

Abstract
Aims
We aim to modulate the renin–angiotensin system (RAS) by active immunization against angiotensin I hormone (AI), potentially providing a novel conjugate vaccine treatment for hypertension in man.
Methods
Immunization studies in rat and human subjects compare the effectiveness of tetanus toxoid (TT) and keyhole limpet haemocyanin (KLH) vaccines for immunotherapy following conjugation with an AI peptide analogue (AI). Cardiovascular responses were assessed in immunized rats and human subjects (two-dose trial only), following increasing i.v. infusions of either AI or angiotensin II hormone (AII).
Results
The AI–TT and AI–KLH conjugate vaccines induced an equivalent immune response, and inhibition of the pressor effects to exogenous AI in rats. Single-dose clinical trials with both conjugate vaccines only resulted in an immune response to the KLH carrier protein. A two-dose clinical trial of AI–KLH conjugate vaccine resulted in a significant immune response to AI. A shift in diastolic blood pressure (DBP) dose–response was demonstrated following challenge with AI and AII for the study volunteer showing the largest anti-AI IgG induction.
Conclusion
KLH was shown to be a suitable alternative to TT as a carrier protein for AI, thus supporting continued evaluation of our AI–KLH conjugate vaccine for treatment of hypertension in man.
Keywords: angiotensin conjugate vaccines, epitopic suppression, hypertension
Introduction
The renin–angiotensin system (RAS) commands an important physiological role, influencing normal cardiovascular status and contributing to diseases such as hypertension and heart failure [1]. Consequently, the RAS has been a target for clinical control, either by angiotensin-converting enzyme (ACE) inhibitors [2], or angiotensin II hormone (AII)-receptor antagonists [3]. Due to undesirable side-effects resulting from these treatment forms such as dry cough, first dose hypotension, diuretic interactions and angioneurotic oedema [4, 5] as well as poor oral drug compliance, immunization against angiotensins has been suggested as an attractive alternative for RAS-targeted control of hypertension [6]. Besides these clinical advantages over ACE inhibitors and AII-receptor antagonists, there are other potential benefits to a vaccine treatment for controlling hypertension. Potential benefits include improvements in compliance management (especially mild/moderate hypertension), smooth and progressive onset of action for patients with mild hypertension or left ventricular dysfunction, improved diurnal control of blood pressure and reduction in drug interactions associated with conventional drug polypharmacy. These advantages are due to the vaccine mode of action, where infrequent doses induce a biological response, overcoming the need for one or more daily tablets. Therefore, vaccination could provide a treatment for controlling hypertension having improved patient acceptability and compliance, with a smooth onset of action.
The vaccines evaluated in this paper induce blocking immunoglobulins against angiotensin I hormone (AI), the ACE substrate, preventing generation of AII and subsequent increase of blood pressure. The immunoglobulins are raised to a peptide analogue (AI) having projected structural similarity to the native AI hormone. To increase immunogenicity the AI is conjugated to a carrier protein and adjuvanted with aluminium hydroxide gel. Similar methods of inducing immunoglobulins for a number of biological and clinical applications have been documented previously [7, 8, 9, 10, 11, 12, 13, 14, 15, 16]. Described are the design considerations [7–11] and use [12–16] of small molecules to elicit induction of immunoglobulins to a range of targets including hormones, coenzymes, drugs, toxins, protein fragments, carbohydrates, cholesterol and nucleic acids.
We have shown that rats treated with a conjugate vaccine containing AI, tetanus toxoid (TT) carrier protein and aluminium hydroxide adjuvant, demonstrated highly significant reductions in the pressor response to challenge with exogenous AI [17]. Similar earlier studies but in spontaneously hypertensive rats (SHRs), demonstrated that blood pressure-lowering effects of AI-receptor antagonists (valsartan and frusemide) were comparable to the AI–TT conjugate vaccine immunized SHRs [18]. However, successful clinical use of such a conjugate vaccine may have limited application, due to the potential for epitopic suppression: a phenomenon demonstrated in rodents [19–21] and man [22, 23]. Epitopic suppression results when a subject is vaccinated with a compound to which they have been previously exposed. So this is important to question, since TT is a common immunogen in man. Epitopic suppression is due to antigenic competition between an expanded population of carrier specific B-cell clones, and the unexpanded population of naive, peptide analogue-specific B-cell clones [24, 25]. Thus, the possibility of epitopic suppression limits the use of common immunogens as peptide carriers for novel conjugate vaccines, due to the difficulty of predicting the effectiveness of a response following vaccination.
To avoid this potential problem, we have assessed keyhole limpet haemocyanin (KLH) as an alternative peptide carrier protein to TT for a novel conjugate vaccine treatment. KLH is a potent immunogen that is prepared from the mollusc, Megathura crenulata, in a form suitable for use as peptide carrier in a conjugate vaccine. This large, respiratory glycoprotein has been characterized for biochemical [26, 27] and immunological function [28–31]. KLH has been used in a range of immunotherapeutic studies, both unconjugated [32–34], and as a carrier conjugated with peptide analogues [35–37]. The commercial availability, and its use as a carrier for a range of peptide immunotherapeutics, supports the suitability of KLH for development of our antihypertensive conjugate vaccine.
