

Abstract
Aims
Women experience more adverse drug reactions (ADR) to antiretroviral therapy than men. This may be attributed to higher plasma concentrations of protease inhibitors due to pharmacokinetic interactions with hormonal preparations. Thus, in the present study we aimed to investigate the influence of oral contraceptives (OC) on the pharmacokinetics of the protease inhibitor saquinavir.
Methods
Saquinavir was administered in a hard gelatin capsule formulation (Invirase®) to rule out confounding by pharmaceutical aids of the more frequently used soft gelatin capsule. After an overnight fast, eight healthy female participants ingested a single oral dose of 600 mg saquinavir immediately before and after the 19th dose of a combined, low dose OC (0.03 mg ethinylestradiol, 0.075 mg gestodene) in a prospective, fixed sequence study design. The first saquinavir application was scheduled on day 1, 2, or 3 of the individual menstrual cycle. Plasma concentrations of saquinavir and relative concentrations of its M2&M3-hydroxy metabolites were determined by LC/MS/MS for 48 h.
Results
Intake of OC resulted in a significant decrease in morning serum concentrations (before intake of OC, compared to day 19 of OC therapy) of 17β-estradiol by −23.4 pg ml−1 (57%, 95%CI: −76% to −37.4%); progesterone by −0.25 ng ml−1 (33%, 95%CI: −45.3% to −21.5%); follicle-stimulating hormone by −4.06 U l−1 (82%, 95%CI: −96.5% to −67.7%); and luteinizing hormone by −3.49 U l−1 (74%, 95%CI: −93 to −54.6%). Conversely, sexual hormone binding globulin serum concentrations increased by 83.6 nmol l−1 (205%, 95%CI: 32.2% to 377%). Pharmacokinetic parameters of saquinavir (AUC, Cmax, tmax, t1/2, CLR) were not affected by OC, nor was the relative metabolic ratio of saquinavir/M2&M3-hydroxy saquinavir. Furthermore, there was no association of serum hormone concentrations or MDR1-polymorphisms (C3435T and G2677T) with pharmacokinetic parameters of saquinavir.
Conclusions
There was no effect of OC on saquinavir pharmacokinetics. Thus, pharmacokinetic interactions of synthetic sexual steroids with saquinavir are not likely to account for the increased ADR to antiretroviral therapy seen in women.
Keywords: Oral contraception, saquinavir, drug interaction, CYP3A, P-glycoprotein
Introduction
Adverse drug reactions (ADR) are more frequent in women than in men, and this difference is particularly evident in the 15- to 44-year age group [1]. One factor by which the sex difference can be explained is a disproportionate rate of medication use in women [1]. In Germany 34% of women in reproductive years are current users of oral contraceptives (OC), and the cumulative incidence of ever taking OC is 91%[2]. OC may contribute to poly pharmacotherapy, mainly in the above mentioned age group, and so increase the risk of drug interactions. Accordingly, several studies have shown that women experience more ADR to antiretroviral therapy [3–5], including protease inhibitors [6], such as saquinavir. However, none of these studies reported on the use of OC in affected women.
Controlled studies and clinical case reports provide evidence for interactions of OC with the pharmacokinetics of other drugs. Among others, cytochrome-P450 (CYP) 3A-substrates, such as midazolam [7], nifedipine [8], ciclosporin [9], and prednisolone [10] are involved. But with regard to midazolam – a frequently used probe drug for CYP3A – there are also reports, which failed to show significant interactions [11]. Combined OC usually contain ethinylestradiol and a progestin, which by themselves are metabolized by CYP3A [12, 13]. Synthetic steroids with acetylenic groups, such as ethinylestradiol and gestodene, have been shown to be irreversible inhibitors of CYP3A in vitro [14–17]. The enzyme inactivation is mechanism-based [16, 17] and thus merely affects the enzyme involved in the biotransformation of the compound. More recently the drug-transporter P-glycoprotein (PGP) has been recognized as another target of clinically relevant drug interactions (e.g. digoxin-quinidine) [18]. Several progestins are competitive inhibitors of PGP in vitro [19], however, just as for the mechanism-based inactivation of CYP3A, it is unclear whether local concentrations of synthetic steroids achieved in vivo are sufficient to induce the phenomena described in vitro.
In some [20], but not all reports [21], a higher saquinavir exposure has been reported for women, compared to men, which may account for their increased risk of ADR to protease inhibitors. Saquinavir is both, a substrate for CYP3A [22] and PGP [23], and inhibition of CYP3A and PGP increases saquinavir concentrations [24]. Thus, in the present study we intended to evaluate the effect of a combined OC, containing 0.03 mg ethinylestradiol and 0.075 mg gestodene, on the pharmacokinetics of saquinavir in young healthy females, and to assess the potential contribution of CYP3A and PGP.
