

Abstract
Kv1.3, a member of the Shaker family of potassium channels, has been found to play diverse roles in immunity, metabolism, insulin resistance, sensory discrimination, and axonal targeting in addition to its traditional role in the stabilization of the resting potential. We demonstrate that the neurotrophin B receptor (TrkB) causes an upregulation of Kv1.3 ion channel protein expression in the absence of the preferred ligand for the receptor (brain-derived neurotrophic factor; BDNF) and oppositely downregulates levels of Kv1.5. Although the effect occurs in the absence of the ligand, Kv1.3 upregulation by TrkB is dependent upon the catalytic domain of the TrkB kinase as well as Y residues in the N and C terminus of the Kv1.3 channel. Using pulse-chase experiments we find that TrkB alters the half-life residence of the channel by approximately 2X and allows it to sustain activity as reflected in an increased current magnitude without alteration of kinetic properties. TrkB and Kv1.3 co-immunoprecipitate from tissue preparations of the olfactory bulb and olfactory cortex, and by immunocytochemical approaches, are found to be co-localized in the glomerular, mitral cell, and internal plexiform layers of the olfactory bulb. These data suggest that Kv1.3 is not only modulated by direct phosphorylation in the presence of BDNF-activated TrkB kinase, but also may be fine tuned via regulation of surface expression while in the proximity of neurotrophic factor receptors. Given the variability of TrkB expression during development, regeneration, or neuronal activation, modulation of surface expression and turnover of Kv channels could significantly impact neuronal excitability, distinct from that of tyrosine kinase phosphorylation.
Keywords: neurotrophins, olfactory bulb, potassium channel, tyrosine kinase, Shaker, trafficking
Within the human genome, almost half of the approximately 400 ion channel genes are annotated to encode potassium (K) channels (Sun et al., 2004). Traditionally K channels are critical for regulating neuronal excitability through stabilization of the resting potential, however, recent evidence has uncovered unexpected functions for a particular Shaker family member, Kv1.3, which includes such diverse roles in immunity, metabolism (weight maintenance), insulin resistance, sensory discrimination, and axonal targeting (Cahalan et al., 2001; Xu et al., 2003; Chandy et al., 2004; Fadool et al., 2004; Xu et al., 2004; Biju et al., 2006). Due to the oligomeric composition of K channels, the recruitment of these channels into multiprotein regulatory scaffolds, and the presence of auxiliary subunits, adaptor proteins, or modulatory kinases that can influence post-translational events, the number of cell surface copies of ion channels can be manipulated by the cell to dramatically impact neuronal excitability (Misonou and Trimmer, 2004; Heusser and Schwappach, 2005). Kv1.3 has a discreet and restricted expression in the central nervous system to include the olfactory cortex, the olfactory bulb, and the dentate gyrus of the hippocampus (Kues and Wunder, 1992). Gene-targeted deletion approaches have demonstrated an unusual cadre of cellular and behavioral phenotypes in Kv1.3-null mice. The mice have irregular ingestive behaviors and although they consume the same daily caloric intake as wildtype mice, they weigh less and show resistance to obesity when challenged with a high fat diet (Xu et al., 2003; Fadool et al., 2004). Although mice display only a modest perturbation in metabolism and locomotion (Xu et al., 2003; Fadool et al., 2004), Kv1.3 ion channels have been reported in the mitochondria supportive of a role in energy homeostasis (Szabo et al., 2005). In the olfactory system, Kv1.3-null mice have anatomical changes in the glomeruli of the olfactory bulb (Fadool et al., 2004). These sites of synaptic contact between the peripheral olfactory sensory neurons and those of the first order output neurons, the mitral cells (where Kv1.3 is expressed in wildtype animals), are smaller and more numerous than wildtype mice; a structural change that may be responsible for an increased olfactory ability that these mice have as assessed by behavioral tests of olfactory discrimination and threshold (Fadool et al., 2004). The Kv1.3-null mice additionally have a marked increase (7 fold) in the expression of the neurotrophic factor receptor, TrkB, in the olfactory bulb (Fadool et al., 2004). Due to the well-characterized activity dependence of TrkB signaling cascades, our previous studies that demonstrated BDNF-evoked Kv1.3 current suppression in olfactory bulb neurons, combined with anatomical changes in the olfactory bulb of the null mice that suggested different axonal targeting of the sensory neurons to the central glomerular targets, we became curious to explore a suspected reciprocal regulation between TrkB and Kv1.3. To what degree could TrkB modulate Kv1.3 expression, trafficking, and biophysical properties that appeared to developmentally establish proper olfactory bulb anatomy, olfactory sensitivity, and electrical properties?
All three neurotrophic receptors, TrkA, TrkB, and TrkC, are expressed in the olfactory bulb; but only TrkB is expressed predominately as the full length (145 kDa) protein as opposed to the truncated form of the tyrosine receptor kinase (95 kDa) that lacks catalytic activity (Tucker and Fadool, 2002). In strong correlation to expression patterns in the olfactory bulb, the preferred ligand for the TrkB receptor, brain-derived neurotrophic factor (BDNF), can acutely suppress Kv1.3 current in mitral cells of the olfactory bulb without changes in inactivation or deactivation kinetics of the whole cell current (Tucker and Fadool, 2002; Colley et al., 2004). In contrast, NGF and NT3, the preferred ligands of TrkA and TrkC, respectively, do not modulate Kv1.3 currents in the olfactory bulb neurons (Tucker and Fadool, 2002). We have previously reported that the mechanism of BDNF-evoked current suppression of Kv1.3 is via tyrosine phosphorylation of three sites in the N and C termini of the channel, Y111–113, Y137 and Y449, determined through site-directed mutagenesis of the channel in combination with functional patch-clamp studies and immunoprecipitation assays of Y phosphorylated states of the channel following BDNF acute stimulation (minutes)(Colley et al., 2004). We now explore the mechanism of TrkB kinase-mediated channel upregulation, whether the regulation is a general principle of other receptor tyrosine kinases, and complete confocal microscopy studies that demonstrate the ability of this receptor tyrosine kinase to alter surface expression and trafficking of the channel. These data suggest that in light of the activity dependence of TrkB signaling cascades, the functioning of ion channels may depend not only on the modulation of their biophysical properties via biochemical processes such as phosphorylation but also fine-tuning of their surface expression that might change neuronal excitability upon presence and proximity of neurotrophic factor receptors.
EXPERIMENTAL PROCEDURES
Solutions and Reagents
Human embryonic kidney cell (HEK 293) patch pipette solution contained (in mM): 30 KCl, 120 NaCl, 10 HEPES, and 2 CaCl2 (pH 7.4). HEK 293 cell recording bath solution contained (in mM): 150 KCl, 10 HEPES, 1 EGTA, and 0.5 MgCl2 (pH 7.4). Cell lysis buffer contained (in mM): 25 tris (hydroxymethyl) aminomethane (pH 7.5), 150 NaCl, 150 NaF, 0.5 EDTA, and 1.0% Triton X-100 (pH 8.0). Homogenization buffer (HB) contained in (mM): 320 sucrose, 10 Tris Base, 50 KCl, and 1 EDTA (pH 7.8). Protease and phosphatase inhibitor (PPI) solution was added to the lysis or HB just prior to use for a final concentration as follows: 1 μg/ml pepstatin A, 1 μg/ml leupeptin, 2 μg/ml aprotinin, 10 μg/ml phenylmethylsulfonyl fluoride, and 10 mM Na3VO4. Nonidet-NP40 protease and phosphatase inhibitor (NP40 PPI) solution was prepared in the same PPI solution as above but contained (in mM): 20 Tris base (pH 7.5), 150 NaCl, 1 % nonidet-NP40 and 10% glycerol. Wash buffer contained (in mM): 25 Tris base (pH 7.5), and 150 NaCl, 150 NaF, 0.5 EDTA and 0.1% Triton X-100. Tris stripping buffer (TSB) contained (in mM): 10 Tris, 10 β-mercaptoethanol, with 1% SDS (pH 8.8). Sodium citrate stripping buffer (SCSB) contained (in mM): 100 Na3C6H5O7.· 2H20, 10 β-mercaptoethanol, with 1% SDS; pH 3.0. Human recombinant brain-derived neurotrophic factor (rhBDNF) was purchased from Promega (Madison, WI). Human recombinant insulin was purchased from Roche (Indianapolis, IN). Cycloheximide was purchased from Calbiochem (San Diego, CA). Tissue culture reagents were purchased from Gibco/BRL (Gaithersburg, MD). All salts and other reagents were purchased from Sigma Chemical Co. (St. Louis, MO) or Fisher Scientific (Suwanne, GA).
cDNA Constructs and Antibodies
All channel and kinase coding regions were downstream from a cytomegalovirus (CMV) promoter. IR cDNA was a gift from Dr. R. Roth, Stanford University, in the pECE vector. The entire IR-coding region was removed using the unique restriction sites Sal I and Xba I, and ligated into pcDNA3 between the Xho I and Xba I sites in the multiple cloning region (Fadool et al., 2000). TrkB cDNA was a generous gift from Dr. P. Barker, McGill University, in a CMX vector. Human Kv1.5 channel, tagged at the C-terminal end with the C-peptide antigen and cloned into pCS2, was a gift from Dr. Todd C. Holmes, New York University (Nitabach et al., 2001). Kv1.3 channel was subcloned into the multiple cloning region of pcDNA3 (Invitrogen) at the unique restriction HindIII site of the multiple cloning region. The ten amino acid c-myc epitope (EQKLISEEDL) was inserted into the S1/S2 loop between residues 226–227 of Kv1.3 channel via two consecutive polymerase chain reactions (PCRs) using the Expand Long Template PCR System (Roche, Indianapolis, IN). All Kv1.3 channel mutants were constructed with the use of two sequential PCRs with oligonucleotides designed to introduce a conservative Y to F point mutation as previously described (Fadool et al., 1997). Kv1.3 α-subunit fused to the C-terminus of green fluorescent protein (GFP) in pEGFP-C1 was a generous gift of Dr. J. Kupper, Max-Planck Institute (Kupper, 1998).
AU13, a rabbit polyclonal antiserum, was generated against a 46 amino acid sequence (478MVIEEGGMNHSAFPQTPFKTGNSTATCTTNNNPNDCVNIKKIFTDV523) representing the unique coding region of Kv1.3 on the C- terminus. Genmed Synthesis (San Francisco, CA) purified the peptide and Cocalico Biologicals (Reamstown, PA) the antisera as previously characterized (Tucker and Fadool, 2002). This antibody was used for immunoprecipitation (1:1000) and Western blot detection (1:4000) of Kv1.3. Tyrosine phosphorylated proteins were visualized by the anti-phosphotyrosine antibody 4G10 (Upstate Biotechnology; Lake Placid, NY) and used at 1:1000 for Western blots. Tyrosine phosphorylated proteins were immunoprecipitated with 4G10 antibody (3 μg per 600–1200 μg of whole lysate protein). Monoclonal antiserum directed against amino acids 156–322 of human TrkB was purchased from Transduction Laboratories (San Diego, CA) and used at 1:1000 for Western blots. Rabbit polyclonal Insulin Receptor (IR) antibody was purchased from BD Biosciences Pharmingen (San Jose, CA) and used for Western blots at 1:1000. Rabbit anti-Kv1.5 purified polyclonal antibody against an intracellular, C-terminal portion of mouse Kv1.5 protein (amino acids 513–602) was purchased from Chemicon International (Temecula, CA) and used for Western blots at 1:1000. Monoclonal actin antibody was purchased from Sigma and used at 1:750 for Western blots as a secondary confirmation for equivalent protein loading. Anti-c-myc mouse monoclonal (clone E910; antigenetic peptide EQKLISEEDL against from the human myc protein) was purchased from Roche and used at 1:800 for Western blots.