In this paper we describe a series of studies with AI, conjugated with either TT or KLH carrier protein, coded PMD2850 and PMD3117, respectively. These conjugates were mixed with adjuvant to form vaccines and used to immunize rats, and, subsequently, healthy human volunteers (single-dose clinical trial). The degree of inhibition of the pressor response to either AI or AII was assessed in the rats. These studies compared each peptide–carrier protein conjugate vaccine with respect to the effect on the anti-AI immune response, and any subsequent control of experimentally induced hypertension. Thereafter, a two-dose clinical trial was initiated using an AI–KLH conjugate vaccine. The degree of inhibition of the pressor response to either AI or AII was assessed as part of this second clinical trial. The results indicated that it might be possible to produce a peptide–carrier protein conjugate vaccine with a potential to control hypertension in man. Some of the results herein have been presented to the British Pharmacological Society [38].
Methods
Angiotensin vaccine preparation
The AI peptide analogue (AI), was purchased from Cambridge Research Biochemicals (Zeneca, UK). The appropriate amount of peptide was weighed out, dissolved in phosphate buffered saline (PBS buffer), and mixed with either of the two activated carrier proteins. The carrier proteins, purchased in solution, were TT (Chiron Behring, Germany) and KLH (Biosyn Arzneimittel, Germany). To activate for conjugation, an appropriate amount of each carrier protein was mixed with an excess of m -maleimidobenzoyl-N -hydroxysulphosuccinimide ester, a bivalent linker (Pierce, Rockford, IL, USA). Following activation, the carrier proteins were separated from the remaining reaction components by size exclusion chromatography on Sephadex G-25 matrix columns (Pharmacia, Uppsala, Sweden). The level of maleimide activation of each carrier protein was determined using an assay developed in-house (PMD, Runcorn, UK), before being mixed with an excess of AI to conjugate. Following the reaction, conjugates were separated from the remaining free peptide by size exclusion chromatography on Sephadex G-25 matrix columns (Pharmacia). The conjugates were sterilized using 0.2-µm filters (Millipore, UK) and the AI–carrier protein components identified using an ELISA developed in-house (PMD). The peptide concentration of each was determined following protein concentration analysis using a bicinchoninic acid (BCA) kit (Pierce) and utilizing the level of maleimide activation determined for each carrier protein. The conjugates were then formulated into vaccines, yielding the appropriate AI concentration, using Alhydrogel® (Superfos, Denmark) as adjuvant and 0.9% w/v saline (Flowfusor®; Fresenius, UK) as the conjugate vaccine vehicle. The conjugate vaccines were formulated to dose recipients with AI equivalents (µg).
Ethical considerations
The clinical trials described were performed at good clinical practice (GCP) compliant clinical research organizations (DDS and GDRU) in the UK with approval of the local ethics committee at each study centre. Written consent was obtained from all study subjects following a full explanation of what was involved in the study. Materials for the clinical trials were produced to current good manufacturing practice (GMP) under international conference on harmonization (ICH) guidelines.
Preclinical toxicology
Preclinical toxicological safety was demonstrated following evaluation based on regulatory (ICH) guidelines for a new chemical entity, adapted to incorporate specific issues applicable to a peptide linked to a conjugate and formulated with an adjuvant. Both TT and KLH conjugate vaccine formulations were assessed in the toxicology studies which included: acute (for systemic indications), subchronic (including clinical chemistry, haematology, macroscopic and histopathological assays), mutagenic (including bacterial-AMES, mouse lymphoma and micronucleus assays), local tolerance and safety pharmacology (Irwin behavioural screen) protocols. The toxicology studies were carried out at recognized contract research organizations (CTL, Alderley Park, UK and IRI, Tranent, UK) according to the principles of Good Laboratory Practice (GLP).
Immunization protocol
The four studies described are referred to as Study A, B, C or D having treatment, vaccine formulation, and experimental regimes as indicated in Table 1. Each of the study subjects was injected with either a placebo control (saline or Alhydrogel), or a conjugate vaccine in volumes as indicated. In Study A, male, Sprague-Dawley rats (Harlan Olac, UK), with a body weight of 200–250 g were used. The sample number (n) for all treatment groups and saline control was n = 6; the injection volume for all treatment groups, and the saline control group was 0.5 ml. In Studies B, C and D, healthy, male, human volunteers of body weight 65–90 kg, body mass index 18–28 kg m−2 and aged 18–45 years were chosen. In Study B, for all treatment groups n = 2, and for the saline control n = 8. The injection volumes for all treatment groups and the saline control group were between 1 and 2 ml. In Study C, for all treatment groups n = 4, and for the saline control n = 6. The injection volumes for all treatment groups and the saline control group were between 0.5 and 2 ml. In Study D, for the treatment group and Alhydrogel control, n = 8. The injection volume for the treatment group and the adjuvant (Alhydrogel®) control group was 1 ml.
Table 1.