Methods
The study protocol was approved by the Ethics Committee of the Medical Faculty, University of Heidelberg and conducted in accordance with the Declaration of Helsinki, as amended in Somerset West 1996. Written informed consent was obtained from each participant before entering the study.
Participants
Eight healthy, nonsmoking female individuals, with no menstrual cycle abnormalities were included into the study. All were of Caucasian origin. Mean (± s.d.) age was 23.8 (± 2.25) years, and mean body mass index 21.1 (± 1.82) kg/m2. All participants were ascertained to be healthy by physical examination; gynaecological examination, comprising a PAP smear; electrocardiography; and routine laboratory testing. Pregnancy was excluded by a qualitative β-human chorionic gonadotropin (βHCG)-test in urine. None of the participants received any regular drug treatment, including OC and herbal products, within the last 2 months before entering the study. Alcoholic and caffeinated beverages were not allowed from 12 h before until 48 h after saquinavir administration.
Study design
The study was performed in a prospective, open, fixed sequence design (Figure 1). After an overnight fast, all participants received 600 mg saquinavir (3 × 200 mg Invirase® capsules, F. Hoffmann-La Roche AG, Grenzach-Wyhlen, Germany), along with 200 ml water, at approximately 08:00 a.m. on days 1 and 22 of the individual study period. During the first 4 h following drug administration the participants were asked to stay in a semirecumbent position in bed. A standardized meal was served 4 and 8 h after saquinavir administration. From study day 4, participants received a single tablet of a low-dose combination OC, containing 0.03 mg ethinylestradiol and 0.075 mg gestodene (Minulet®, Wyeth-Pharma GmbH, Münster, Germany) once daily for 21 days. The beginning of the study period (day 1) was scheduled on the first three days of the women's menstrual cycle. On day 22 the 19th OC dose was administered along with 100 ml water 15–30 min before saquinavir intake.
Figure 1.
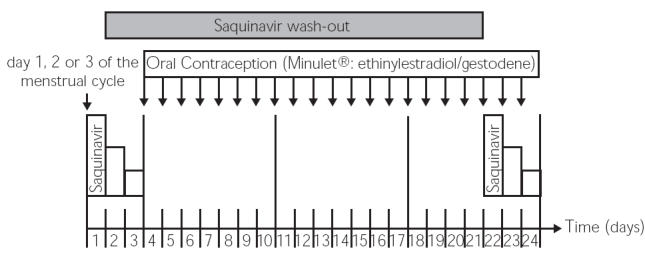
Study design: The first evaluation of saquinavir pharmacokinetics was scheduled on day 1, 2, or 3 of the individual menstrual cycle of each participant (study days 1–3), and evaluation of saquinavir pharmacokinetics was repeated on day 19–21 of therapy with the OC (study days 22–24)
Venous blood samples were drawn through an intravenous catheter into ammonium-heparin coated 7.5 ml tubes. Blood samples were obtained before, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 12, 24, and 48 h after saquinavir administration. They were immediately centrifuged at 3000 g for 10 min at 4 °C, and the plasma aliquoted and stored at −20 °C until analysis. On study days 1 and 22 additional serum samples were taken for hormone and SHBG analysis, and EDTA blood was taken for RNA and DNA isolation. Urine was collected in consecutive fractions 12, 24, and 48 h after administration of saquinavir. After measurement of the urine volume, a 3 ml aliquot of each fraction was stored at −20 °C until analysis.
Saquinavir determination
Plasma and urine concentrations of saquinavir were assessed by a validated LC/MS/MS method, as described previously [25]. Intraassay variability (%CV) obtained by analysing replicate samples (6 replicates in each of 3 batches) at three concentrations varied between 0.10% and 9.50% for plasma and 0.04% to 12.0% for urine, and interassay variability (%CV) varied between 1.70% and 7.70% for plasma. Lower limits of quantification (LLOQ) for saquinavir were 0.1 ng ml−1 for both, plasma and urine. Due to the lack of reference material saquinavir monohydroxylated metabolites M2&M3 [26] were measured indirectly using their theoretical tandem mass spectroscopic transitions and expressed as the signal ratio of metabolites to saquinavir, assuming that the mass spectroscopic response of M2&M3 is nearly equal to saquinavir. Since both metabolites reveal the same retention time and mass transition, M2 and M3 cannot be discriminated. Thus, M2&M3 refers to a combined signal response.
Hormone and SHBG determination
Serum concentrations of 17β-estradiol, progesterone, luteinizing hormone (LH), and follicle-stimulating hormone (FSH) were determined on days 1 and 22 by routine laboratory electrochemiluminescence immunoassays (ECLIA) (Elecsys Systeme®, Roche Diagnostics GmbH, Mannheim, Germany), and sexual hormone binding globulin (SHBG) serum concentration was measured by means of a routine laboratory enzyme immunoassay (ELISA) (DRG Instruments GmbH, Marburg, Germany).