HEK 293 Cell Culture and Transient Transfection
HEK 293 cells were maintained in Minimum Essential Medium (MEM), 2% penicillin/streptomycin, and 10% FBS (Gibco BRL). Before transfection, cells were grown to confluency (7 days), dissociated with trypsin-EDTA (Sigma) and mechanical trituration, diluted in MEM to a concentration of around 600 cells/ml, and replated on Corning dishes (Catalog # 25000, Fisher Scientific). At the time of transfection, cells had reached between 50–60% and 80–95% confluency for electrophysiology or biochemistry respectively. cDNA was introduced into HEK 293 cells with a LipofectamineTM Transfection Reagent (Invitrogen, Carlsbad, CA) 3–5 days after passage as previously described (Fadool et al., 1997). Briefly, cells were transfected for 4–5 hrs using 2.0 μg of each cDNA construct per 60 mm dish for biochemistry or 4–5 hrs using 1.0 μg of each cDNA construct per 35 mm dish for physiology. Cells were then harvested for biochemistry or used for patch-clamp recording approximately 24–48 hrs post transfection.
Cycloheximide-chase assay experiments (Cullen et al., 2004) were used to determine the half-life of Kv1.3 in the presence or absence of TrkB co-transfection. Cycloheximide (30 to 100 μg/ml) or vehicle control (0.03% DMSO) was applied to the culture media 20 – 24 hours post-transfection. Cells were harvested 0, 3, 6, 9 and 12 hours post cycloheximide treatment. Experiments were performed in triplicate.
Electrophysiology
Hoffman modulation contrast optics were used to visualize HEK 293 cells at 40X magnification (Axiovert 135, Carl Zeiss). Macroscopic currents in cell-attached membrane patches were detected using an Axopatch-200B amplifier (Axon Instruments). All electrophysiological data were recorded and analyzed using pClamp 8.0 or 9.0 software in combination with the analysis package of Origin (MicroCal Software, Northampton, MA) and Quattro Pro (Borland International, Scotts Valley, CA). Patch electrodes were fabricated from Jencons glass (Jencons Limited, Bedfordshire, UK), fire-polished to ~1 μm, and coated near the tip with beeswax to reduce the electrode capacitance. Pipette resistances were between 9 and 14 MΩ . Currents were recorded 24–48 hrs after transfection. The diameter of the patch pipette, and hence number of ion channels sampled, was held uniform by checking the bubble number of the pipette immediately after electrode fabrication and polishing (Mittman et al., 1987). In general, currents were recorded from cell-attached patches held at a holding potential (Vh) of −90 mV and stepped to +40 mV depolarizing potentials (Vc) with a pulse duration of 1000 ms for Kv1.3 and 200 ms for Kv1.5. For Kv1.3, voltage pulses were delivered at 45-s intervals in order to prevent cumulative or use-dependent inactivation, which is characteristic of this channel. The mean peak macroscopic current amplitude and kinetic time constants of channel inactivation (τInact) and deactivation (τDeact) were determined by parameter fitting for pre-stimulation (time 0) and 20 min post-growth factor stimulation (BDNF/insulin). The inactivation of the macroscopic current (τInact) was fit to the sum of two exponentials by minimizing the sums of squares. The two inactivation time constants were combined by multiplying each by its weight and summing as described previously {Kupper et al., 1995). The deactivation of the macroscopic current (τDeact) was fit similarly, but to a single exponential. Differences in these calculated biophysical properties were analyzed between control (time 0) and post-growth factor stimulation (time 20 min) groups within single cells by a paired t-test with statistical significance at the 95% confidence level. BDNF stimulation was accomplished by tip-filling (~0.01 mm) the patch-pipette with control solution and then back filling (~35 mm) with BDNF (1 ng/μl). Insulin (10 ng/ml) stimulation was done the same as for BDNF. The dilution of the BDNF/insulin using this procedure would be insignificant due to the ratio of the volume (back-fill/tip-fill) difference that is estimated to be at least 1 million.
Immunocytochemistry
For cryosections, whole OBs were fixed in 4% paraformaldehyde for 3 h followed by overnight infiltration with 10% sucrose, then 4 h with 30% sucrose. OBs were cut to 9- to 12-μm thickness on a Leica CM1850 microtome-cryostat (Leica, Meyer Instruments, Houston, TX). Sections were transferred to 1% gelatin-coated glass slides (Sigma) and stored at −20oC until use. For human embryonic kidney cells, twenty-four to 48 hours after transfection, cells were lightly fixed with 1% paraformaldehyde in phosphate-buffered saline (PBS) for 10 minutes at room temperature (rt). Nonspecific binding was blocked using 1–2% bovine serum albumin (BSA) in PBS. For determining the surface expressed channel fraction, mycKv1.3 transfected cells were incubated with α-c-myc at rt for 90 minutes using a 1:800 dilution in 2% BSA in PBS. The HEK 293 cells containing the antibody bound c-myc-epitope tagged channels were then washed three times in PBS and then were permeabilized using 1–2% BSA in 0.1% Triton X-100 and PBS (PBST) for 45 minutes. For determining total channel fraction, cells were first permeabilized and then incubated with α-AU13 (Kv1.3 antibody) in 1–2% BSA in PBST. Total channel protein or surface labeled protein was subsequently visualized via incubation with species-specific fluorescent secondary antisera, namely Alexa Fluor 488-conjugated goat anti-rabbit (1:200) or Texas Red-conjugated goat anti-mouse (1:100). Some cells were additionally counter stained with a five minute incubation with the nuclear stain diamidino-phenyindole (DAPI) in PBS (1:5000) with a final wash in PBS. Coverslips were mounted onto slides using Vectashield (Vector Labs, Burlingame, CA) or 70/30% glycerol/PBS, containing p-phenylenediamine to prevent photobleaching.
Confocal Microscopy and Image Analysis
Microscopic analysis of fluorescently-labeled cells was performed on a Zeiss Axioplan 2 Microscope attached to a Zeiss LSM510 confocal system. Samples were excited at 488 nm (for GFP) and 595 nm (for Texas Red) through a 63x oil-immersion objective (NA=1.40) and fluorescence between 500–545 and 565–615 nm was detected from <0.8 μm thick optical sections. Images (1024 x 1024 x 8 bit) were taken from the cells at their maximal cross section. Cell outlines were identified using the Texas Red staining. Green channels of the images were analyzed using Metamorph software (Molecular Devices, Sunnyvale, CA). After removing background pixels by thresholding, integrated pixel intensity was calculated both from the total cross section of the cell (PixelT) and from an area that encompassed the cross section of the cell excluding the outermost region (Pixel I). The plasma membrane fraction (PM) was approximated as (PixelT - PixelI)/(PixelT) and compared by Student’s t-test across transfection condition (control Kv1.3 vs. TrkB co-transfected cells).
Immunoprecipitation and Electrophoretic Separation
Olfactory bulbs were quickly harvested from C57/BL6 mice (Jax Laboratories, Jacksonville, FL) at postnatal day 20 (P20) after decapitation. Strict adherence was kept to American Veterinary Medical Association- and National Institutes of Health-approved methods. For immunoprecipitation, olfactory bulbs or olfactory cortex was immediately homogenized with a Kontes tissue grinder (size 20; 50 strokes) in ice-cold HB containing 0.5% Nonidet P-40 detergent in PPI solution (see "Solutions and Reagents"). Lysis was continued on a Roto-Torque slow speed rotary (model 7637; Cole Palmer, Vernon Hills, IL) for 30 minutes at 4oC. The lysates were clarified by centrifugation at 14,000 X g for 10 minutes at 4oC and precleared for 1 hour with 3 mg/ml protein A-sepharose (Amersham-Pharmacia, Newark, NJ), which was followed by another centrifugation step to remove the protein A-sepharose. For standard transfections, whole-cell lysates were similarly obtained from transfected HEK 293 cells but as previously described (Colley et al., 2004). Kv1.3 or Kv1.5 channel proteins, or TrkB and IR receptors were immunoprecipitated from native tissue or transfected HEK 293 cells. Clarified lysates were incubated overnight at 4oC with 3 μg of respective antiserum per 600–1200 μg of HEK 293 cell protein or 2000–3000 μg native tissue protein lysate. For experiments in which surface expression of c- myc Kv1.3 was quantified, transfected HEK 293 cells were first live labeled with anti-myc as the initial immunoprecipitation step prior to cell lysis. All immunoprecipitates (native, standard lysis, or surface labeled) were collected by 2 hours incubation with protein A-sepharose and centrifugation as above and washed 4 times with ice-cold NP40 PPI solution for native tissue or with ice-cold wash buffer for HEK 293 cells. Lysates and washed immunoprecipitates were diluted 1:1 in sodium dodecyl sulfate (SDS) gel loading buffer (Sambrook et al., 1989) containing 1 mM Na3VO4 and stored at −20oC for subsequent analysis. Immunoprecipitated proteins were separated on 10% acrylamide gels by SDS-PAGE and electro-transferred to nitrocellulose blots. Blots were blocked with 5% nonfat milk and incubated overnight at 4oC in primary antiserum. Blots were then incubated with species-specific horseradish peroxidase (HRP)-conjugated secondary antibody (1:6000) (Amersham-Pharmacia or Sigma). Enhanced chemiluminescence (ECL; Amersham-Pharmacia) exposure on Fugi Rx film (Fisher) was used to visualize labeled proteins. The film autoradiographs were analyzed by quantitative densitometry using a Hewlett-Packard PhotoSmart Scanner (model 106–816, Hewlett Packard, San Diego, CA) in conjunction with Quantiscan software (Biosoft, Cambridge, England). The raw densitometry values were always normalized to a control transfection condition within the same autoradiograph in order to reduce variance introduced by any difference in length of exposure across individual films.
RESULTS
We demonstrate that the expression and surface retention of a voltage-dependent channel, Kv1.3, can be modulated by the neurotrophin receptor, TrkB, by a mechanism that appears to require basal phosphorylation and co-localization, yet in the absence of ligand stimulation (brain-derived neurotrophic factor, BDNF) and without a direct protein-protein interaction between the kinase and the channel.
TrkB selectively upregulates Kv1.3 channel protein expression that is linked to increased voltage-activated current
Co-transfection of Kv1.3 plus TrkB cDNA (KT) in HEK293 cells caused a significant increase in total Kv1.3 channel expression compared with Kv1.3 alone (K) as determined in whole cell lysates by SDS-PAGE followed by Western blot analysis (Figure 1; Student t-test, n= 18, α≤0.05). To determine whether this upregulation of channel protein expression was dependent upon the basal catalytic activity of TrkB, we truncated the kinase by eliminating all residues C terminal to amino acid 485 (TrkB dead kinase, D). As shown in Figure 1, co-transfection of Kv1.3 plus TrkB dead kinase (KD) was ineffective in upregulating Kv1.3 channel expression above that of Kv1.3 alone (K) conditions (Student t-test, n=4, α≤0.05), indicating that basal activity of TrkB kinase was necessary to upregulate Kv1.3 channel protein expression in the presence of TrkB. To test whether Kv1.3-upregulated channel protein translated into an increase in the functional abundance of channels, we performed patch-clamp electrophysiology on singly and Kv1.3 plus TrkB co-transfected HEK 293 cells and compared the generation of Kv1.3 currents in response to depolarizing voltage steps in cell-attached patches (Figure 2C, D). There was a significant increase in Kv1.3 current magnitude when the channel was co-transfected with TrkB (Figure 2D, Student t-test, n= 23–33, α≤0.05) without any change in inactivation (τinact) or deactivation (τdeact) kinetics, voltage at half-activation (V1/2), or voltage-dependence (k) (see Table 1); implying that there was an increase in channel expression without modulation of channel properties. This effect of increased channel expression was not generally applicable to other receptor tyrosine kinases that are known to be highly expressed and co-localized with Kv1.3 in the olfactory bulb (Fadool et al., 2000). Co-transfection of IR kinase (KI) compared with that of Kv1.3 transfection alone (K) actually demonstrated a modest, but statistically significant, decrease in Kv1.3 channel protein expression (Figure 2A, B) that was not correlated to any change in total current magnitude in response to depolarizing voltage steps (Figure 2C, D). It was possible to demonstrate a dose-dependent type of upregulation of the Kv1.3 channel expression by increasing the amount of transfected TrkB cDNA from 0 to 1.7 μg while holding Kv1.3 at a fixed concentration (Figure 3A). Likewise an increase in TrkB concentration from 0 to 2.0 μg was able to upregulate channel expression when a range of transfection concentrations of Kv1.3 cDNA from 0.5 to 4.0 μg (Figure 3B) was used. These data combined with the fact that use of different promoters as well as fac sorting of Kv1.3 vs. Kv1.3 plus TrkB co-transfected conditions (data not shown) did not alter the ability of TrkB to upregulate channel protein expression, make it unlikely that TrkB was modifying the transfection efficiency of Kv1.3 channel protein. We were curious if TrkB could modulate channel expression of other Shaker family members that were expressed in the olfactory bulb or if the effect were selective to Kv1.3. Kv1.5 co-transfected with TrkB kinase did not show an increased channel protein expression in comparison to Kv1.5 alone conditions, and in fact, contrarily, was significantly decreased by 50% (Figure 4A, B). This downregulation of Kv1.5 was not analogously produced upon co-transfection of IR kinase (Figure 4A, B) nor was co-transfection with either receptor kinase capable of functional modulation of peak current amplitude of Kv1.5 (Figure 4C, D). These data suggested the presence of a pool of Kv1.5 ion channels that were regulated by TrkB and which may not actively contribute to voltage-activated macroscopic current in these cells.