Study treatment groups, their respective conjugate vaccine formulation, AI equivalent dose and experimental regime.
| Days | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Days Injection | Blood sample | Challenge AI/AII | ||||||||||||
| Study | PMD code | Formulation | AI dose (µg) | 0 | 21 | 42 | 0 | 7 | 14 | 21 | 28 | 42 | 49 | |
| A | None | Saline control | 0 | • | • | • | • | • | • | 63 | ||||
| PMD2850 | AI–TT, Al(OH)3 | 25 | • | • | • | • | • | • | 63 | |||||
| PMD3117 | AI–KLH, Al(OH)3 | 10, 50 | • | • | • | • | • | • | 63 | |||||
| B | None | Saline control | 0 | • | • | • | • | • | • | |||||
| PMD2850 | AI–TT, Al(OH)3 | 10, 20, 50, 100 | • | • | • | • | • | • | ||||||
| C | None | Saline control | 0 | • | • | • | • | • | • | |||||
| PMD3117 | AI–KLH, Al(OH)3 | 25, 50, 100 | • | • | • | • | • | • | • | |||||
| D | None | Al(OH)3 | 0 | • | • | • | • | • | − 1 & 49 | |||||
| PMD3117 | AI–KLH, Al(OH)3 | 50 | • | • | • | • | • | − 1 & 49 | ||||||
AI/AII, Angiotensin I or II hormone; AI, peptide analogue of AI; PMD, Protherics plc.; TT, tetanus toxoid peptide carrier protein; Al(OH)3, Alhydrogel® gel adjuvant; KLH, keyhole limpet haemocyanin peptide carrier protein; •, treatment.
Study A: rat immunoglobulin class and subclass response
To measure immunoglobulin class and subclass response, sera collected 42 days after three immunizations with either AI–TT (25 µg) or AI–KLH (10 µg) conjugate vaccine was used (see Table 1). Sera samples were diluted with 0.9% w/v saline (Flowfusor®; Fresenius) and measured for three immunoglobulin classes (IgA, IgM and IgG) and four IgG subclasses (IgG1, IgG2a, IgG2b, IgG2c), by ELISA at the National Institute for Biological Standards and Control (Potters Bar, UK). Whole IgG was measured using an antirat IgG–horseradish peroxidase conjugate. All other immunoglobulins were measured using biotinylated IgG conjugates and a streptavidin-based amplification system.
Study A: in vivo angiotensin pressor testing
Sixty-two days after the first immunization, rats were anaesthetized using sodium methohexitone (40–60 mg kg−1 i.p. supplemented as required) and catheters were implanted in the abdominal aorta (via the ventral caudal artery) and the right jugular vein. Catheters ran subcutaneously to exit at the back of the neck, and then through a flexible spring (for protection) attached to a harness fitted to the rat. The spring was supported by a freely moving, counterbalanced lever. The arterial catheter was connected to a swivel system to allow continuous infusion of saline to maintain patency [39]. The following day, when animals were conscious, unrestrained and with free access to food and water, cardiovascular responses were assessed. Increasing i.v. bolus (0.1 ml) doses of either AI (3, 6, 18, 30 and 60 pmol/rat) or AII (0.5, 5 and 25 pmol/rat) were given with sufficient intervals (at least 15 min) between doses to allow the acute pressor effects to wane. Measurements of mean arterial blood pressure (MBP) were made immediately before, and 0.25, 0.5, 0.75, 1, 2, 3, 4 and 5 min after each injection of AI or AII. In each animal, the peak pressor response to AI and AII occurred within the first minute after injection, and this value was used in the assessment of response. Animals were randomized to receive AI or AII as the first challenge. At the end of the experiment rats were terminally anaesthetized (sodium pentobarbitone, 100 mg i.v.) and a blood sample was taken by cardiac puncture for the measurement of anti-AI IgG by ELISA.
Study D: in vivo angiotensin pressor testing
On days −1 and 49 of the protocol, a series of ascending i.v. infusions lasting 5 min each were administered to the supine volunteers via an indwelling cannula. The doses were AI (4, 20, 40, 60 and 80 ng min−1 kg−1) followed by AII (1, 5, 10, 15 and 30 ng min−1 kg−1), until an increase of at least 25 mmHg in diastolic blood pressure (DBP) was achieved. There was a wash-out period of at least 30 min between the end of the last AI infusion and the first infusion of AII. A single blood pressure measurement was made during the final 2 min of each infusion using a Datascope Accutorr 2A monitor (Datascope, USA). This methodology is based on that of Erb et al. [40] and Essig et al. [41].
