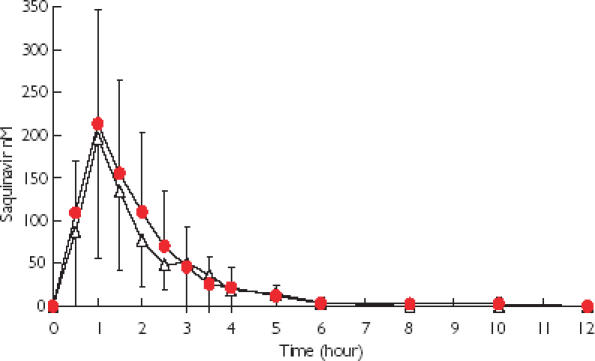
Mean plasma concentration of saquinavir after a single dose of saquinavir (400 mg) administered on day 8 of dosing with stiripentol ( ) (1000 mg) or placebo ▵.
) (1000 mg) or placebo ▵.
Table 4.
Mean ± SD pharmacokinetic parameters for saquinavir during the placebo and the stiripentol phases.
| Period Placebo | Stiripentol | 95% confidence interval on differences | ||
|---|---|---|---|---|
| Tmax (h) | 1.2 ± 0.6 | 0.92 ± 0.2 | NS | 0, 0* |
| Cmax (ng ml−1) | 149.3 ± 83.2 | 149.7 ± 82.4 | – | |
| log Cmax | 4.86 ± 0.58 | 4.84 ± 0.65 | NS | −39.8, +39.8† |
| AUC (ng h ml−1) | 229.4 ± 131.8 | 269.0 ± 189.0 | – | |
| log AUC | 5.23 ± 0.75 | 5.31 ± 0.84 | NS | −33.2, +112† |
| CL/F (l h−1) | 1.90 ± 1.12 | 2.43 ± 1.90 | NS | −0.873, +1.59† |
| t1/2 (h) | 1.12 ± 0.65 | 1.44 ± 0.93 | NS | −0.33, +0.97† |
AUC, Area under the plasma concentration-time curve from 0 to 12 h; Cmax, maximum plasma concentration; tmax, time to reach Cmax; CL/F, apparent clearance; t1/2, half-life; NS, not significant.
Binomial confidence interval.
Student's confidence interval.
Trough stiripentol plasma concentrations ranged from 2.44 to 6.70 mg l−1 (10.4–28.6 µm). No stiripentol was measurable in plasma after the 6-day washout period. No correlation was found between the area under the plasma concentration-time curve for saquinavir (AUCsqv) and that of stiripentol (AUCstp), suggesting that the disposition of saquinavir was not influenced by the systematic exposure to stiripentol.
Discussion
The inhibitory effect of stiripentol on the CYP3A4-dependent metabolism of CBZ and saquinavir has been studied in vitro and in vivo.
In order to saturate CBZE formation, the final concentration of microsomal protein used in our in vitro studies (0.02 nmol P450 ml−1) was 10 times lower than that used previously. Under these experimental conditions, unlike those of Kerr et al. [9], who found a nonlinearity in the biotransformation of CBZ as a function of the concentration of CBZ, we were able to show this linearity and to determine values for Michaelis-Menten kinetic parameters. Using cDNA-expressed CYP3A4 and 2C8, our results were similar to those previously published by Kerr et al. [9] (for CYP3A4, apparent Km = 442 µm and Vmax = 1.73 nmol min−1 nmol−1 P450) and showed that CBZ had a higher affinity for CYP3A4 than for CYP2C8.
The inhibitory effect of stiripentol on the in vitro formation of CBZE was much weaker than that of ritonavir, a potent inhibitor of CYP3A4. Apparent Ki and IC50 values obtained with cDNA-expressed CYPs showed that stiripentol inhibited CYP3A4- preferentially to CYP2C8-dependent biotransformation of CBZ. Stiripentol inhibited the former competitively and the latter noncompetitively. These CYPs are the major enzymes responsible for CBZ metabolism [9]. In human microsomes, stiripentol inhibited the activity of both CYP3A4 and 2C8. The data were best described by a mixed inhibition model.
The interaction between stiripentol and carbamazepine has already been reported in vitro and in vivo by Tran et al. [2]. However, an apparent Ki could not be calculated because CBZ was not used at saturating concentration due to its poor solubility. In the present work, the problem of CBZ solubility was overcome by using a small concentration of microsomal protein in the incubation medium. An apparent in vivo Ki was determined in 13 epileptic patients. These results were similar to those published by Tran et al. [2] in a preliminary study comprising five patients. The model used to calculate apparent Kiin vivo for stiripentol assumes that the kinetics of CBZ biotransformation is linear and that the inhibition of CBZ metabolism by stiripentol is competitive. This hypothesis is consistent with the experimental data, as the minimum therapeutic plasma concentrations of CBZ (15–60 µm) were much smaller than apparent Km (362 µm) and below the Km the enzyme kinetics are linear. Moreover, the inhibition of CYP3A4 by stiripentol is competitive and this CYP metabolizes 60–80% of CBZ [9]. Inhibition of CYP2C8 by stiripentol was weak compared with that of CYP3A4 (apparent Ki = 35 vs. 2.7 µm). Therefore, the estimates of apparent Kiin vivo appeared appropriate. Moreover, the values calculated in children in the present study (10.5–41.4 µm) were similar to those reported by Tran et al. [2] in two adults and in three 10–16-year-old children (12–34 µm).