MDR1 genotyping
C3435T polymorphisms in exon 26 of the multidrug resistance gene 1 (MDR1) was detected by rapid-cycle polymerase chain reaction (PCR) and fluorescent melting point analysis using fluorogenic hybridization probes on the LightCyclerTM (Roche Applied Sciences, Mannheim, Germany) [27]. G2677T polymorphism in exon 21 of the MDR1 gene was genotyped by PCR-restriction fragment length polymorpism (RFLP) according to Cascorbi et al. [28].
Pharmacokinetic analysis
The peak plasma concentrations of saquinavir (Cmax) and the time to reach Cmax (tmax) were taken from the raw data. All other pharmacokinetic parameters were determined by noncompartmental analysis using WinNonlin 3.1 (Pharsight Corporation, Mountain View, CA, USA). The area under the plasma concentration time curve during a time interval t1–t2 (AUCt1,t2) was calculated by using the linear trapezoideal rule. The slope of the terminal part of the plasma concentration-time curve (lz) was obtained by linear regression after semilogarithmic transformation. The terminal plasma half-life (t1/2) was calculated as t1/2 = ln 2 lz−1. Renal clearance (CLR) of saquinavir was calculated as amount excreted into urine (Ae) during the time interval t1–t2 divided by the respective AUC(t1,t2) interval. The relative metabolic ratio of saquinavir and its hydroxy metabolites M2&M3 in plasma was calculated as AUCSQV(0–48)/AUCM2&M3(0–48), and in urine as AeSQV(0–48)/AeM2&M3(0–48).
Statistical analysis and sample size
The results are expressed as median (range) in the table. Because of the highly asymmetrical distribution of pharmacokinetic parameters we decided to use nonparametric analyses throughout the study. Differences in serum hormone and SHBG concentrations and pharmacokinetic parameters of saquinavir in women with and without OC were compared by the nonparametric Wilcoxon signed rank test for paired data. Spearman rank correlation analyses were performed between hormone levels and pharmacokinetic parameters of saquinavir. Pharmacokinetic parameters of saquinavir in the different MDR1 genotype groups CC vs CT + TT for the C3435T polymorphism [29], and GG vs GT + TT for the G2677T polymorphism [30] were compared by the nonparametric Wilcoxon rank sum test (Mann–Whitney U-test) for unpaired data. A P-value < 0.05 was considered statistically significant.
A sample size of 16 was determined to achieve a 90% power of demonstrating a statistically significant difference of 33% in saquinavir AUC at the 5% alpha level. The data are based on estimates of intraindividual variability of the AUC of saquinavir (40%) observed in our previous study in healthy males [31]. The present study was early terminated because an interim analysis did not reveal a trend towards different saquinavir concentrations before and after therapy with OC. A post hoc power calculation from the present data (N = 8, intraindividual variability = 29.7%) resulted in a power of 87% of detecting a statistically significant difference of 33% in saquinavir AUC at the 5% alpha level.
Results
Intake of OC resulted in a statistically significant decrease in 17β-estradiol, progesterone, FSH, and LH serum concentrations, and in a significant increase in SHBG serum concentrations (Table 1).
Table 1.
Median (range) of sexual hormones and sexual hormone binding globulin (SHBG) in eight women before and during therapy with oral contraceptives (OC)
| Relative change† | |||||
|---|---|---|---|---|---|
| Variable | Before OC | During OC | P* | Mean(±SD) | Median (95% CI, %) |
| 17β-estradiol [pg/ml] | 31(20-65) | 14(8-19) | 0.012 | −56.7(±23.1) | −63.8 (−76 to −37.4) |
| Progesterone [ng/ml] | 0.65(0.3-1) | 0.5(0.2-0.6) | 0.011 | −33.4(±14.2) | −33.3 (−45.3 to −21.5) |
| FSH [U/l] | 5.25(0.7-8.1) | 0.5(0.1-3.5) | 0.012 | −82.1(±17.3) | −87.8 (−96.5 to −67.7) |
| LH [U/l] | 4.3(0.2-7.9) | 0.35(0.1-4.5) | 0.011 | −73.8(±23.1) | −81.4 (−93 to −54.6) |
| SHBG [nmol/l] | 54(19-79) | 151.5(47-190) | 0.012 | 205(±207) | 130 (32.2 to 377) |
P: Wilcoxon signed rank test for paired data, comparing women not taking OC with women taking OC.
: Percentage ratio of the means during OC (study day 22, after the 19th OC dose) relative to before OC (study day 1).
Despite the highly standardized study procedure, pharmacokinetic parameters of saquinavir revealed a high interindividual variability. Furthermore, the distribution of the pharmacokinetic parameters is highly asymmetrical which accounts for the large differences seen between mean and median relative changes of the variables during OC relative to before OC. Treatment with OC had no effect on saquinavir pharmacokinetics, and there was no difference in the relative ratio of saquinavir and its M2&M3-hydroxy metabolites, indicating no effect of OC on CYP3A activity (Table 2).