ELISA analysis of the sera
ELISA plate wells (Nunc, UK) were coated for 1 h at 20–22 °C with either TT, KLH, or AI which had been conjugated to bovine serum albumin (BSA; Sigma, Poole, UK), as a carrier; control wells were coated with BSA. The coated wells were washed throughout with PBS buffer containing 0.2% v/v Tween 20 (Sigma). Any remaining well space was blocked using PBS buffer containing 3% w/v dried milk powder (Study A sera analysis only). Diluted sera from the vaccinated subjects were then incubated in their respective wells. For detection of the AI–BSA conjugates, the rat sera (Study A) were diluted over a range from 1000- to 10 000-fold in PBS buffer (Sigma). The human sera (Studies B, C and D) were diluted over a range from 1000- to 5000-fold in PBS buffer containing 0.2% v/v Tween 20. For detection of TT and KLH, the human sera (Studies B, C and D) were diluted over a range from 1000- to 25 000-fold in PBS buffer containing 0.2% v/v Tween 20. Following incubation (1 h at 20–22 °C) and subsequent plate washing, immobilized IgG were detected using either rabbit antirat IgG–horseradish peroxidase (Sigma) or sheep antihuman IgG–horseradish peroxidase conjugate (Sigma) diluted in PBS buffer containing 0.2% v/v Tween 20. The peroxidase chromogenic substrate, 3,3′-5,5′-tetramethyl benzidine (TMB; Sigma), was mixed with 0.5% v/v hydrogen peroxide (Sigma) in a sodium acetate buffer and incubated in the ELISA plate wells. The reaction was terminated by the addition of 10% v/v sulphuric acid (Sigma). Colour generated following reaction between the peroxidase and TMB substrate was determined by absorbency at 450 nm, using a Packard plate reader (Packard, USA). Test A450-nm readings were adjusted for background IgG interactions by deducting the control BSA absorbency reading for each study sera. For Studies A and D the adjusted absorbency readings were then analysed by a statistical package (SAS Institute, USA) to determine the sera anti-AI IgG titre resulting from each conjugate vaccine treatment. A titre being the estimated serum dilution corresponding to an increase of 0.1 nm in the adjusted absorbency reading.
Study A and D: blood pressure data analysis
The maximum change in MBP relative to the value immediately prechallenge was calculated for each conjugate vaccine-treated subject and each AI or AII challenge dose. The AI and AII dose–response for each subject was modelled by fitting a three-parameter logistic:
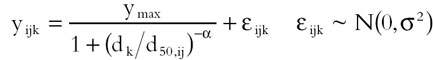 |
In this model, the maximum response, ymax, and slope parameter, α, are assumed to be the same for all subjects, and d50,ij is the estimated median effective dose, ED50 (i.e. challenge dose giving a half-maximal increase in MBP) for subject j in treatment group i; yijk is the peak change in MBP following challenge with dose dk of AI and AII. values were analysed by anova to test for significant differences between study groups. This model is similar to one used by Belz et al. [42].
Results
In Study A, active immunization produced anti-AI IgG titres and shifts in the pressor responses to AI and AII, as shown in Table 2. The IgG titres were similar for the groups actively immunized with AI–TT (PMD2850, 25 µg) and AI–KLH (PMD3117, 10 or 50 µg) conjugate vaccines, irrespective of the AI dose. There was a significant reduction in MBP response to AI for the actively immunized groups compared with the saline control group (P < 0.05). However, there were no significant decreases in MBP responses to AII for any of the active treatment groups in this study.
Table 2.
Effects of immunization on both the anti-(AI) IgG titres and mean arterial blood pressure (MBP) response to AI or AII challenge in Study A rats.
| Peak change in MBP (mmHg) over all doses of agonist | |||
|---|---|---|---|
| Treatment and AI dose | Anti-AI IgG titre | AI | AII |
| Saline control (0 µg) | 0 | 25.5 ± 2.8 | 24.3 ± 1.6 |
| PMD2850 (25 µg) | 55629 ± 14414 | 15.4 ± 1.7* | 22.1 ± 1.5 |
| PMD3117 (10 µg) | 66121 ± 13264 | 13.3 ± 2.4* | 15.7 ± 3.0 |
| PMD3117 (50 µg) | 67410 ± 7393 | 13.8 ± 2.8* | 19.0 ± 3.3 |
Significance probabilities (P) were adjusted for the multiple MBP comparisons by Dunnett's method,
P < 0.05 vs. the saline control treatment. Values are mean ± SEM, n = 6.
Classifying the anti-AI immunoglobulin subclasses in rat sera following active immunization demonstrated a similar response profile to both AI–TT and AI–KLH conjugate vaccines (see Table 3). The highest immunoglobulin titre was shown for whole IgG. Dividing the IgG response into four subclasses showed that IgG2a, followed by IgG1, had the highest titres. A similar low titre was detected for IgM, whilst no IgA resulted for either conjugate vaccine.
Table 3.
Immunoglobulin class response to (AI) in sera from Study A rats treated with either AI–tetanus toxoid (TT) or AI–keyhole limpet haemocyanin (KLH) conjugate vaccine.
| Class or subclass | AI–TT conjugate vaccine Ig titre | AI–KLH conjugate vaccine Ig titre |
|---|---|---|
| IgG (whole molecule) | 10 000 | 12 000 |
| IgG1 | 900 | 700 |
| IgG2a | 1 600 | 2 500 |
| IgG2b | 800 | 200 |
| IgG2c | 400 | 300 |
| IgA | 0 | 0 |
| IgM | 120 | 100 |
Immunoglobulin (Ig) titres shown are the mid-point values from duplicate analysis.