Fig. 1.
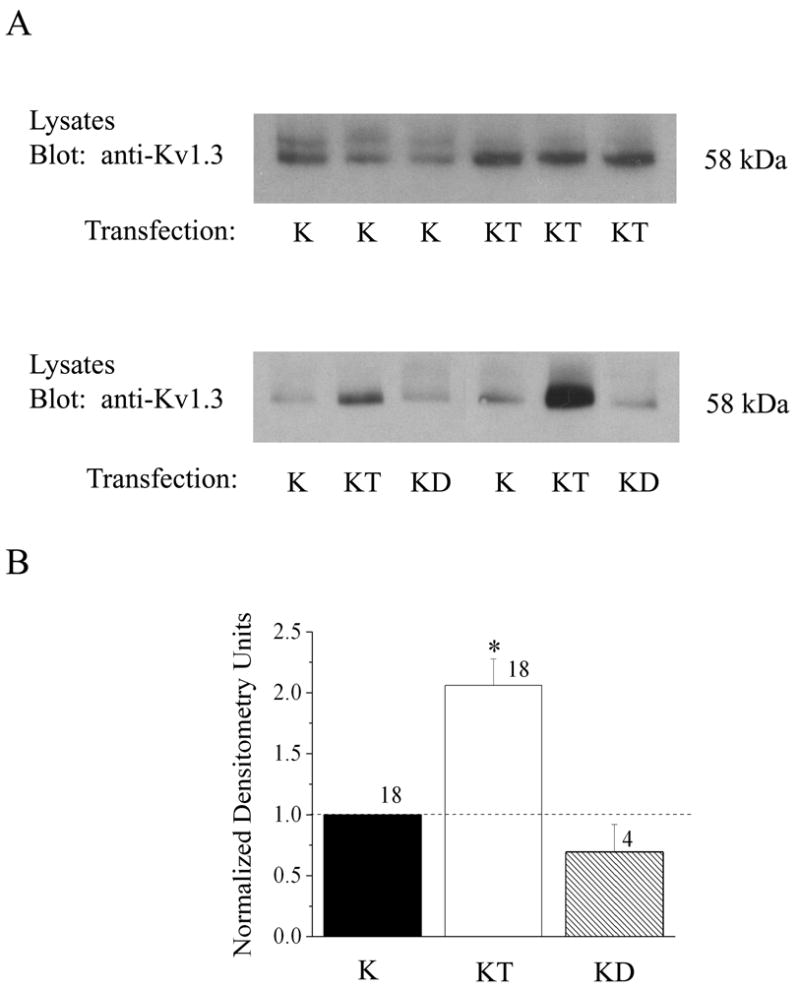
Kv1.3 channel protein expression in the presence of TrkB kinase. (A) Western blot of human embryonic kidney (HEK 293) cells transiently transfected with cDNA encoding Kv1.3 (K), Kv1.3 plus TrkB kinase (KT), or Kv1.3 plus TrkB mutant kinase lacking catalytic activity (dead kinase, KD). Five to ten μg whole cell lysates from each respective transfection condition, were separated by SDS-PAGE, electro-transferred to nitrocellulose, and probed with anti-Kv1.3. Upper panel shows representative expression bands for three such experiments comparing K vs. KT transfection conditions. Lower panel shows representative expression bands for two experiments in which K, KT, and KD are compared. (B) Histogram plot of the mean ± standard error of the mean (s.e.m.) normalized immunodensity values for experiments demonstrated in A. Pixel density ratios (dashed line) were generated by normalizing to control K transfection within a single autoradiographic film to eliminate variability introduced by differential exposure times. * = Significantly different from control K transfection, α≤ 0.05.
Fig. 2.
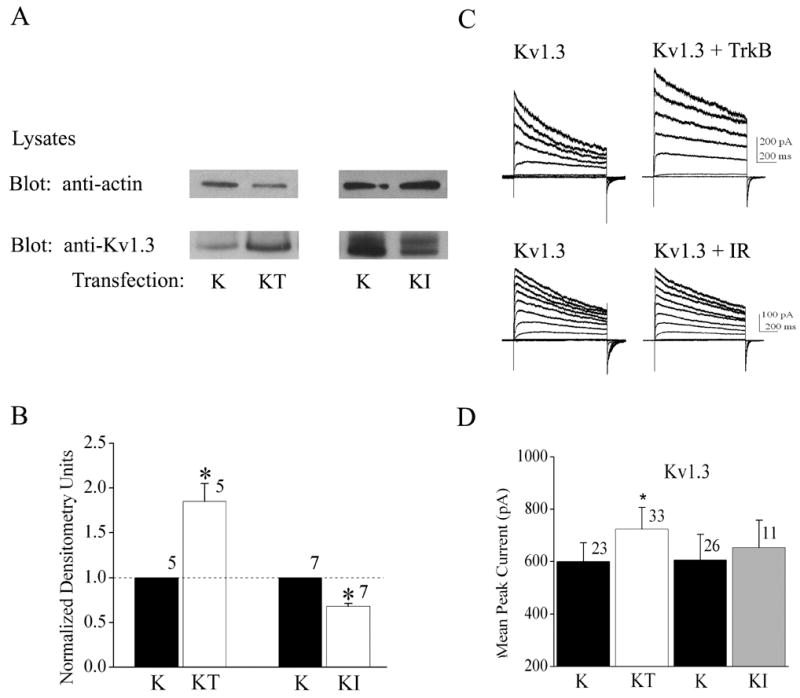
Differential effects of co-expression of TrkB and insulin receptor (IR) kinase on Kv1.3 expression and voltage-activated current. (A) HEK293 cells were transiently transfected with cDNA encoding Kv1.3 minus (K) or plus TrkB (KT) or insulin receptor (KI) kinase; lysates prepared and separated by SDS PAGE as in Fig. 1. Top panel was blotted with anti-actin to confirm equal loading. Bottom panel was blotted with anti-Kv1.3. (B) Histogram plot of the mean ± standard error of the mean (s.e.m.) normalized immunodensity values for experiments demonstrated in (A). Quantification, statistical analysis, and notation as in Fig. 1B. (C) Representative Kv1.3 current recorded from HEK 293 cells under the specified transfection conditions using the cell-attached configuration. Patches were held at −90 mV (Vh) and stepped from -80 to +40 mV in 10 mV increments (Vc) for a duration of 400 ms using 45 s interpulse intervals between voltage steps. (D) Histogram plot of the peak current amplitude for various transfection conditions and recordings as in A. Data are expressed as mean ± s.e.m., sample size as indicated, * = Statistically significant by Student’s t-test at α≤ 0.05.
TABLE 1.
The Effect of Receptor Tyrosine Kinases on Shaker Current Properties
| Channel Construct and Co-transfected Kinase | Peak Current (pA) | τInact (ms) | τDeact (ms) |
|---|---|---|---|
| WT Kv1.3 (n= 23) | 600 ± 71 | 739 ± 105 | 15.5 ± 1.6 |
| WTKv1.3 +TrkB (n=33) | *724 ± 63 | 668 ± 44 | 16.9 ± 1.4 |
| WTKv1.3 + IR (n=11) | 653 ± 55 | 737 ± 64 | 15.4 ± 1.2 |
|
| |||
| WTKv1.5 (n=20) | 690 ± 82 | 108 ± 12 | 8.8 ± 1.3 |
| WTKv1.5 +TrkB (n=11) | 682 ± 84 | 104 ± 10 | 9.1 ± 1.2 |
| WTKv1.5 +IR (n=10) | 634 ± 133 | 83 ± 6 | 7.7 ± 1.1 |
HEK 293 cells were transfected with cDNAs encoding Kv1.3 or Kv1.5 and co-transfected with either TrkB receptor or IR kinase. Cells were held in the cell-attached configuration and stepped from -90 mV to +40 mV for 1000 msec using a 45 s interpulse interval. Peak current amplitudes were acquired approximately 5 minutes after achieving seal formation. Mean values (± s.e.m.) for the inactivation time constant (τInact), deactivation time constant (τDeact ), and peak current amplitude were compared across transfection conditions. Data for each respective channel (subfamily member 1.3 or 1.5) were analyzed by completely randomized one-way ANOVA with Student-Newman-Keuls’ (SNK) followup test with significance (*) determined at the 95% confidence level (α ≤ 0.05).
Fig. 3.
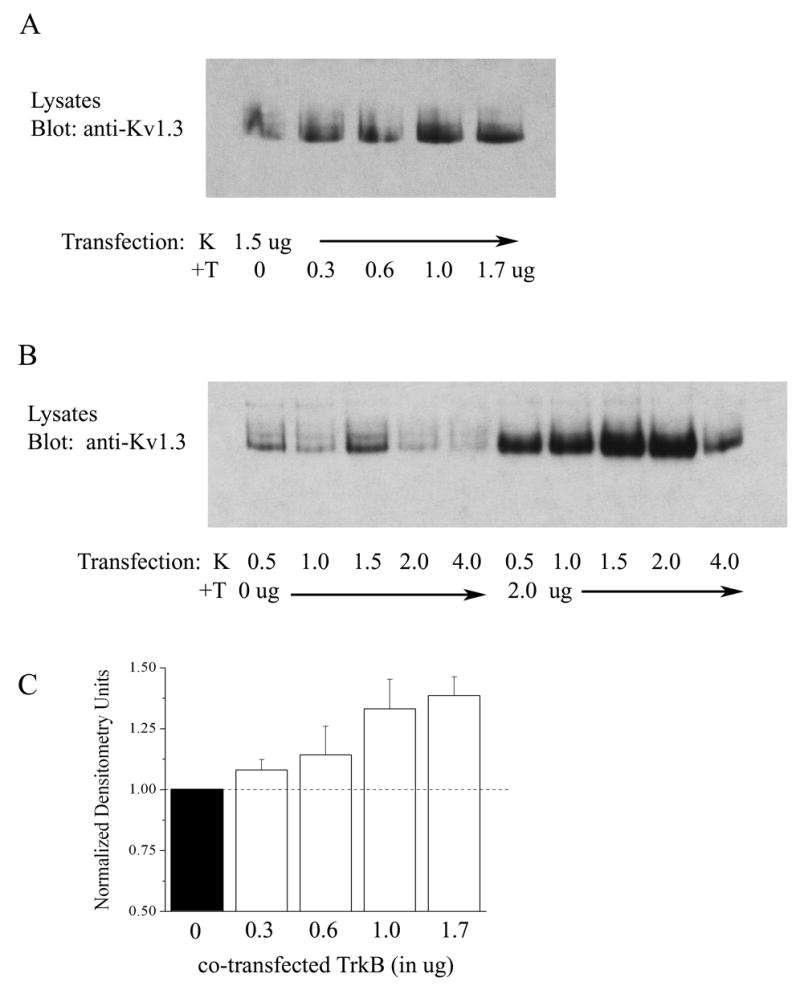
Dose-dependent upregulation of Kv1.3 protein expression in the presence of TrkB. (A) HEK293 cells were transiently transfected with cDNA encoding Kv1.3 (K) plus increasing concentrations of TrkB (+T) ranging from 0 to 1.7 μg. Lysates were prepared and separated by SDS PAGE as in Fig. 1. Nitrocellulose was blotted with anti-Kv1.3. Gel is representative of three such experiments testing increasing transfection concentrations of TrkB. (B) As in (A) but varying transfection concentrations of Kv1.3 cDNA in the absence (0 μg) and presence (2.0 μg) of a fixed concentration of TrkB (+T) cDNA. (C) Histogram plot of the mean ± standard error of the mean (s.e.m.) normalized immunodensity values for 3 such experiments demonstrated in A. Pixel density ratios (dashed line) were generated by normalizing each value with that of the density for control K transfection using 0 μg TrkB cDNA.