Table 2.
Median (range) of saquinavir after a single oral 600 mg dose in eight women before and during therapy with oral contraceptives (OC)
| Relative change† | |||||
|---|---|---|---|---|---|
| Variable | Before OC | During OC | P* | Mean(±SD) | Median (95% CI, %) |
| tmax[h] | 1.75(0.75–6) | 1.5(0.75–5) | 0.48 | −61.5(±182) | −10 (−312 to 90.6) |
| Cmax[ng/ml] | 5.42(1.49–19.9) | 5.98(1.54–35.2) | 0.48 | −37.4(±165) | 10.3 (−175 to 100) |
| t1/2[h] | 19.9(11–26.3) | 20.1(5.89–34.6) | 0.58 | −10.7(±50) | 7.23 (−52.4 to 31.0) |
| AUC0–4[ng x h/ml] | 6.6(2.36–15) | 7.19(2.46–17) | 0.67 | −27.6(±135) | 10.2 (−140 to 85.2) |
| AUC0–48[ng x h/ml] | 23.2(15.9–98.6) | 25.4(17.1–101) | 0.89 | 1.8(±21.3) | −5.65 (−16.0 to 19.6) |
| AUC0–∞[ng x h/ml] | 24.5(18.1–121) | 27.5(19.7–111) | 0.40 | 1.56(±20.8) | −6.46 (−15.8 to 18.9) |
| CLR (0–24)[ml/min] | 12.8(7.16–14.2) | 11.1(5.56–13.7) | 1 | −20(±66.4) | 2.76 (−81.5 to 41.5) |
| Saquinavir/M2&M3Plasma | 140(47.5–2158) | 198(52.8–1403) | 0.92 | 6.43(±51.1) | 3.92 (−47.2 to 60.1) |
| Saquinavir/M2&3Urine | 36.2(19.7–67.1) | 35.7(14.7–75.4) | 0.61 | −16.9(±28) | −12 (−42.8 to 9.10) |
P: Wilcoxon signed rank test for paired data, comparing women not taking OC vs. women taking OC.
: Relative change of the variables during OC (study day 22, after the 19th OC dose) relative to before OC (study day 1).
In Figure 2 concentration-time curves of saquinavir and its M2&M3-hydroxy metabolites in women without and with OC are shown. Saquinavir was absorbed rapidly, and a second smaller peak occurred after 4 h, reflecting the known influence of food intake on the bioavailability of the substance [32].
Figure 2.
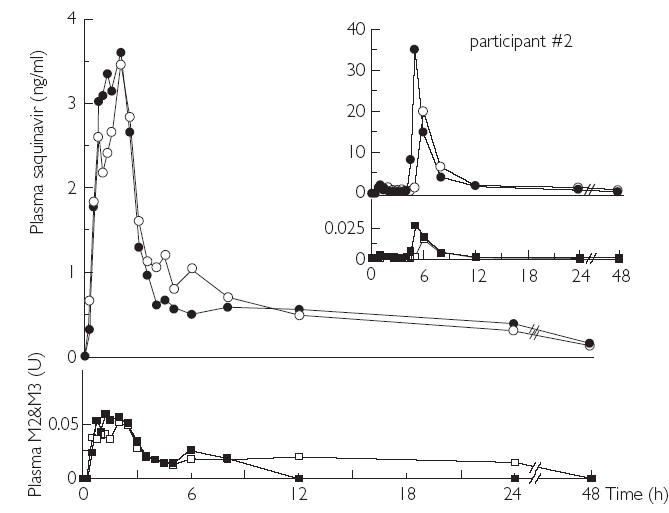
Concentration-time curves of saquinavir (dots) and its M2&M3-hydroxy metabolites (squares) in women without (open symbols) and with (solid symbols) OC. Concentration-time curves of participant #2 are depicted separately (insert). M2&M3 plasma concentrations are described as relative units (see Methods)
MDR1-genotypes were distributed as follows: C3435T: CC 1/8, CT 6/8, TT 1/8; G2677T: GG 2/8, GT 6/8, TT 0/8. There was no association of MDR1-polymorphisms or hormone concentrations with pharmacokinetic parameters of saquinavir (data not shown).