No anti-AI IgG induction was detected in humans given single doses of AI–TT or AI–KLH conjugate vaccine in Studies B or C, respectively (data not shown). The ELISA optical densities neither increased nor differed greatly for any of the actively immunized treatments, compared with the saline control group (0 µg AI dose). Immunoglobulins were only generated against TT (see Figure 1) at the highest conjugate vaccine dose of 100 µg AI. However, immunoglobulins were generated against KLH (see Figure 2) for all actively immunized treatment groups. The two highest dose groups showed maximum IgG induction against KLH at 21 days after the single conjugate vaccine dose. The largest increase in IgG against KLH resulted with the conjugate vaccine dose of 50 µg AI.
Figure 1.
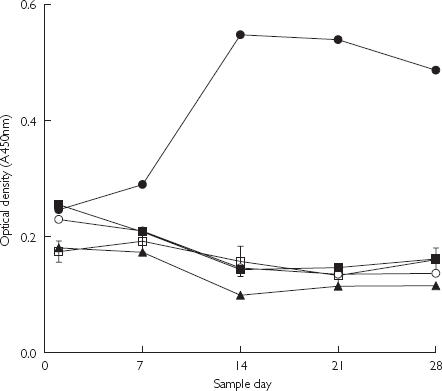
Results from ELISA showing the binding to tetanus toxoid (TT) carrier protein of human antiserum IgG raised against (AI)–TT conjugate vaccine encoded PMD2850 (see Table 1; Study B). The AI dose was 10 µg (^), 20 µg (▪), 50 µg (♦), 100 µg (•) and 0 µg (□). Values are mean for each group and error bars show standard error of the mean (n = 8), for the 0-µg AI dose only.
Figure 2.
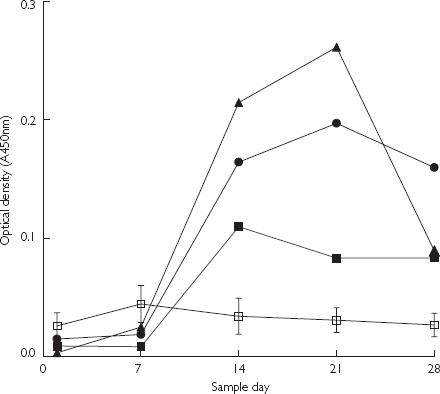
Results from ELISA showing the binding to keyhole limpet haemocyanin (KLH) carrier protein of human antiserum IgG raised against (AI)–KLH conjugate vaccine encoded PMD3117 (see Table 1; Study C). The AI dose was 25 µg (▪), 50 µg (•), 100 µg (•) and 0 µg (□). Values are mean for each group and error bars show standard error of the mean (n = 6), for the 0-µg AI dose only.
A two-dose immunization with AI–KLH conjugate vaccine (Study D) of healthy, male, human volunteers caused production of anti-AI IgG molecules. At 21 days after the second AI–KLH conjugate vaccination (50 µg) the range of anti-AI IgG titres obtained was 2381–18 651, with a mean value of 6777 ± 1822 SEM. No statistically significant effect of treatment was found with either AI or AII challenge on MBP, the combined mean of systolic blood pressure (SBP) and DBP (data not shown). None of the AI or AII challenge-treatment effects reach conventional levels of statistical significance (P ≤ 0.05) for either mean change in SBP or DBP (see Table 4). However, it was encouraging that for the study subject showing the largest anti-AI IgG induction there was a shift of up to two-fold in the challenge dose of exogenous AI and AII required to cause a 15-mmHg rise in DBP.
Table 4.
Effects of immunization on the systolic blood pressure (SBP) and diastolic blood pressure (DBP) response to angiotensin I hormone (AI) or angiotensin II hormone (AII) challenge in humans (Study D).
| Change in SBP and DBP (mmHg) at each dose of agonist | ||
|---|---|---|
| AI or AII challenge & dose | SBP | DBP |
| AI (4 ng min−1 kg−1) | −5.7 ± 3.4 (P = 0.12) | −1.1 ± 3.7 (P = 0.76) |
| AI (20 ng min−1 kg−1) | −7.4 ± 4.3 (P = 0.11) | −2.4 ± 2.7 (P = 0.39) |
| AII (1 ng min−1 kg−1) | −1.3 ± 4.3 (P = 0.76) | −0.8 ± 1.8 (P = 0.67) |
| AII (5 ng min−1 kg−1) | −1.8 ± 4.8 (P = 0.72) | −2.3 ± 2.0 (P = 0.29) |
Results shown are from analyses with mean change in SBP and DBP seen in the study subjects to the infused AI or AII doses. Significance probabilities (P) were adjusted for the multiple blood pressure comparisons by a two-sided t-test method vs. the Al(OH)3 control treatment using day −1 response as covariant. Values are mean ± SEM, n = 8.