Stiripentol concentrations observed in epileptic patients (10–60 µm) [3–5] were in the range of its apparent in vivo Ki (10.5–41.4 µm). As the concentration of protein in the incubation system was low (0.06 mg ml−1), nonspecific microsomal binding of stiripentol was assumed to be negligible and the Ki of stiripentol observed in vitro was considered to be an unbound in vitro Ki. As stiripentol is 95% bound in plasma, unbound Kiin vivo values (5% of total Ki) would range from 0.5 to 2.1 µm. These values were similar to the Kiin vitro observed in microsomes (3.7 µm). Therefore, the in vivo data are consistent with the in vitro data. The availability of in vivo inhibition constants provides a new framework for in vitro-in vivo comparisons and emphasizes the relevance of in vitro data to the clinical setting.
Apparent Km values for the formation of M2 and M7 from saquinavir in human liver microsomes were low (0.8–1.6 µm), showing a high affinity of the drug for the enzymes involved. Km values were similar to those reported by Fitzsimmons et al. [12] using human small intestine (apparent Km 0.3–0.5 µm). Stiripentol appeared to be a noncompetitive inhibitor of the biotransformation of saquinavir to M7in vitro. The apparent Ki and IC50 (86 and 163 µm, respectively) were consistent with a weak inhibition in comparison with that seen with ritonavir (IC50 = 0.0135 µm). Because inhibition of saquinavir by stiripentol was weak, no further experiments were conducted in vitro with cDNA-expressed CYP3A4.
In vivo, no change in the pharmacokinetics of saquinavir during the stiripentol treatment period was observed. Stiripentol plasma concentrations observed in the healthy volunteers were lower than apparent Ki and IC50 values calculated in vitro, which may explain the lack of effect of stiripentol on saquinavir kinetics. The differences in the magnitude of the inhibitory effect of stiripentol and ritonavir in vivo on saquinavir pharmacokinetics [22] are consistent with the results of our in vitro experiments.
The increase of saquinavir AUC by ritonavir is attributed in part to the inhibition of saquinavir intestinal transfer by P-glycoproteine, NDR1 gene product (P-gp). The differences we observed between CBZ and saquinavir might also be related to a differential inhibitory effect of stiripentol on P-gp activity. The difference in the effect of stiripentol may as well be related to a differential inhibitory effect of stiripentol at the intestinal level (P-gp, CYP3A4, CYP3A5), because of differences in intestinal compared with plasma concentrations. Taken together these mechanisms may explain on one hand the difference in the effect of stiripentol in the present sl in vivo study and ritonavir [22] on the pharmacokinetics of saquinavir, and the potent effect of stiripentol on the pharmacokinetics of CBZ.
Since stiripentol inhibits CBZ metabolism, the autoinduction of CBZ metabolism may not be as pronounced in patients treated with these two drugs together. The decrease in CBZ concentrations that occurs after the onset of CBZ treatment might be prevented by stiripentol coadministration, thus shortening the time to reach the optimum dose of CBZ.
In conclusion, the effect of stiripentol was studied on the CYP3A4-mediated biotransformation of CBZ and saquinavir. Stiripentol was a poor inhibitor of saquinavir metabolism both in vitro and in vivo and probably would not prove to be useful as an adjunctive treatment to saquinavir by decreasing its first pass effect in HIV-infected patients. In epileptic patients stiripentol was a potent inhibitor of CBZ transformation and could be used to prolong the dosing interval of CBZ.
Acknowledgments
Supported by a grant from Laboratoires Biocodex (Montrouge, France).