The concentration-time curves of participant #2 are depicted separately, due to the large deviation of the curves from the others. Her saquinavir concentrations were roughly five-fold higher (Cmax: 19.9 ng ml−1 without OC and 35.2 ng ml−1 with OC) and increased largely only after the meal. The relative ratios of saquinavir to its M2&M3-hydroxy metabolites in plasma were, compared to the 7 other participants, on average 11-fold higher (ratio: 2158 without OC and 1403 with OC), indicating decreased CYP3A activity which may account for reduced saquinavir clearance and higher saquinavir concentrations. Her MDR1-genotype was CT for the C3435T polymorphism and GT for the G2677T polymorphism. Statistical evaluation of the group without participant #2 was similar to the results of the group as a whole (Table 2): (Mean relative change (95%CI) and P-value (Wilcoxon signed rank test for paired data) for tmax: −67,4% (−235% to 100%), p = 0.73; Cmax: −49% (−199% to 101%), p = 0.87; t1/2: −7.8% (−53,4% to 37.9%), p = 0.5; AUC0-4: −32.8% (−157% to 91.6%), p = 0.74; AUC0-48: −1.77% (−18% to 21.5%), p = 0.74; AUC0-¥: −3% (−15.9% to 21.5%), p = 0.61; CLR(0–24): −26% (−93.7% to 41.6%), p = 0.75, and Saquinavir/M3&M3 Plasma: 18.5% (−33.4% to 17.4%), p = 0.35; Saquinavir/M2&M3 Urine: −12.5% (−39.2% to 14.3%), p = 0.92).
Discussion
Saquinavir is a specific HIV protease inhibitor, which is widely used in highly active antiretroviral therapy (HAART). Its therapeutic [33] effects are concentration-dependent and – although generally well tolerated – also adverse effects [34] appear to be linked to systemic concentrations; thus, an altered bioavailability may necessitate dose-adjustments. Due to their extensive phase I metabolism by CYP3A in the liver and gut, protease inhibitors are prone to drug interactions. Inhibitors of CYP3A such as ritonavir, nelfinavir, indinavir, ketoconazole, clarithromycin [24, 35], and grapefruit juice [36] increase the AUC of saquinavir up to 50-fold. There is an intriguing overlapping substrate specificity of CYP3A and PGP [37] and many of the above mentioned CYP3A-inhibitors such as ritonavir, ketoconazole, and grapefruit juice also inhibit PGP. Indeed, recent data of our group demonstrated an up to 5-fold increase of the AUC of saquinavir, when coadministered with the pharmaceutical aid cremophor EL [30]. The effect of cremophor EL on saquinavir pharmacokinetics occurred during the absorption phase, indicating an effect of the pharmaceutical aid on gastrointestinal dissolution or inhibition of intestinal first pass metabolism, e.g. by inhibition of mucosal PGP [38]. Thus, these data support, although they do not prove, a contribution of PGP to drug interactions with saquinavir.
In vitro data indicated a potent inhibition of both, CYP3A and PGP, particularly by gestodene and to a lesser extent by ethinylestradiol [14–17]. Hence, in the present study we investigated the pharmacokinetic interaction of an ethinylestradiol/gestodene combination OC with saquinavir. This may be of particular practical relevance when HIV infected women on saquinavir therapy start OC treatment. In all participants a clear decrease in LH, FSH, 17β-estradiol, and progesterone and an increase in SHBG concentrations during the 21-day period were observed, indicating reliable intake of OC. However, therapy with OC had no effect on any of the parameters of saquinavir pharmacokinetics, and there was also no effect on the relative ratio of saquinavir to its M2&M3-hydroxy metabolites, a biotransformation step in which CYP3A is largely involved [26]. Thus, no relevant inhibition of either CYP3A or PGP occurred in vivo.
In general saquinavir pharmacokinetics revealed a high interindividual variability, and particularly the AUC of one participant showed a large deviation from the others. Compared to the other participants, in this individual the relative ratio of saquinavir to its M2&M3-hydroxy metabolites was on average 11-fold higher, indicating decreased CYP3A activity. CYP3A activity is regulated by transcriptional control of its expression [39], and it is known that up to 400-fold variations in CYP3A expression and activity contribute to the interindividual differences in the bioavailability and systemic clearance of CYP3A substrates [40], including protease inhibitors [41], which could explain this finding.
As known from other studies with saquinavir administration in the fasted state [31, 36], maximal plasma concentrations (Cmax) of saquinavir were quite low also in the present study, although they were just within the IC50 values of HIV protease inhibition of 1–10 nmol l−1 (∼0.67–6.7 ng ml−1) reported in vitro [42]. Saquinavir was administered in the fasted state to lower the known inter- and intraindividual variability in pharmacokinetics. Furthermore, despite a more than 3-fold higher bioavailability of saquinavir from the soft gelatin capsule (SGC) [43], in the present study we deliberately decided to administer saquinavir in a hard gelatin (HGC) preparation, because we could not rule out confounding of the results by pre-existent PGP-inhibition by pharmaceutical aids of the SGC [31]. Saquinavir HGC is still used in boosted regimens in which its tolerability seems to be more favourable than that of the SGC [34, 44]. Unlike to the present study, in this setting saquinavir is usually combined with lopinavir or ritonavir which largely increase its bioavailability [45]. Thus, application of a saquinavir HGC without ritonavir is unlikely to occur in clinical practice which potentially limits the generalisability of our results. However, we assumed that this design would have the best chances for detecting an OC–saquinavir interaction, because we selected a progestin with a high interaction potential to which participants were exposed for a sufficiently long period of time (19 days). Concurrently we minimized the influence of other factors which possibly affect the bioavailability of saquinavir (food [32], pharmaceutical aids [31], ritonavir [45]). Under these conditions the achieved systemic saquinavir concentrations (∼15 nmol l−1) were quite similar to those expected with gestodene (∼13–41 nmol l−1) [13], thus qualifying this design for the evaluation of pharmacokinetic drug interactions.