Discussion
Active immunization of rats with a novel immunogen consisting of AI conjugated with either TT or KLH (Study A) demonstrated equivalent anti-AI IgG titres and shift in the dose–response to exogenous AI challenge. Conversely, no shift in the dose–response to exogenous AII challenge was observed with either AI–carrier protein conjugate vaccine treatment in rats. The results observed with the AI–TT conjugate vaccine are consistent with a previously study [18]. Similar immunoglobulin classification profiles were observed for both AI–carrier protein conjugate vaccine treatments. The highest immunoglobulin titre was observed for whole IgG, whilst subclassification demonstrated the same response profiles for both AI–TT and AI–KLH conjugate treatments. These results show that AI–TT and AI–KLH conjugate vaccines had the same effect on the generation of anti-AI immunoglobulins in rats, and caused similar inhibition of the pressor effects of AI. Therefore, KLH provides a suitable alternative to TT as a peptide carrier protein in a novel conjugate vaccine for suppressing AI responses, in rats.
A single treatment dose in a Phase Ia clinical study of healthy, male, human volunteers with both AI–carrier protein conjugate vaccines (Studies B and C) did not cause an anti-AI IgG response. However, an anti-carrier protein IgG response resulted with the highest AI–TT conjugate vaccine dose (100 µg AI) and each of the AI–KLH conjugate vaccine doses investigated. The poor anti-carrier protein response observed after a single dose of AI–TT conjugate vaccine in volunteers potentially demonstrates epitopic suppression resulting from a previous exposure to TT. Hence, subjects immunized with AI–TT conjugate vaccine may not be expected to have any anti-AI IgG molecules in their sera samples. In contrast, the anticarrier protein response observed in Study C volunteers demonstrated their immune naivety to KLH. The poor anti-AI IgG levels generated in volunteers vaccinated with AI–KLH conjugate vaccine may have been due to the single dose administered.
A two-dose treatment (Phase Ib clinical study) of healthy, male, human volunteers, with AI–KLH conjugate vaccine using a 50-µg AI equivalent dose, resulted in active immunization to AI. However the anti-AI IgG titres (≤ 18 700) that resulted with the Study D subjects were lower than in the three-dose rat studies (Study A). This may explain the lack of effect of immunization on responses to exogenous AI throughout the treatment group. It is encouraging that, for the subject with the largest anti-AI IgG titre, a shift in the DBP response to exogenous AI and AII was observed. Presumably, sufficient anti-AI IgG molecules are required to have a functional effect upon AI-induced blood pressure changes. However, the shift in AI and AII dose–response required to cause a rise in DBP for this subject could have been the result of random variability.
In summary, KLH has been demonstrated to be a suitable alternative to TT as a carrier protein for AI in a potential conjugate vaccine treatment for control of hypertension in man. Rats immunized with both conjugate vaccines demonstrated similar immunoglobulin class profiles and anti-AI immunoglobulin titres. Additionally, the anti-AI immunoglobulins appeared to cause a shift in the MBP response following challenge with exogenous AI. The single-dose clinical study failed to show statistically significant evidence of anti-AI IgG generation. However, the AI–KLH conjugate vaccine demonstrated a qualitative anti-carrier protein response that identified it to be of preference to the AI–TT conjugate vaccine for further studies. The two-dose clinical study yielded anti-AI IgG titres of 2381–18 651, but this response appeared to be insufficient to cause a statistically significant shift in either the SBP or DBP response, following either an AI or AII challenge of the study volunteers.
However, the clear anti-AI IgG response observed with the two-dose trial indicates that further clinical studies to consider other treatment regimes are worthwhile. Future studies with AI–KLH conjugate vaccine aim to improve the anti-AI IgG titres and evaluate the blood pressure-lowering effect in patients with essential hypertension. Further clinical trials were initiated in 2001 under a UK clinical trials certificate exemption (CTX).