Fig. 4.
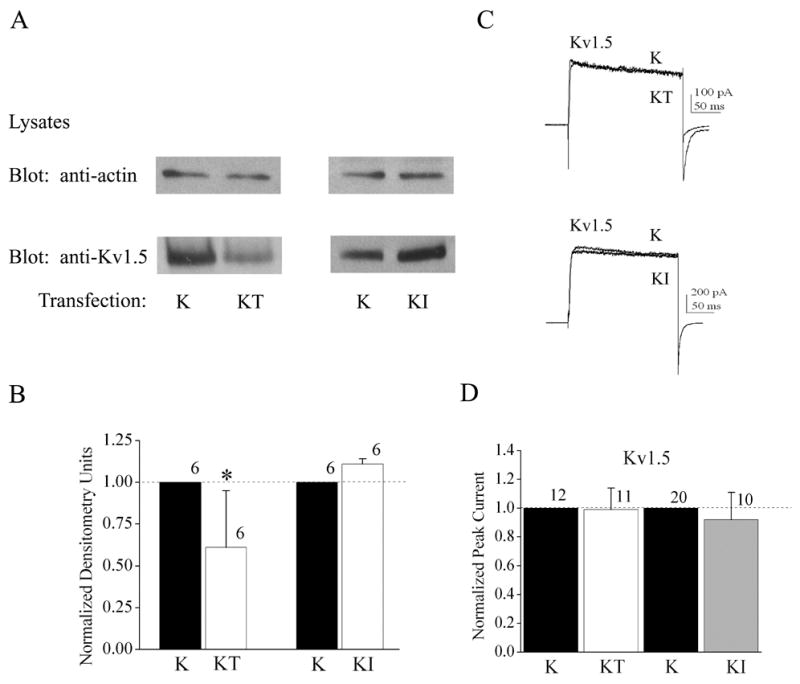
Differential effects of co-expression of TrkB and insulin receptor (IR) kinase on Kv1.5 expression and voltage-activated current. (A-D) As in Fig. 2, but for Kv1.5 as opposed to Kv1.3. (C) As in Fig. 2C, but with a different voltage paradigm for Kv1.5. Patches were held at -90 mV (Vh) and stepped to a single depolarizing voltage of +40 mV (Vc) for a duration of 200 ms using 10 s interpulse intervals between voltage steps.
TrkB upregulation of Kv1.3 channel protein acts to redistribute an increased fraction of channels in the plasma membrane
We next took the subsequent approach to design epitope-tagged Kv1.3 channel constructs in order to track changes in surface expression of the channel in the presence of the TrkB receptor (Figure 5A). The strategy was to utilize an intracellular, N terminal GFP tag and an extracellular Myc tag placed between residues 226–227 (S1/S2 loop) to optically measure cytoplasmic- and plasma membrane-inserted channel using confocal microscopy (Figure 5A). It was important to first determine if either epitope tag perturbed the upregulation effect of TrkB or any basal biophysical properties of the channel. As demonstrated in Figure 5, the extracellular loop myc tag (Myc) did not alter TrkB ability to upregulate Kv1.3 channel expression (Figure 5B, E) or functional increase in voltage-activated currents (Figure 5C, D), however, channels tagged with both Myc and gfp (Mycgfp) or gfp alone (gfp) were not functionally (Figure 5C, D) or biochemically upregulated (Figure 5B, E) by co-transfection with TrkB. Because the myc-tagged channel also did not modulate any other biophysical property compared with the untagged Kv1.3 channel (data not shown), we only used the myc-tagged construct under non-permeabilizing conditions, to optically track changes in surface expression in the presence of TrkB kinase. Quantification of approximately forty cells collected over six individual transfection experiments, demonstrated that the proportion of Kv1.3 channel at the membrane surface (or closely juxtaposed) significantly increases 2X in the TrkB co-transfected condition compared with Kv1.3 alone (Figure 6A, B). Kv1.3 channel tagged with both myc and gfp (gfpMyc), which functionally failed to show an increase in current magnitude or increase in protein expression in the presence of TrkB, likewise demonstrated no increase in pixel density at the cell surface when co-transfected with TrkB (Figure 6C). In order to more strongly suggest that the upregulation was localized to the plasma membrane as opposed to close juxtaposition in neighboring membrane structures such as lipid rafts, we designed the parallel experiments in Figure 6D. Thus, to biochemically track changes in surface expression in the presence of TrkB kinase, lives cells were immunoprecipitated with anti-Myc following transfection with the myc-tagged channel construct prior to cell lysis (Figure 6D, right). As shown in Figure 6D and in keeping with the confocal imaging data, the fraction of Kv1.3 channel expressed on the surface was significantly upregulated in the presence of TrkB or TrkB plus an adaptor protein Shc (LEFT Panel) in comparison to the total fraction of expressed Kv1.3 in the presence of TrkB (RIGHT Panel).
Fig. 5.
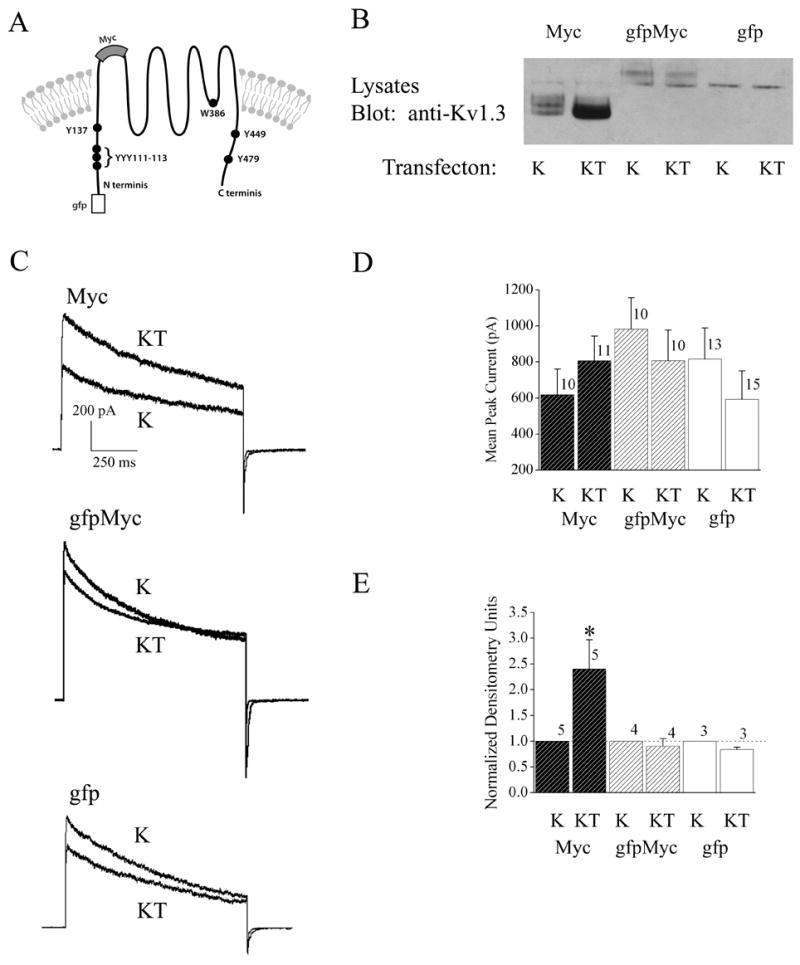
Epitope-tagged Kv1.3 channels vary in ability to be upregulated by TrkB co-expression. (A) Schematic of N terminal location for the gfp epitope channel construct (gfp) and the myc channel construct (Myc), where myc was inserted into the S1/S2 loop between residues 226-227 to establish an extracellular location. Myc was also identically inserted into the gfp construct to create a double epitope tag (gfpMyc). (B) HEK293 cells were transiently transfected with cDNA encoding one of the three epitope tagged channels (Myc, gfpMyc, or gfp) using the Kv1.3 minus (K) or plus TrkB (KT) paradigm. Lysates were prepared and separated by SDS PAGE as in Fig. 1. Nitrocellulose was blotted with anti-Kv1.3. (C) Representative Kv1.3 current recorded from HEK 293 cells under the specified transfection conditions using the cell-attached configuration. Patches were held at −90 mV (Vh) and stepped to a single depolarizing voltage of +40 mV (Vc) for a duration of 1000 ms using 60 s interpulse intervals between voltage steps. (D) Histogram plot of the peak current amplitude for various transfection conditions and recordings as in (C). Data are expressed as mean ± s.e.m., sample size as indicated, * = Statistically significant by Student’s t-test at α≤ 0.05.
Fig. 6.