In vitro both, competitive inhibition [46] and a potent mechanism-based inhibition of CYP3A- particularly by synthetic steroids with an acetylenic group at the C17 position [17]– have been described. Two studies [16, 47] reported an inhibition of the microsomal nifedipine oxidation by different steroids, such as gestodene, 3-keto-desogestrel, 17α-ethinylestradiol, desogesrel, levonorgestrel, 11-CH2-Δ15-norethisterone, norethisterone, and 11-CH2-norethisterone, in descending order of potency. Gestodene (Ki = 4.8 µm) exhibited a 3–8 fold greater inhibitory effect on CYP3A than other synthetic steroids, including ethinylestradiol (Ki∼30 µm) [48]. This order of inhibitor potency was confirmed in experiments assessing the effects of these steroids on the 2-hydroxlation of 17α-ethinylestradiol, but this reaction was generally less affected. In another study inhibition of CYP by progestins was confirmed, however, gestodene revealed similar inhibitor potency, compared to the other progestins [48]. Besides CYP inhibition, progestins, such as progesterone and medroxyprogesterone acetate are also inhibitors of PGP-mediated vinblastine efflux [19], with a potency comparable to that of verapamil. Concentrations of steroids in these in vitro studies were 30–100 µm which is approximately 1000–10000-fold higher than plasma concentrations reported in vivo [13]. However, one may expect higher concentrations at the site of absorption (intestinal epithelium) and the major site of metabolism (liver), the place where the interaction is most likely to occur.
In summary, these in vitro data give evidence for an inhibitory interaction of progestins with CYP3A mediated biotransformation and for an inhibition of active drug transport by PGP. Because gestodene was found to be one of the most potent CYP mechanism-based inactivators ever described [16], we decided to select a gestodene containing OC for our study protocol.
In contrast to the present study other clinical studies reported significant interactions of OC with substrates of CYP3A. For example, the t1/2 of i.v. prednisolone was reported to be 107% longer in women on OC [10], and a case report described doubled ciclosporin plasma-concentrations after introduction of an OC [9] which caused typical ADR to this substance. Moreover, there was a slight, but nonsignificant increase of the nifedipine AUC after intake of OC for 21 days, and the AUC of its hydroxymethyl metabolite decreased significantly by 25%[8]. Finally, the AUC of orally administered alprazolam was 35% higher [49], and the AUC of orally administered midazolam was 21% higher [7] in women taking OC, compared to women not taking OC. With respect to the in vitro data it is remarkable that a further study failed to show a significant interaction of OC and midazolam [11]. Interestingly, in the positive interaction study [7] an OC containing gestodene as the progestagenic component was used, whereas in the negative study norgestrel was administered which is roughly 5 times less potent an inhibitor of CYP3A in vitro [16].
Hence, the available in vivo data suggest that substrates with a significant interaction with OC are compounds with a lower affinity to CYP3A than saquinavir (Ki = 0.7 µm) [50]. The apparent Kivalues of nifedipine are 10–22 µm, of midazolam 40–63 µm, and of ciclosporin 1–37 µm[50]. Furthermore, the dose of saquinavir (600 mg) administered in the present study was rather higher than the dose of midazolam (7.5 mg) [8], nifedipine (10 mg) [7], and prednisolone (0.53 mg kg−1) [10]. Therefore, the concentration of saquinavir, relative to the concentrations of OC steroids at the intestinal epithelium, is higher than in the positive interaction studies, which may limit the likelihood of an interaction during the absorption phase, and thus explain the negative findings in the present study.
In conclusion this study revealed that therapeutic contraceptive doses do not increase saquinavir plasma concentrations. Because the progestin used in this study is the most potent CYP3A inhibitor of all evaluated progestins, OC combination therapy generally appears safe with respect to concentration-dependent ADR of saquinavir in women.
Acknowledgments
This work was supported in part by grant #01EC9902 from the German Ministry of Education and Research (BMBF) and by F. Hoffmann-La Roche Ltd, Basel, Switzerland. Saquinavir (Ro 31–8959/008) and 2H5-saquinavir (Ro 31–8959/048) standards were kindly provided by Roche Products Limited, Hertfordshire, UK. The authors thank Mrs Brigitte Tubach for her thorough assistance in planning and performing the study, and Mrs Stephanie Fuchs and Mrs Andrea Deschlmayr for their excellent technical assistance. Furthermore we thank Dr Kristina Unnebrink, Coordination Center for Clinical Trials (KKS) Heidelberg, for her statistical support.