References
- 1.Gavras I, Gavras H. The renin–angiotensin system in hypertensive cardiac disease. In: Ulfendahl HR, Aurell M, editors. Renin–angiotensin, a centenary symposium of the discovery of the renin–angiotensin system. London: Portland Press; 1997. pp. 265–272. [Google Scholar]
- 2.Brown NJ, Vaughan DE. Angiotensin-converting enzyme inhibitors. Circulation. 1998;97:1411–1420. doi: 10.1161/01.cir.97.14.1411. [DOI] [PubMed] [Google Scholar]
- 3.Messerli FH, Weber A, Brunner HR. Angiotensin II receptor inhibition. Arch Intern Med. 1996;156:1957–1965. [PubMed] [Google Scholar]
- 4.Sunman W, Sever PS. Non-angiotensin effects of angiotensin-converting enzyme inhibitors. Clin Sci. 1993;85:661–670. doi: 10.1042/cs0850661. [DOI] [PubMed] [Google Scholar]
- 5.Horiuchi M, Akishita M, Dzau VJ. Recent progress in angiotensin II type 2 receptor research in the cardiovascular system. Hypertension. 1999;33:613–621. doi: 10.1161/01.hyp.33.2.613. [DOI] [PubMed] [Google Scholar]
- 6.Michel JB, Guettier C, Reader R, Sayah S, Corvol P, Ménard J. Immunologic approaches to blockade of the rennin–angiotensin system: a review. Am Heart J. 1989;117:756–767. doi: 10.1016/0002-8703(89)90767-9. [DOI] [PubMed] [Google Scholar]
- 7.Lerner RA. Tapping the immunological repertoire to produce antibodies of predetermined specificity. Nature. 1982;299:592–596. doi: 10.1038/299592a0. [DOI] [PubMed] [Google Scholar]
- 8.Sutcliffe JG, Shinnick TM, Green N, Lerner RA. Antibodies that react with predetermined sites on proteins. Science. 1983;219:660–666. doi: 10.1126/science.6186024. [DOI] [PubMed] [Google Scholar]
- 9.Newman MJ, Powell MF. Immunological and formulation design considerations for subunit vaccines. In: Powell MF, Newman MJ, editors. Vaccine design: the subunit and adjuvant approach. New York: Plenum Press; 1995. pp. 1–42. [DOI] [PubMed] [Google Scholar]
- 10.Craig L, Sanschagrin PC, Rozek A, Lackie S, Kuhn LA, Scott JK. The role of structure in antibody cross-reactivity between peptides and folded proteins. J Mol Biol. 1998;281:183–201. doi: 10.1006/jmbi.1998.1907. [DOI] [PubMed] [Google Scholar]
- 11.Jackson DC, Fitzmaurice CJ, Brown LE, Zeng W. Preparation and properties of totally synthetic immunogens. Vaccine. 2000;18:355–361. doi: 10.1016/s0264-410x(99)00205-4. [DOI] [PubMed] [Google Scholar]
- 12.Butler VP, Beiser SM. Antibodies to small molecules: biological and clinical applications. Adv Immunol. 1973;17:255–311. doi: 10.1016/s0065-2776(08)60734-8. [DOI] [PubMed] [Google Scholar]
- 13.Beale J. Synthetic peptides as the basis for future vaccines. Nature. 1982;298:14–15. doi: 10.1038/298014a0. [DOI] [PubMed] [Google Scholar]
- 14.Bahraoui EM, Granier C, Rietschoten JV, Rochat H, Ayeb ME. Specificity and neutralizing capacity of antibodies elicited by a synthetic peptide of scorpion toxin. J Immunol. 1986;136:3371–3377. [PubMed] [Google Scholar]
- 15.Long L, Glover RT, Kaufman HL. The next generation of vaccines for the treatment of cancer. Curr Opin Mol Ther. 1999;1:57–63. [PubMed] [Google Scholar]
- 16.Ellis R. Product development plan for new vaccine technologies. Vaccine. 2001;19:1559–1566. doi: 10.1016/s0264-410x(00)00352-2. [DOI] [PubMed] [Google Scholar]
- 17.Gardiner SM, Auton TR, Downham MR, et al. Active immunisation with angiotensin I peptide analogue vaccines selectively reduces the pressor effects of exogenous angiotensin I in conscious rats. Br J Pharmacol. 2000;129:1178–1182. doi: 10.1038/sj.bjp.0703178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smits JF, Auton TR, Sharp H, Downham MR, Glover JF, Janssen BJ. Anti-hypertenive properties of angiotensin immuno-therapeutic in spontaneous hypertension in rats. FASEB J. 1999;13(4):A483. [Google Scholar]
- 19.Herzenberg LA, Tokuhisa T, Herzenberg LA. Carrier priming leads to hapten-specific suppression. Nature. 1980;285:664–667. doi: 10.1038/285664a0. [DOI] [PubMed] [Google Scholar]
- 20.Schutze M-P, LeClerc C, Jolivet M, Audibert F, Chedid L. Carrier-induced epitopic suppression, a major issue for future synthetic vaccines. J Immunol. 1985;135:2319–2322. [PubMed] [Google Scholar]
- 21.Sad S, Gupta HM, Talwar GP, Raghupathy R. Carrier induced suppression of the antibody response to a ‘self’ hapten. Immunology. 1991;74:223–227. [PMC free article] [PubMed] [Google Scholar]
- 22.Di John D, Wasserman SS, Torres JR, et al. Effects of priming with carrier on response to conjugate vaccine. Lancet. 1989;2:1415–1418. doi: 10.1016/s0140-6736(89)92033-3. [DOI] [PubMed] [Google Scholar]
- 23.Herrington DA, Clyde DF, Davis JR, et al. Human studies with synthetic peptide sporozoite vaccine (NANP) 3-TT and immunisation with irradiated sporozoites. Bull WHO. 1990;68:S33–S37. [PMC free article] [PubMed] [Google Scholar]