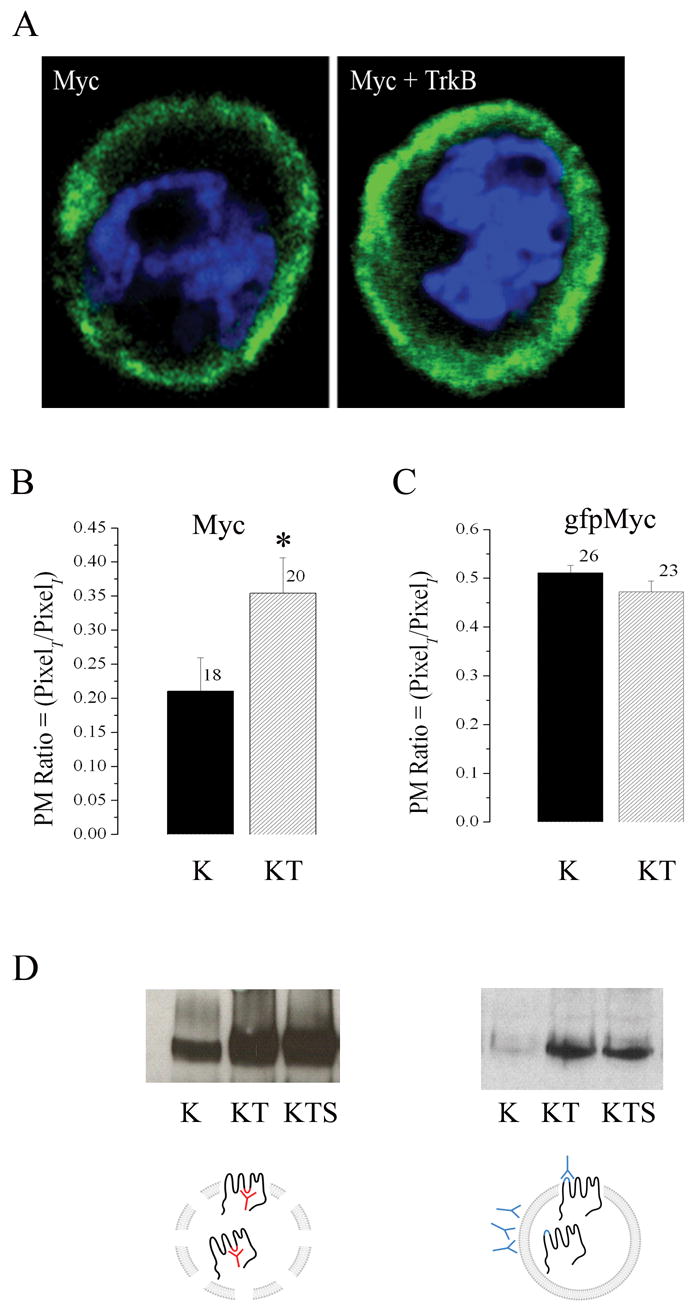
TrkB co-expression increases surface distribution of Kv1.3 channel. (A) Representative HEK 293 cell transiently transfected with myc tagged Kv1.3 (Myc) alone or plus TrkB (Myc + TrkB). Channel protein was detected by incubation with anti-myc under non-permeabilizing conditions and then visualized with species appropriate fluorescent-conjugated secondary antiserum (green). Cells were also stained with DAPi (blue) to visualize the nuclei and general integrity of the cells. Stacks of confocal images were acquired and projected as a z-series. (B–C) Pixel immunodensity was calculated (see text) for the Myc and gfpMyc Kv1.3 constructs under K and KT transfection conditions using blind assessment of a population of cells across multiple plates and transfection batches. Histogram represented data are expressed as mean ± s.e.m., sample size as indicated, * = Statistically significant by Student’s t-test at α≤ 0.05. (D) HEK293 cells were either transiently transfected (LEFT PANEL) with cDNA encoding Kv1.3 (K), Kv1.3 plus TrkB (KT), or Kv1.3, TrkB, and the adaptor protein Shc (KTS) or the same series of constructs (RIGHT PANEL) substituting MycKv1.3 construct for untagged Kv1.3. Lysates were either immunoprecipitated (IP) with an antiserum to Kv1.3 (anti-Kv1.3) as previously described (LEFT PANEL SCHEMATIC), or surface labeled as live cells with anti-Myc prior to cell lysis (RIGHT PANEL SCHEMATIC. Both preparations were separated by SDS PAGE and nitrocellulose was probed with anti-Kv1.3 as in Fig. 1. Autoradiographs are representative of four such experiments.
TrkB increases the half-life of channel turnover at the plasma membrane
It is well supported that channels have signaling sequences that direct the rate and regulation of their synthesis and release from the endoplasmic reticulum to govern their insertion into the plasma membrane and channels exist in the plasma membrane for a quantifiable time period prior to degradation and new protein synthesis (Zhu et al., 2003; Sun et al., 2004; Misonou and Trimmer, 2004; Zarel et al., 2004;). We questioned whether TrkB might be stabilizing the channel in the plasma membrane; retaining it longer prior to degradation. To test this hypothesis Kv1.3 singly and co-transfected Kv1.3 plus TrkB HEK 293 cells were treated with an inhibitor of protein synthesis, cycloheximide, approximately 25–30 hours post-transfection. Cell lysates were harvested prior to inhibitor and two, four, six, or eight hours post treatment and probed with anti-Kv1.3. The observed pattern of decreased channel protein expressed over time in the Kv1.3 only condition following cycloheximide treatment (Figure 7A) reveals a defined turnover of Kv1.3 protein, which when normalized, provided a half-life of 3.8 ± 1.4 hours (n=3). Under the Kv1.3 plus TrkB co-transfection conditions, however, cyclohexmide treatment reveals a significantly slower rate of channel turnover and a calculated half-life of 6.6 ± 1.4 (n=5) as plotted in Figure 7B. The mean half-life of Kv1.3 was 1.7 times greater in the presence of TrkB and increased on average 2.5 ± 1.1 (N=3). A t-test on the log-transformed ratios of the ½ lives plus and minus TrkB demonstrated a significant difference (Student’s t-test, α≤0.05).
Fig. 7.
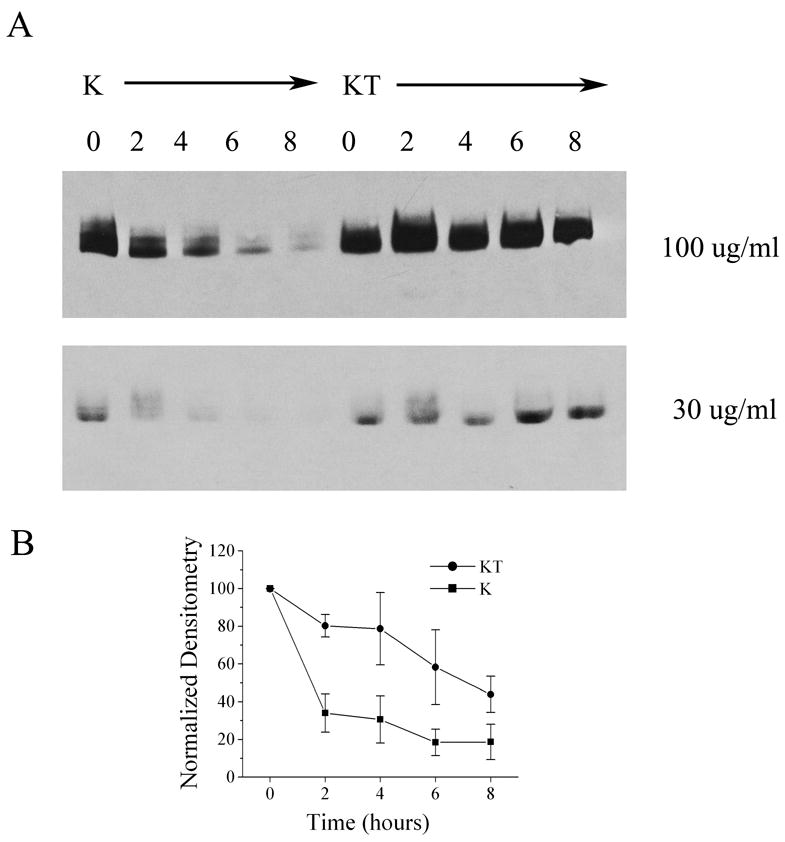
TrkB increases the half-life of Kv1.3 channel protein. (A) HEK293 cells were transiently transfected with cDNA encoding Kv1.3 (K) or Kv1.3 plus TrkB (KT). Twenty-eight hours following transfection, cells were treated with two different concentrations of cycloheximide and then lysates were harvested at varying time intervals (in hours) as noted. Lysates were separated by SDS PAGE and nitrocellulose was blotted with anti-Kv1.3 as in Fig. 1. (B) Line graph of the normalized immunodensity values for the Kv1.3 band versus time following 100 μg/ml cycloheximide treatment as in (A). Data are expressed as mean ± s.e.m., n = 3–4, ■ = Kv1.3 transfected cells (K), ● = Kv1.3 plus TrkB transfected cells (KT).
TrkB upregulation of Kv1.3 is dependent upon channel basal phosphorylation and channel activity
Data demonstrated that catalytic activity of TrkB was essential for upregulation of Kv1.3 channel protein (Figure 1), but it was not clear whether the increase in channel expression was dependent upon basal tyrosine phosphorylation. To address this question, we utilized four Y to F single point mutations of the channel (Figure 5A) that targeted strong recognition motifs for tyrosine-dependent phosphorylation (Songyang et al., 1994; Songyang et al., 1995). Additionally we were curious as to whether ionic conduction was a requirement for the upregulation, thus we also tested W386F Kv1.3; a non-conducting channel mutant (Holmes et al., 1997). Our previous work has demonstrated that acute BDNF stimulation of TrkB kinase, causes Kv1.3 current suppression by multiple phosphorylation of residues Y111–113,Y137, and Y449 (Colley et al., 2004). Using a similar stimulation by BDNF or control vehicle (10 minute bath application to Kv1.3 vs. Kv1.3 + TrkB co-transfected HEK293 cells)(Tucker and Fadool, 2002; Colley et al., 2004), Kv1.3 channel was immunoprecipitated and blotted with an antiserum that detects tyrosine phosphorylated residues (anti-PY) to determine the phosphorylated state of the channel in conjunction with quantified upregulation of channel protein in cell lysates (Figure 8A). Acute stimulation by BDNF to activate TrkB kinase and increase Y phosphorylation of Kv1.3, did not alter channel expression between the basally phosphorylated (-BDNF) plus BDNF-phosphorylated state (+BDNF) for the Kv1.3 + TrkB co-transfection condition. Likewise, insulin activation of IR kinase, that we have previously demonstrated to increase tyrosine phosphorylation of Kv1.3 at Y111–113, Y137, and Y479 (Fadool et al., 2000), did not alter channel expression between the basally phosphorylated (-insulin) plus insulin-phosphorylated state (+insulin)(Figure 8A) under the co-transfected condition. However, basal phosphorylation of the channel, as opposed to kinase-induced Y phosphorylation of the channel, was very important for both upregulation by TrkB (Figure 8B, C) as well as basal protein expression (Figure 8D). As demonstrated in Figure 8B and quantified through densitometry in the histogram contained in Figure 8C, loss of ANY of the four targeted Y residues, resulted in loss of the upregulation of channel protein in the presence of TrkB. Likewise, the inability of the channel to conduct K ions, even in the presence of all four Y residues intact, prevented TrkB upregulation of channel protein. Lastly, mutation of any Y residue decreased total protein expression compared to the WT channel, which reached statistical significance with the Y111–113F Kv1.3 mutant, in which three Y residues were simultaneously substituted with F (Figure 8D). Note also that prevention of channel conduction (W386F Kv1.3), unexpectedly increased protein expression of the channel by 3X in comparison to the wildtype Kv1.3 channel.
Fig. 8.
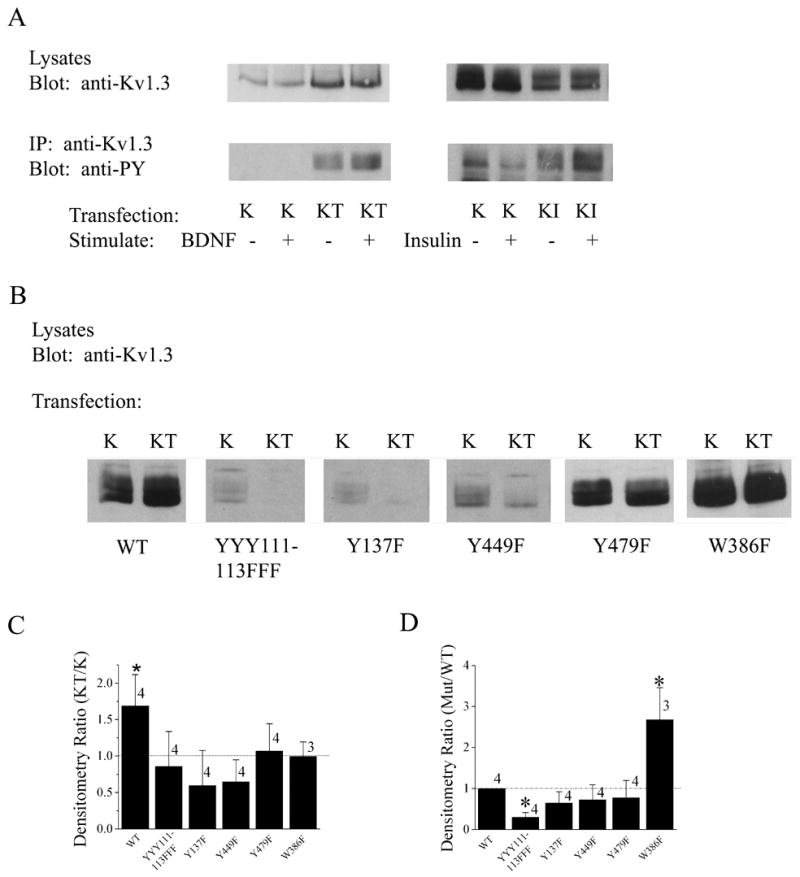
Receptor tyrosine activation phosphorylates Kv1.3, which does not affect channel expression, however, basal phosphorylation of Kv1.3 is required for TrkB upregulation of channel expression and general expression of Kv1.3. (A) HEK293 cells were transiently transfected with various transfection conditions as in Fig. 2 but were acutely stimulated with vehicle control (−) or BDNF/Insulin (+) for 10 minutes prior to cell lysis. Lysates were immunoprecipitated (IP) with an antiserum to Kv1.3 (anti-Kv1.3), separated by SDS PAGE and nitrocellulose was probed with an antiserum that recognizes Y phosphorylated residues (anti-PY). (B) Same as in Fig. 1, but using Kv1.3 Y to F point mutants as diagrammed in Fig. 5A. (C) Histogram graph of the ratio of KT:K immunodensity values generated for the various channel mutants in comparison to the wildtype Kv1.3 channel (WT). (D) Histogram graph of the ratio of mutant:WT immunodensity values generated for the various channel mutants. (C–D) Data are expressed as mean ± s.e.m., dashed line represents a ratio of unity (no difference). * = Statistically significant (K vs. KT or Mutant vs. WT) by Student’s t-test at α≤0.05.