References
- 1.Hsu A, Granneman R, Bertz R. Ritonavir, clinical pharmacokinetics and interactions with other anti-HIV agents. Clin Pharmacokinet. 1998;35:275–291. doi: 10.2165/00003088-199835040-00002. [DOI] [PubMed] [Google Scholar]
- 2.Tran A, Rey E, Pons G, et al. Influence of stiripentol on cytochrome P450-mediated metabolic pathways in humans: in vitro and in vivo comparison and calculation of in vivo inhibition constants. Clin Pharmacol Ther. 1997;62:490–504. doi: 10.1016/S0009-9236(97)90044-8. [DOI] [PubMed] [Google Scholar]
- 3.Tran A, Vauzelle-Kervroedan Rey E, Pons G, et al. Effect of stiripentol on carbamazepine plasma concentration and metabolism in epileptic children. Eur J Clin Pharmacol. 1996;50:497–500. doi: 10.1007/s002280050147. [DOI] [PubMed] [Google Scholar]
- 4.Perez J, Chiron C, Musial C, et al. Stiripentol: efficacy and tolerability in children with epilepsy. Epilepsia. 1999;40:1618–1626. doi: 10.1111/j.1528-1157.1999.tb02048.x. [DOI] [PubMed] [Google Scholar]
- 5.Chiron C, Tran A, Rey E, et al. California, USA: American Epilepsy Society Los Angeles; Stiripentol in childhood partial epilepsy: a placebo-controlled trial. 1–6 December 2000; abstract. [Google Scholar]
- 6.Tran A, Treluyer JM, Rey E, et al. Prospective effect of stiripentol on acetaminophen-induced hepatotoxicity in rat. Toxicol Appl Pharmacol. 2001;170:145–152. doi: 10.1006/taap.2000.9091. [DOI] [PubMed] [Google Scholar]
- 7.Cresteil T, Flinois JP, Pfister A, Leroux JP. Effect of microsomal preparations and induction on cytochrome P450 dependent monooxygenases in fetal and neonatal rat liver. Biochem Pharmacol. 1979;28:2057–2063. doi: 10.1016/0006-2952(79)90224-7. [DOI] [PubMed] [Google Scholar]
- 8.Greim H. Synthesesteigerung und Abbauhemung bei der Vermchrung der Mikrosomalen Cytochrome P450 und b5 durch Phenobarbital. Arch Pharmakol. 1970;266:260–275. [PubMed] [Google Scholar]
- 9.Kerr BM, Thummel KE, Wurden CJ, et al. Human liver carbamazepine metabolism, role of CYP3A4 and 2C8 in 10,11-epoxide formation. Biochem Pharmacol. 1994;47:1969–1979. doi: 10.1016/0006-2952(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 10.Chauret N, Gauthier A, Nicoll-Griffith DA. Effect of common organic solvents on in vitro cytochrome P450 mediated metabolic activities in human liver microsomes. Drug Metab Dispos. 1998;26:1–4. [PubMed] [Google Scholar]
- 11.Busby WF, Jr, Ackermann JM, Crespi CL. Effect of methanol, ethanol, dimethylsulfoxide and acetonitrile on in vitro activities of cDNA-expressed cytochromes P450. Drug Metab Dispos. 1999;27:246–249. [PubMed] [Google Scholar]
- 12.Fitzsimmons M, Collins J. Selective biotransformation of the HIV protease inhibitor saquinavir by human small-intestinal cytochrome P4503A4, potential contribution to high first pass metabolism. Drug Metab Dispos. 1997;25:256–267. [PubMed] [Google Scholar]
- 13.Yamaoka K, Nakagawa T, Uno T. Application of Akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. 1978;6:165–175. doi: 10.1007/BF01117450. [DOI] [PubMed] [Google Scholar]
- 14.Shaw PN, Houston JB. Kinetics of drug metabolism inhibition: use of metabolite concentration-time profiles. J Pharmacokinet Biopharm. 1987;15:497–510. doi: 10.1007/BF01061759. [DOI] [PubMed] [Google Scholar]
- 15.Adedoyin A, Aarons L, Houston JB. Plasma concentration-response relationship for cimetidine inhibition of drug metabolism in the rat. Drug Metab Dispos. 1987;15:127–132. [PubMed] [Google Scholar]
- 16.Kunze KL, Trager WF. Warfarin-fluconazole. III. A rational approach to management of a metabolically based drug interaction. Drug Metab Dispos. 1996;24:429–435. [PubMed] [Google Scholar]
- 17.Ha HR, Follath F, Bloemhard Y, Krahenbuhl S. Determination of saquinavir in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1997;694:427–433. doi: 10.1016/s0378-4347(97)00165-5. [DOI] [PubMed] [Google Scholar]
- 18.Gex-Fabry M, Balant LP. Considerations on data analysis using computer methods and currently available software for personal computers. Handbooks Exp Pharmacol. 1992;110:507–527. [Google Scholar]
- 19.Mayhew BS, Jones DR, Hall SD. An in vitro model for predicting in vivo inhibition of cytochrome P450 3A4 by metabolic intermediate complex formation. Drug Metab Dispos. 2000;28:1031–1037. [PubMed] [Google Scholar]
- 20.Voorman RL, Maio SM, Payne NA, Zhao Z, Koeplinger KA, Wang X. Microsomal metabolism of delavirdine: evidence for mechanism-based inactivation of human cytochrome P4503A. J Pharmacol Exp Ther. 1998;287:381–388. [PubMed] [Google Scholar]
- 21.Yao C, Kunze KL, Kharasch ED, et al. Fluvoxamine-theophylline interaction: gap between in vitro and in vivo inhibition constants toward cytochrome P4501A2. Clin Pharmacol Ther. 2001;70:415–424. doi: 10.1067/mcp.2001.119724. [DOI] [PubMed] [Google Scholar]
- 22.Hsu A, Granneman R, Cao G, et al. Pharmacokinetic interaction between two human immunodeficiency virus protease inhibitors, ritonavir and saquinavir. Clin Pharmacol Ther. 1998;63:453–464. doi: 10.1016/S0009-9236(98)90041-8. [DOI] [PubMed] [Google Scholar]