- 24.Schutze M-P, Deriaud E, Przewlocki G, LeClerc C. Carrier-induced epitopic suppression is initiated through clonal dominance. J Immunol. 1989;142:2635–2640. [PubMed] [Google Scholar]
- 25.LeClerc C, Schutze M-P, Deriaud E, Przewlocki G. The in vivo elimination of CD4+ cells prevents the induction but not the expression of carrier-induced epitopic suppression. J Immunol. 1990;145:1343–1349. [PubMed] [Google Scholar]
- 26.Swerdlow RD, Ebert RF, Lee P, Bonaventura C, Miller KI. Keyhole limpet haemocyanin: structural and functional characterisation of two different subunits and multimers. Comp Biochem Physiol. 1996;113B:537–548. doi: 10.1016/0305-0491(95)02091-8. [DOI] [PubMed] [Google Scholar]
- 27.Harris JR, Markl J. Keyhole limpet haemocyanin (KLH): a biomedical review. Micron. 1990;30:597–623. doi: 10.1016/s0968-4328(99)00036-0. [DOI] [PubMed] [Google Scholar]
- 28.Salvaggio J, Castro-Murillo E, Kundur V. Immunologic response of atopic and normal individuals to keyhole limpet haemocyanin. J Allergy. 1969;44:344–354. doi: 10.1016/0021-8707(69)90026-4. [DOI] [PubMed] [Google Scholar]
- 29.Curtis JE, Hersh EM. The human secondary immune response to keyhole limpet haemocyanin. Clin Exp Immunol. 1972;10:171–177. [PMC free article] [PubMed] [Google Scholar]
- 30.Herscowitz HB, Harold WW, Stavitsky AB. Immunochemical and immunogenic properties of a purified keyhole limpet haemocyanin. Immunology. 1972;22:51–61. [PMC free article] [PubMed] [Google Scholar]
- 31.Bird P, Calvert JE, Amlot PL. Distinctive development of IgG4 subclass antibodies in the primary and secondary responses to keyhole limpet haemocyanin in man. Immunology. 1990;69:355–360. [PMC free article] [PubMed] [Google Scholar]
- 32.Olsson CA, Chute R, Rao CN. Immunologic reduction in bladder cancer recurrence rate. J Urol. 1974;111:173–176. doi: 10.1016/s0022-5347(17)59919-x. [DOI] [PubMed] [Google Scholar]
- 33.Jurincic CD, Engelmann U, Gasch J, Klippel KF. Immunotherapy in bladder cancer with keyhole-limpet haemocyanin: a randomised study. J Urol. 1988;139:723–726. doi: 10.1016/s0022-5347(17)42610-3. [DOI] [PubMed] [Google Scholar]
- 34.Wishahi MM, Otto T, Ruebben H. Keyhole limpet haemocyanin (KLH) immunotherapy of pT1, G2–3 transitional cell carcinoma of the bladder: a phase III clinical trial. Brit J Urol. 1997;80:142. [Google Scholar]
- 35.Naylor PH, Sztein MB, Wada S, et al. Preclinical and clinical studies on immunogenicity and safety of the HIV-1 p17-based synthetic peptide aids vaccine – HGP-30-KLH. Int J Immunol. 1991;13(Suppl 1):117–127. doi: 10.1016/0192-0561(91)90133-r. [DOI] [PubMed] [Google Scholar]
- 36.Livingston P, Zhang S, Adluri S, et al. Tumour cell reactivity mediated by IgM antibodies in sera from melanoma patients vaccinated with GM2 ganglioside covalently linked to KLH is increased by IgG antibodies. Cancer Immunol Immunother. 1997;43:324–330. doi: 10.1007/s002620050340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddish MA, MacLean GD, Koganty RR, et al. Anti-MUC1 class I restricted CTLs in metastatic breast cancer patients immunised with a synthetic MUC1 peptide. Int J Cancer. 1998;76:817–823. doi: 10.1002/(sici)1097-0215(19980610)76:6<817::aid-ijc9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 38.Gardiner SM, Auton TR, Downham MR, et al. Active immunisation with two carrier proteins conjugated with an angiotensin I (AI) analogue in conscious rats: pressor effects of AI and anti-AI antibody response. Br J Pharmacol. 1999;128:79P. [Google Scholar]
- 39.Waller J, Gardiner SM, Jose J, Bennett T. Lack of effect of TNF antibodies on the cardiovascular sequel of lipopolysaccharide infusion in conscious rats. Br J Pharmacol. 1995;116:2487–2495. doi: 10.1111/j.1476-5381.1995.tb15100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erb KA, Essig J, Breithaupt K, Belz GG. Clinical pharmacodynamic studies with cilazapril and a combination of cilazapril and propranolol. Drugs. 1991;41:11–17. doi: 10.2165/00003495-199100411-00004. [DOI] [PubMed] [Google Scholar]
- 41.Essig J, Belz GG, Wellstein A. The assessment of ACE activity in man following angiotensin I challenges: a comparison of cilazapril, caprotpril and enalapril. Br J Clin Pharmacol. 1989;27:217S–223S. doi: 10.1111/j.1365-2125.1989.tb03485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belz GG, Essig J, Wellstein A. Hemodynamic responses to angiotensin I in normal volunteers and the antagonism by the angiotensin-converting enzyme inhibitor cilazapril. J Cardiovasc Pharm. 1987;9:219–224. doi: 10.1097/00005344-198702000-00015. [DOI] [PubMed] [Google Scholar]