Footnotes
With the assistance of Dr. M. M. Hoffmann (University of Freiburg, Germany) the whole CYP3A4 gene of participant #2 was sequenced to evaluate whether the substantial pharmacokinetic differences are caused by a polymorphism of this gene. PCR amplification of exons 1 to 13 was carried out using intronic primers flanking each of the CYP3A4 exons. Oligonucleotide sequences were created based on the published sequence (GenBank Accession No. AF209389), excluding sequences with homology to the genes of CYP3A5, CYP3A7, and CYP3A43. Purified PCR products (20 ng) were directly sequenced in two directions using the amplification primers and the dideoxy chain termination method (DYEnamic ET Kit, Amersham Biosciences, Freiburg, Germany) on a MegaBACE500 sequencer (Amersham Biosciences, Freiburg, Germany). No sequence variations were found thus excluding polymorphisms in the CYP3A4 gene as the underlying cause for the different saquinavir kinetics in participant #2.
Walter E. Haefeli, M.D.
Heidelberg, December 11, 2003
References
- 1.Kando JC, Yonkers KA, Cole JO. Gender as a risk factor for adverse events to medications. Drugs. 1995;50:1–6. doi: 10.2165/00003495-199550010-00001. [DOI] [PubMed] [Google Scholar]
- 2.Heinemann K, Moehner S, Lewis M, Assmann A, Garbe E, Heinemann LA. Trends of OC use 1980–99 in a German cohort of women. Zentralbl Gynakol. 2002;124:128–31. doi: 10.1055/s-2002-24241. [DOI] [PubMed] [Google Scholar]
- 3.Wit FW, Weverling GJ, Weel J, Jurriaans S, Lange JM. Incidence of and risk factors for severe hepatotoxicity associated with antiretroviral combination therapy. J Infect Dis. 2002;186:23–31. doi: 10.1086/341084. [DOI] [PubMed] [Google Scholar]
- 4.Lucas GM, Chaisson RE, Moore RD. Highly active antiretroviral therapy in a large urban clinic: risk factors for virologic failure and adverse drug reactions. Ann Intern Med. 1999;131:81–7. doi: 10.7326/0003-4819-131-2-199907200-00002. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen ML, Nagy GS, Hernandez I, Holtzberg J, Del Rio C, Lennox J. Use of HAART in women: similar response but greater toxicity. XIV. International AIDS Conference; Barcelona. 2002. Abstract WePeB5968. [Google Scholar]
- 6.Bonfanti P, Valsecchi L, Parazzini F, et al. Incidence of adverse reactions in HIV patients treated with protease inhibitors: a cohort study. J Acquir Immune Defic Syndr. 2000;23:236–45. doi: 10.1097/00126334-200003010-00004. [DOI] [PubMed] [Google Scholar]
- 7.Palovaara S, Kivisto KT, Tapanainen P, Manninen P, Jeuvonen PJ, Laine K. Effect of an oral contraceptive preparation containing ethinylestradiol and gestodene on CYP3A4 activity as measured by midazolam 1′-hydroxylation. Br J Clin Pharmacol. 2000;50:333–7. doi: 10.1046/j.1365-2125.2000.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balogh A, Gessinger S, Svarovsky U, et al. Can oral contraceptive steroids influence the elimination of nifedipine and its primary pyridine metabolite in humans? Eur J Clin Pharmacol. 1998;54:729–34. [Google Scholar]
- 9.Deray G, le Hoang P, Cacoub P, Assogba U, Grippon P, Baumelou A. Oral contraceptive interaction with cyclosporin. Lancet. 1987;1:158–9. doi: 10.1016/s0140-6736(87)91988-x. [DOI] [PubMed] [Google Scholar]
- 10.Gustavson LE, Legler UF, Benet LZ. Impairment of prednisolone disposition in women taking oral contraceptives of conjugated estrogens. J Clin Endocrinol Metab. 1986;62:234–7. doi: 10.1210/jcem-62-1-234. [DOI] [PubMed] [Google Scholar]
- 11.Belle DJ, Callaghan JT, Gorski JC, et al. The effects of an oral contraceptive containing ethinyloestradiol and norgestel on CYP3A activity. Br J Clin Pharmacol. 2002;53:67–74. doi: 10.1046/j.0306-5251.2001.01521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orme MLE, Back DJ, Breckenridge AM. Clinical pharmacokinetics of oral contraceptive steroids. Clin Pharmacokinet. 1983;8:95–136. doi: 10.2165/00003088-198308020-00001. [DOI] [PubMed] [Google Scholar]