Kv1.3 is co-localized and co-immunoprecipitates with TrkB in native olfactory bulb tissue
Previously we had demonstrated that gene-targeted deletion of Kv1.3 channel unbalances the expression of various kinases in the olfactory bulb, including TrkB, which shows a marked 7X increase in the absence of the channel (Fadool et al., 2004). Thus we anticipated that the channel and the kinase should be in close proximity to one another in the olfactory bulb, if our demonstrated modulation of protein expression, surface expression, and channel half-life by TrkB had potential physiological relevance. Co-transfection of TrkB and Kv1.3 in HEK293 cells do NOT support a direct protein-protein interaction as demonstrated by lack of co-immunoprecipitation, except in the presence of the adaptor protein nShc, in which they become easily co-immunoprecipitated (Fadool unpublished data). Knowing that nShc and other adaptor proteins are expressed in various levels of the olfactory bulb neurolamina (Colley unpublished data), we asked whether we could immunoprecipitate Kv1.3 and TrkB from native olfactory bulb or olfactory cortex whole lysates (Figure 9, Part I). Curiously we could co-immunoprecipitate Kv1.3 and TrkB from both the bulb and the cortex; in the olfactory bulb the greater portion of the complex was with the 95kDa or truncated TrkB, whereas in the olfactory cortex the greater portion of the complex was with the full length, 145kDa TrkB protein. Kv1.5 was exclusively precipitated with the truncated TrkB fraction in the olfactory bulb and more predominately complexed with the truncated TrkB fraction in the cortex. Interestingly IR kinase could be co-immunoprecipitated with the TrkB receptor in both tissues. Figure 9, Part II demonstrates immunolabeling for Kv1.3 ion channel protein that was evident starting from the exterior circumference of the OB in fibers of the glomerular layer (GL), external plexiform layer (EPL), mitral cell layer (MCL), internal plexiform layer (IPL), and the granule cell layer (GCL); no signal was observed in the outer nerve layer. Kv1.3 and TrkB labeling distribution patterns overlapped (Figure 9, Part III) mainly in fibers passing through the internal plexiform layer (III, C), slightly in some fibers surrounding the glomeruli in a closer magnification of the GL (III F), and in the granule cell layer (III, I).
Fig. 9.
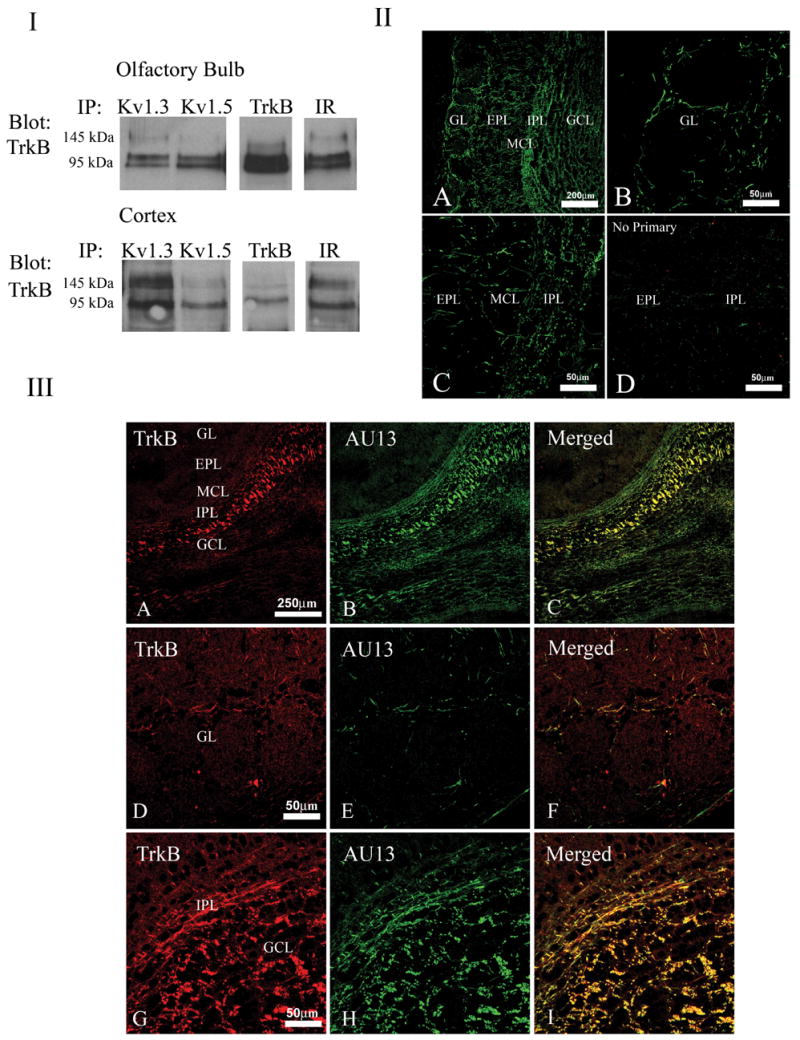
Shaker family members expressed in the olfactory bulb co-immunoprecipitate and co-localize with TrkB kinase. (I) Native lysates were prepared from postnatal day 20 (P20) mouse olfactory bulb (top panel) or cortex (bottom panel) and immunoprecipitated with various channel/kinase antisera as noted. Immunoprecipitated proteins (IP) were separated by SDS-PAGE and nitrocellulose was blotted with anti-TrkB. The molecular weight for the full length kinase (145 kDa) and truncated isoform (95 kDa) are noted. (II) Light micrograph of low (A) and higher power (B–D) magnification of a ten micrometer coronal cryosection of a P20 mouse olfactory bulb labeled with anti-Kv1.3 and visualized with FITC-conjugated secondary antiserum. Kv1.3 labeling was observed throughout the neurolamina of the olfactory bulb, including the glomerular layer (GL), external plexiform layer (EPL), mitral cell layer (MCL), internal plexiform layer (IPL), and granule cell layer (GCL) but was absent from the outer nerve layer. (D) Same but with the omission of primary antibody. (III) Same as in II. TrkB labeling is seen in the red channel (A, D, G), Kv1.3 is seen in the green channel (B, E, H), with merged image in yellow (C, F, I). Note Kv1.3 and TrkB overlapping distribution mainly in fibers passing through the internal plexiform layer (IPL)(C) and slightly in fibers, at higher magnification, surrounding the glomeruli (F) and in the GCL as shown in (I).
DISCUSSION
Our study introduces a yet undescribed role for neurotrophins modulating ion channel expression and retention at the membrane through low or basal levels of phosphorylation in contrast to the well-described modulatory functions of ligand-initiated, kinase-activated phosphorylation of voltage-gated ion channels. The activity of voltage-gated ion channels is changed in response to rapid and chronic stimulation with neurotrophins that induce robust phosphorylation of the channel at mapped residues probed through mutagenesis studies (Rose et al., 2004). The expression of receptors for neurotrophins and related growth factors is linked to activity-dependence of a neuron, is programmed during neural development, and can be perturbed during nerve injury, regeneration, or metabolic disease such as diabetes {Bonhoeffer, 1996; Lu and Figurov, 1997; Zweifel et al., 2005). Receptor tyrosine kinases, TrkB and IR kinase, have the ability to differentially modulate the expression of two Shaker family members, Kv1.3 and Kv1.5. The fact that TrkB can increase expression of Kv1.3 and oppositely decrease expression of Kv1.5 suggests a specificity that could afford changes in excitability for a cell given the differences in inactivation kinetics of these two potassium channels. Microglia are reported to ‘switch’ from principally a Kv1.5 to a Kv1.3 current during activation (Kotecha and Schlichter, 1999); it would be interesting to ascertain if this change of potassium current driven by a transition to a different Shaker ion channel was governed by increased expression of TrkB.
Certainly the differential regulation and expression of ion channels by neurotrophins during development or activity is not uncommon (Urbano and Buno, 2000; Suzuki, et al., 2005), but what is notable in our finding is that the presence of the receptor in the absence of applied trophic factor can mediate changes in channel expression. It is highly probable that TrkB is basally phosphorylated in the absence of applied BDNF, however, our methods of Y phosphorylation detection cannot quantify such low level basal phosphorylation, but deletion of the catalytic activity (TrkB dead kinase construct) of the receptor did block upregulation of Kv1.3 channel expression, implying that such basal or low level phosphorylation was essential for the upregulation mechanism. Although our data do not indicate how TrkB might be activated in our preparation, Rajagopel et al. (2004) report that Trk neurotrophin receptors can be activated by GPCR ligands in the absence of neurotrophins. Given the presence of the neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) reported in the olfactory epithelium and olfactory bulb (Hegg et al., 2003), such ligands could activate TrkB in vivo in the absence of BDNF (Rajagopal et al., 2004). The change in Shaker channel expression in the presence of TrkB or IR was not directly correlated to a concomitant change in functional current measured electrophysiologically. This suggests, particularly in the case for Kv1.5, that there may be a pool of docked channels in the membrane for which a given fraction do not functionally contribute to current generation for the cell. Such a pool of silent or “sleeping” channels has been reported for specific Kv subfamily members (Kv6 and Kv9 family members) (Stocker et al., 1999; Ottschytsch et al., 2005) and uncovered for other potassium channels during FRAC (function recovery after chemobleaching) studies to explore kinetic rates of functional reinsertion of potassium channels (Sun et al., 2004). Our data and that reported by Sun et al. (2004) support that molecular/protein density of a channel may not always exhibit linearity with functional density. An alternative interpretation of our data is that there may be contribution of Kv1.3-based current from mitochondria as recently reported by Szabo et al., 2005. Based upon surface area calculations for a typical mitochondria and the approximate unitary currents recorded as mitochondrial Kv1.3 in native lymphocytes (1.5 pA/0.79 μm), the amount of current generated could be fairly significant, however, we feel that contribution by organelle current would likely be prevented in our recording configuration, that is cell-attached at the plasma membrane rather than whole-cell.
The mechanism for TrkB upregulation of Kv1.3 channel expression is tied with parallel results in our study. First, treatment with the protein inhibitor cycloheximide, suggests that TrkB may stabilize Kv1.3 channel protein in the membrane for a greater half-life, delaying normal degradation pathways by several hours compared to the channel alone. By comparison, in non-neuronal cells, such as mouse promonocytoid cells (U937), pulse-chase experiments have determined much slower rates of turnover of cell-surface Kv1.3 on the order of 18 h (Chandy et al., 1997). Retention of surface expressed Kv1.3 that contributed functionally to potassium current generation in neuronal cells, would establish a dampening of excitability by driving the cell towards its EK, close to the resting potential. Upon TrkB activation by its preferred ligand BDNF, we know that Kv1.3 is Y phosphorylated, current magnitude is suppressed, driving the excitability of the cell in the opposite direction. Positioning TrkB receptors in close proximity to Kv1.3, as suggested by our native co-immunoprecipitation and co-localization data in the olfactory bulb, would thus facilitate this type of channel regulation by neurotrophic signaling pathways to increase or decrease excitability as dictated by physiological condition.
Secondly, removal of any of four principle Y residues in the channel contained within strong recognition motifs for Y specific phosphorylation, prevented TrkB upregulation of channel expression. Mechanistically this implies that both basal phosphorylation of TrkB and the channel are essential for the upregulation. Additionally it was noted that loss of these residues also affected protein expression in terms of pixel density (ratiometric comparison to that of wildtype channel), however, functionally these channel Y to F point mutants do not have reduced current in comparison to that of the wildtype channel (Fadool et al., 1997). This again, suggests a fraction of the Kv1.3 channels visualized by SDS-PAGE do not contribute to functional current generation in the cell. The structural importance of Y residues at discreet sites in potassium channels was elegantly demonstrated in a series of uncaging experiments using Kir2.1 by Tong et al. (Tong et al., 2001). They showed that decaging of residue 242 in this inward rectifier channel led to massive clathrin-mediated endocytosis; membrane trafficking regulated by a single Y residue.
Third, and lastly, our results utilizing epitope-tagged channel constructs, suggest that the N terminus of the channel is mechanistically important for upregulation of channel expression by TrkB. Folding prediction plots for Shaker channels reveal that the N and C terminal segments are intrinsically disordered or unfolded, providing considerable movement of residues in these regions (Magidovich et al., 2006). As such, incorporation of the bulky or poorly positioned GFP tag at the N terminus may introduce unexpected structural constraints that could affect channel modulation by TrkB. Kupper et al. (1998) report that fusing GFP to either the N or C terminal of Kv1.3 did not interfere with localization or biophysical properties of the channel in terms of current magnitude, slow C-type inactivation, or recovery from inactivation, however functional accessment of modulation was not explored. Thus fluorescing fusion constructs at the N terminal may function properly, but may not either traffick or respond to basal phosphorylation in the presence of TrkB signaling in keeping with our data. It has been demonstrated that the amount of protein expressed on the cell membrane will depend, at the ER level, on an exquisite balance of protein export and retention mechanisms (Zarel et al., 2004). In Kv1.1, the pore was found to be a major negative determinant to inhibit high surface expression because it induced high partial ER retention and decreased protein stability. It is interesting that our W386F Kv1.3, non-conducting mutant, representing a pore mutation, increased surface expression 3x fold; an effect that we also note in the blots contained in Holmes et al. (1997). While the lack of TrkB induced upregulation of the non-conducting mutant is equally surprising, we cannot exclude the interpretation that detection of further increased channel expression cannot be resolved by our SDS Page/Western methods since it is so highly expressed even in the absence of TrkB . Since different Shaker family members have different determinants for channel trafficking, further mutagenesis would be required to determine why a pore mutation would increase expression. We do know, however, that basic intracellular motifs (RXR, KR, RRKK, and RR) serving as ER retention signals are found at several locations upstream and flanking the triple YYY motif just proceeding the T1 tetramerization domain in Kv1.3 (between residues 85 and 122: RRMR, RNR, RIRR). Desir et al. (2000) have demonstrated that removal of the N terminal 30 amino acids of Kv1.3 causes a 13X increase in current magnitude. One could conjecture that co-expression with TrkB that would perturb these ER retention sites or render them inaccessible, would make Kv1.3 exit the ER more efficiently to insure the physiological need for channels at the plasma membrane. Because folding of the Kv1.3 T1 domain begins in the ribosomal exit tunnel (Kosolapov et al., 2004) and other K channel family members have demonstrated cell surface enhancement of expression by the presence of adaptor proteins and raft associated proteins (Martens et al., 2004; Marble et al., 2005; Roncarati et al., 2005), it is not inconceivable that TrkB could subserve the same function, and fine-tuned by neuronal activity. Future experiments will be needed to determine a role for the N terminal domain of Kv1.3, which when perturbed by epitope tagging, prevents upregulation of expression by TrkB. One could envision that TrkB or scaffolding adaptor proteins (Shc) could serve to release Kv1.3 from the ER and delay degration of the channel from the plasma membrane during periods of activity-dependence aligned with BDNF/neurotrophic signaling.
The co-immunoprecipitation of Kv1.3 and TrkB from native membranes of the olfactory bulb and cortex, as well as co-localization of the proteins to olfactory bulb tissue sections, strongly suggests that such channel-kinase regulation of expression and surface targeting occurs outside the confines of a heterologous expression system. The two proteins exhibit reciprocal regulation in vivo as evidenced by Kv1.3-null mice, where TrkB is expressed in the olfactory bulb at levels 7X that of wildtype mice (Fadool et al., 2004). Our current experimental results, combined with those in Kv1.3-null mice argue in favor of a regulatory relationship between expression levels of TrkB and those of Kv1.3. Kv1.3 and TrkB protein expression levels vary with development (Tucker and Fadool, 2002; Silhol et al.; 2005). Developmental changes in peak TrkB would be predicted to affect the kinetics of observed increases in Kv1.3 expression. Our finding in heterologous expression systems, that the maximum expression levels of Kv1.3 are reached when TrkB and Kv1.3 are in a 1:1 ratio is informative because we do not know the exact levels at which they co-exist in neurons relative to one another.
The fact that Kv1.3 co-immunoprecipitates predominantly with the truncated 95 kDa form of TrkB (lacking catalytic activity) in the olfactory bulb but equivalently co-immunoprecipates with both the full and truncated form of TrkB in the cortex, may impart a different modulatory ability to upregulate channel expression in each of these brain regions. We and others have reported expression of truncated neurotrophin receptors in the olfactory bulb (Deckner et al., 1993; Nef et al., 2001; Tucker and Fadool, 2002; Rajagopal et al., 2004) but the function for these receptors that can bind ligand but without catalytic activity, remains not well-understood in general for the central nervous system (Guthrie and Gall, 1991). Since our data demonstrate TrkB dead kinase fails to upregulate Kv1.3 channel expression, cells that expressed a greater fraction of truncated TrkB at any given physiological condition (developmental or disease) would be predicted to have less Kv1.3 channel protein, as supported by the greater expression of Kv1.3 in the olfactory cortex over that of the olfactory bulb.
In the olfactory bulb, the Kv1.3 and TrkB distribution patterns overlap in a proportion of fibers surrounding the glomeruli and most predominantly in the internal plexiform layer. As such, the colocalized protein could be functionally important for the primary dendrites of mitral cells or periglomerular cells, axon collaterals of tufted cells, or axon collaterals or dendrites of local interneurons in the granule cell layer. Our data demonstrate localization of the channel and kinase to classically defined histological layers but future studies will be required to extend these findings to confirmed individual neuronal cell types that could be influenced by increased K channel expression. Kv channels often show a highly specific subcellular localization (Rivera et al., 2005) that could be explored in context to TrkB, because such compartmentalization permits signal transduction molecules to be near their ion channel substrates (O’Connell and Tamkun, 2005).
K channels can be modified by post-translational events, including covalent modification (phosphorylation) and non-covalent protein-protein interactions (Jonas and Kaczmarck, 1996;Yi et al., 2001; Misonou and Trimmer, 2004). Much attention has been focused on the first half of this cycle: phosphorylation, as witnessed by the discovered number of ligand and voltage-gated ion channels that serve as substrates for Y phosphorylation. The continuous nature of the cycle itself, however, and the basal phosphorylation of a channel may be important to permit regulation of its expression by signaling molecules, which are in turn, varying with neuronal excitability and disease. This would allow the cell a degree of plasticity to respond to its current environment with changes in membrane excitability. The abundance and location of K channels significantly impacts membrane excitability, and in the case of our data, during basal activity of TrkB co-localization.
Acknowledgments
We would like to thank Ms. Danielle Walker for technical assistance with the mouse colonies, Dr. Rani Dhanarajan for her molecular assistance in the epitope-tagged channel construct, and Jon Ekman and Kim Ritter for their technical assistance with the confocal imaging. This work was supported by the National Institutes of Health (DC03387 from the NIDCD) and a Florida Neuroscience Fellowship.
Comprehensive List of Abbreviations
- anti-PY
phosphotyrosine antiserum
- AU13
antiserum directed against Kv1.3 ion channel
- BDNF
brain-derived neurotrophic factor
- BSA
bovine serum albumin
- cDNA
copy DNA
- CMV
cytomegalovirus promoter
- C terminus
carboxyl terminus of a protein
- ECL
epichemiluminescence
- EK
equilibrium potential for potassium
- EPL
external plexiform layer of the olfactory bulb
- ER
endoplasmic reticulum
- F
phenylalanine
- FRAC
function recovery after chemobleaching
- GFP
green florescence protein
- GCL
glomerular cell layer of the olfactory bulb
- GL
granule layer of the olfactory bulb
- HB
homogenization buffer
- HEK 293
human embryonic kidney 293 cells
- HRP
horseradish perioxidase
- IP
immunoprecipitation
- IPL
internal plexiform layer of the olfactory bulb
- IR
insulin receptor kinase
- K Channel
potassium channel
- K
lysine
- k
voltage dependence
- KD
K Channel + dead TrkB kinase
- kDa
kilodalton
- KI
K Channel + Insulin receptor kinase
- KT
K Channel + TrkB kinase
- Kv Shaker
voltage-dependent potassium channel
- Kv1.3
Kv subfamily member 1.3
- Kv1.5
Kv subfamily member 1.5
- MCL
mitral cell layer of the olfactory bulb
- MEM
minimum essential media
- Myc
myc epitope tag
- NGF
nerve growth factor
- NP40
PPI nonidet-NP40 protease and phosphatase inhibitor
- NT3
neurotrophin 3
- N terminus
amino terminus of a protein
- OB
olfactory bulb
- PACAP
pituitary adenylate cyclase-activating polypeptide
- PBS
phosphate buffered saline
- PBST
phosphate buffered saline + tween
- PCR
polymerase chain reaction
- PM
plasma membrane
- PPI
protease and phosphatase inhibitor
- P20
postnatal day 20
- R
arginine
- rt
room temperature
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TrkB
neurotrophin B receptor kinase
- Vh
holding voltage
- Vc
command voltage
- V1/2
voltage at half-maximum activation
- Y
tyrosine
- W
tryptophan
- WT
wildtype
- 2X,3X, etc.
fold
- τDeact
deactivation time constant
- τInact
inactivation time constant
Footnotes
Appropriate Section Editor to Whom the Manuscript Should Be Sent for Handling: Molecular Neuroscience, Dr. Werner Sieghart, Brain Research Institute, University of Vienna, Division of Biochemistry and Molecular Biology, Spitalgasse 4, A-1090 Vienna, AUSTRIA
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Biju KC, Walker DM, Fadool DA. Gene-targeted deletion of Kv1.3 channel alters olfactory receptor gene expression and modifies primary olfactory projections. Chem Senses Abstract 2006 [Google Scholar]
- Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- Cahalan MD, Wulff D, Chandy KG. Molecular properties and physiological roles of ion channels in the immune system. J Clin Immunol. 2001;21:235–252. doi: 10.1023/a:1010958907271. [DOI] [PubMed] [Google Scholar]
- Chandy KG, Strong M, Aiyar J, Gutman GA. Structural and biochemical features of the Kv1.3 potassium channel: an aid to guided drug design. Cell Physiol Biochem. 1997;7(7):135–147. [Google Scholar]
- Chandy KG, Wulff D, Beeton D, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley B, Tucker K, Fadool DA. Comparison of modulation of Kv1.3 channel by two receptor tyrosine kinases in olfactory bulb neurons of rodents. Receptors Channels. 2004;10:25–36. [PMC free article] [PubMed] [Google Scholar]
- Cullen DA, Leigh PN, Gallo J-M. Degradation properties of polyglutamine-expanded human androgen receptor in transfected cells. Neurosci Let. 2004;357:175–178. doi: 10.1016/j.neulet.2003.12.055. [DOI] [PubMed] [Google Scholar]
- Deckner ML, Frisen J, Verge VM, Hokfelt T, Risling M. Localization of neurotrophin receptors in olfactory epithelium and bulb. Neuroreport. 1993;5(3):301–304. doi: 10.1097/00001756-199312000-00030. [DOI] [PubMed] [Google Scholar]
- Fadool DA, Holmes TC, Berman K, Dagan D, Levitan IB. Multiple effects of tyrosine phosphorylation on a voltage-dependent potassium channel. J Neurophysiol. 1997;78:1563–1573. doi: 10.1152/jn.1997.78.3.1563. [DOI] [PubMed] [Google Scholar]
- Fadool DA, Tucker K, Fasciani G, Perkins R, Overton JM, Parsons AM, Koni PA, Flavell RA, Kaczmarek LK. Kv1.3 channel gene-targeted deletion produces "Super-smeller mice" with altered glomeruli, interacting scaffolding proteins, and biophysics. Neuron. 2004;41(3):389–404. doi: 10.1016/s0896-6273(03)00844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadool DA, Tucker K, Phillips JJ, Simmen JA. Brain insulin receptor causes activity-dependent current suppression in the olfactory bulb through multiple phosphorylation of Kv1.3. J Neurophysiol. 2000;83:2332–2348. doi: 10.1152/jn.2000.83.4.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie KM, Gall CM. Differential expression of mRNAs for the NGF family of neurotrophic factors in the adult rat central olfactory system. J Comp Neurol. 1991;313:95–102. doi: 10.1002/cne.903130107. [DOI] [PubMed] [Google Scholar]
- Hegg CC, Au E, Roskams AJ, Lucero MT. PACAP is present in the olfactory system and evokes calcium transients in olfactory receptor neurons. J Neurophysiol. 2003;90:2711–2719. doi: 10.1152/jn.00288.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusser K, Schwappach B. Trafficking of potassium channels. Curr Opin Neurobiol. 2005;15:364–369. doi: 10.1016/j.conb.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Holmes TC, Berman K, Swartz JE, Dagan D, Levitan IB. Expression of voltage-gated potassium channels decreases cellular protein tyrosine phosphorylation. J Neurosci. 1997;17(23):8964–8974. doi: 10.1523/JNEUROSCI.17-23-08964.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Kaczmarck LK. Regulation of potassium channels by protein kinases. Curr Opin Neurobiol. 1996;6(3):318–323. doi: 10.1016/s0959-4388(96)80114-0. [DOI] [PubMed] [Google Scholar]
- Kosolapov A, Tu L, Wang J, Deutsch C. Structure acquisition of the T1 domain of Kv1.3 during biogenesis. Neuron. 2004;44:295–307. doi: 10.1016/j.neuron.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Kotecha SA, Schlichter LC. A Kv1.5 to Kv1.3 switch in endogenouse hippocampal microglia and a role in proliferation. J Neurosci. 1999;19(24):10680–10693. doi: 10.1523/JNEUROSCI.19-24-10680.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kues WA, Wunder F. Heterogeneous expression patterns of mammalian potassium channel genes in developing and adult rat brain. Eur J Neurosci. 1992;4:1296–1308. doi: 10.1111/j.1460-9568.1992.tb00155.x. [DOI] [PubMed] [Google Scholar]
- Kupper J. Functional expression of GFP-tagged Kv1.3 and Kv1.3 channels in HEK293 cells. Eur J Neurosci. 1998;10:3908–3912. doi: 10.1046/j.1460-9568.1998.00441.x. [DOI] [PubMed] [Google Scholar]
- Kupper J, Bowlby MR, Marom S, Levitan IB. Intracellular and extracellular amino acids that influence C-type inactivation and its modulation in a voltage-dependent potassium channel. Pflugers Arch. 1995;430(1):1–11. doi: 10.1007/BF00373833. [DOI] [PubMed] [Google Scholar]
- Lu B, Figurov A. Role of neurotrophins in synapse development and plasticity. Rev Neurosci. 1997;8:1–12. doi: 10.1515/revneuro.1997.8.1.1. [DOI] [PubMed] [Google Scholar]
- Magidovich E, Fleishman SJ, Yifrach O. Intrinsically disordered C-terminal segments of voltage-activated potassium channels: a possible fishing rod-like mechanism for channel binding to scaffold proteins. Bioinformatics Advance Access Online April 6. 2006;2006:1–4. doi: 10.1093/bioinformatics/btl137. [DOI] [PubMed] [Google Scholar]
- Marble DD, Hegle AP, Snyder ED, Dimitratos S, Bryant PJ, Wilson GF. Camguk/CASK enhances Ether-A-Go-Go potassium current by a phosphorylation-dependent mechanism. J Neurosci. 2005;25(20):4898–4907. doi: 10.1523/JNEUROSCI.4566-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JR, O’Connell K, Tamkun M. Targeting of ion channels to membrane microdomains: localization of Kv channels to lipid rafts. Trends Pharmacol Sci. 2004;25(1):16. doi: 10.1016/j.tips.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Misonou H, Trimmer JS. Determinants of voltage-gated potatssium channel surface expression and localization in mammalian neurons. Crit Rev Biochem Mol Biol. 2004;39:125–145. doi: 10.1080/10409230490475417. [DOI] [PubMed] [Google Scholar]
- Mittman SC, Flaming DG, Copenhagen DR, Belgum JH. Bubble pressure measurement of micropipet tip outer diameter. J Neurosci Methods. 1987;22:161–166. doi: 10.1016/0165-0270(87)90010-0. [DOI] [PubMed] [Google Scholar]
- Nef S, Lush ME, Shipman TE, Parada LF. Neurotrophins are not required for normal embryonic development of olfactory neurons. Dev Biol. 2001;234:80–82. doi: 10.1006/dbio.2001.0240. [DOI] [PubMed] [Google Scholar]
- Nitabach MN, Llamas DA, Araneda RC, Intile JL, Thompson IJ, Zhou Y, Holmes TC. A mechanism for combinatorial regulation of electrical activity: Potassium channel subunits capable of functioning as Src homology 3-dependent adaptors. Proc Natl Acad Sci. 2001;98(2):705–710. doi: 10.1073/pnas.031446198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell KMS, Tamkun MM. Targeting of voltage-gated potassium channel isoforms to distinct cell surface microdomains. J Cell Sci. 2005;118:2155–2166. doi: 10.1242/jcs.02348. [DOI] [PubMed] [Google Scholar]
- Ottschytsch N, Raes AL, Timmermans J-P, Snyders DJ. Domain analysis of Kv6.3, an electrically silent channel. J Physiol. 2005;568.3:737–747. doi: 10.1113/jphysiol.2005.090142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal R, Chen Z-Y, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci. 2004;24(30):6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera JF, Chu P-J, Arnold DB. The T1 domain of Kv1.3 mediates intracellular targeting to axons. Eur J Neurosci. 2005;22:1853–1862. doi: 10.1111/j.1460-9568.2005.04384.x. [DOI] [PubMed] [Google Scholar]
- Roncarati R, Decimo H, Fumagalli G. Assembly and trafficking of human small conductance Ca2+-activated K+ channel SK3 are governed by different molecular domains. Mol Cell Neurosci. 2005;28:314–325. doi: 10.1016/j.mcn.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Rose CR, Blum R, Kafitz KW, Kovalchuk Y, Konnerth A. From modulator to mediator: rapid effects of BDNF on ion channels. BioEssays. 2004;26:1185–1194. doi: 10.1002/bies.20118. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatas T. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory; 1989. 1989. [Google Scholar]
- Silhol M, Bonnichon V, Rage F, Tapia-Arancibia L. Age-related changes in brain-derived neurotrophic factor and tyrosine kinase receptor isoforms in the hippocampus and hypothalamus in male rats. Neurosci. 2005;132:613–624. doi: 10.1016/j.neuroscience.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, Cantley LC. July 2, 2006Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr Biol. 1994;4(11):973–982. doi: 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Carraway KL, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng Lorenzo MJ, Ponder BAJ, Mayer BJ, Cantley LC. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- Stocker M, Hellwig M, Kerschensteiner D. Subunit assembly and domain analysis of electrically silent K+ channel alpha-subunits of the rat Kv9 subfamily. J Neurochem. 1999;72(4):1725–1734. doi: 10.1046/j.1471-4159.1999.721725.x. [DOI] [PubMed] [Google Scholar]
- Sun H, Shikano S, Siong Q, Li M. Function recovery after chemobleaching (FRAC) evidence for activity silent membrane receptors on cell surface. Proc Natl Acad Sci. 2004;101(48):16964–16969. doi: 10.1073/pnas.0404178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Sato M, Morishima Y, Nakanishi S. Neuronal depolarization controls brain-derived neurotrophic factor-induced upregulation of NR2C NMDA receptor via calcineurin signaling. J Neurosci. 2005;25(41):9535–9543. doi: 10.1523/JNEUROSCI.2191-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Bock J, Jekle A, Soddemann M, Adams C, Lang F, Zoratti M, Gulbins E. A novel potassium channel in lymphocyte mitochondria. J Biol Chem. 2005;280(13):12790–12798. doi: 10.1074/jbc.M413548200. [DOI] [PubMed] [Google Scholar]
- Tong Y, Brandt GS, Li M, Shapovalov G, Slimko E, Karshcin A, Dougherty DA, Lester HA. Tyrosine decaging leads to substantial membrane trafficking during modulation of an inward rectifier potatssium channel. J Gen Physiol. 2001;117(2):103–118. doi: 10.1085/jgp.117.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker K, Fadool DA. Neurotrophin modulation of voltage-gated potassium channels in rat through TrkB receptors is time and sensory-experience dependent. J Physiol. 2002;542.2:413–429. doi: 10.1113/jphysiol.2002.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbano FJ, Buno W. Neurotrophin regulation of sodium and calcium channels in human neuroblastoma cells. Neurosci. 2000;96(2):439–443. doi: 10.1016/s0306-4522(99)00552-7. [DOI] [PubMed] [Google Scholar]
- Xu J, Koni PA, Wang P, Li G, Kaczmarek LK, Wu Y, Li Y, Flavell RA, Desir GV. The voltage-gated potassium channel Kv1.3 regulates energy homeostasis and body weight. Hum Mol Genet. 2003;12(5):551–559. doi: 10.1093/hmg/ddg049. [DOI] [PubMed] [Google Scholar]
- Xu J, Wang P, Li Y, Li G, Laczmarek LK, Wu Y, Koni PA, Flavell RA, Desir GV. The voltage-gated potassium channel Kv1.3 regulates peripheral insulin senstivitiy. Proc Natl Acad Sci. 2004;101(9):3112–3117. doi: 10.1073/pnas.0308450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Liu W, Tian S, Rafi H, Segal AS, Desir GV. Close association of the N terminus of Kv1.3 with the pore region. J Biol Chem. 2000;275(15):10859–10863. doi: 10.1074/jbc.275.15.10859. [DOI] [PubMed] [Google Scholar]
- Yi BA, Minor DL, Lin YF, Jan YN, Jan LY. Controlling potassium channel activities: interplay between the membrane and intracellular factors. Proc Natl Acad Sci. 2001;98:11016–11023. doi: 10.1073/pnas.191351798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarel MM, Eghbali M, Alioua A, Song M, Knaus H-G, Stefani E, Toro L. An endoplasmic reticulum trafficking signal prevents surface expression of a voltage- and Ca2+-activated K+ channel splice variant. Proc Natl Acad Sci. 2004;101(27):10072–10077. doi: 10.1073/pnas.0302919101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Watahabe I, Gomez B, Thornhill WB. Heteromeric Kv1 potassium channel expression. J Biol Chem. 2003;278(28):25558–25567. doi: 10.1074/jbc.M207984200. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of retrograde neurotrophin signaling. Nat Rev Neurosci. 2005;6(8):615–625. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]