- 13.Kuhl H. Comparative pharmacology of newer progestogens. Drugs. 1996;51:188–215. doi: 10.2165/00003495-199651020-00002. [DOI] [PubMed] [Google Scholar]
- 14.Testa B, Jenner P. Inhibitors of cytochrome P450s and their mechanism of action. Drug Metab Rev. 1981;12:1–117. doi: 10.3109/03602538109011082. [DOI] [PubMed] [Google Scholar]
- 15.Guengerich FP. Oxidation of 17a-ethynylestradiol by human liver cytochrome P-450. Mol Pharmacol. 1988;33:500–8. [PubMed] [Google Scholar]
- 16.Guengerich FP. Mechanism-based inactivation of human liver microsomal cytochrome P-450 IIIA4 by gestodene. Chem Res Toxicol. 1990;3:363–71. doi: 10.1021/tx00016a015. [DOI] [PubMed] [Google Scholar]
- 17.Ortiz de Montellano PR, Correia MA. Suicidal destruction of cytochrome P-450 during oxidative drug metabolism. Ann Rev Pharmacol Toxicol. 1983;23:481–503. doi: 10.1146/annurev.pa.23.040183.002405. [DOI] [PubMed] [Google Scholar]
- 18.Fromm MF, Kim RB, Stein CM, Wilkinson GR, Roden DM. Inhibition of P-glycoprotein-mediated drug transport: a unifying mechanism to explain the interaction between digoxin and quinidine. Circulation. 1999;99:552–7. doi: 10.1161/01.cir.99.4.552. [DOI] [PubMed] [Google Scholar]
- 19.Barnes KM, Dickstein B, Cutler GB, Fojo T, Bates SE. Steroid transport, accumulation, and antagonism of p-glycoprotein in multidrug-resistant cells. Biochemistry. 1996;35:4820–7. doi: 10.1021/bi952380k. [DOI] [PubMed] [Google Scholar]
- 20.Brundage RC, Acosta E, Haubrich R, Katzenstein D, Gulick R, Fletcher CV. Quantitation of sex differences and drug interactions. pharmacologic studies of saquinavir (SQV) in ACTG 2002, 359. 9th Conference on Retroviruses and Opportunistic Infections; (Abstract) 779–W. [Google Scholar]
- 21.CDER New and Generic Drug Approvals. 1998–2003: Product Label Invirase® (Saquinavir Mesylate) Capsules, Hoffman-La Roche, Inc.; [July 2003, 08]. Available at URL. http://www.fda.gov/cder/approval/index.htm. [Google Scholar]
- 22.Eagling VA, Wiltshire H, Whitcombe IW, Back DJ. CYP3A4-mediated hepatic metabolism of the HIV-1 protease inhibitor saquinavir in vitro. Xenobiotica. 2002;32:1–17. doi: 10.1080/00498250110085845. [DOI] [PubMed] [Google Scholar]
- 23.Kim RB, Fromm MF, Wandel C, et al. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 1998;101:289–94. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buss N, Snell P, Bock J, Hsu A, Jorga K. Saquinavir and ritonavir pharmacokinetics following combined ritonavir and saquinavir (soft gelatine capsules) administration. Br J Clin Pharmacol. 2001;52:255–64. doi: 10.1046/j.0306-5251.2001.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burhenne J, Riedel K-D, Martin-Facklam M, Mikus G, Haefeli WE. Highly sensitive determination of saquinavir in biological samples using liquid chromatography-tandem mass spectrometry. J Chromatogr B. 2003;784:233–42. doi: 10.1016/s1570-0232(02)00803-6. [DOI] [PubMed] [Google Scholar]
- 26.Fitzsimmons ME, Collins JM. Selective biotransformation of the human immunodeficiency virus protease inhibitor saquinavir by human small-intestinal cytochrome P4503A4. Potential contribution to high first-pass metabolism. Drug Metab Dispos. 1997;25:256–66. [PubMed] [Google Scholar]
- 27.Nauck M, Stein U, von Karger S, März W, Wieland H. Rapid detection of the C3435T polymorphism of multidrug resistance gene 1 using fluorogenic hybridization probes. Clin Chem. 2000;46:1995–7. [PubMed] [Google Scholar]
- 28.Cascorbi I, Gerloff T, Johne A, et al. Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther. 2001;69:169–74. doi: 10.1067/mcp.2001.114164. [DOI] [PubMed] [Google Scholar]
- 29.Hoffmeyer S, Burk O, von Richter O, et al. Functional polymorphisms of the human multidrug-resistance gene: Multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA. 2000;97:3473–3347. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim RB, Leake BF, Choo EF, et al. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther. 2001;70:189–99. doi: 10.1067/mcp.2001.117412. [DOI] [PubMed] [Google Scholar]
- 31.Martin-Facklam M, Burhenne J, Ding R, et al. Dose-dependent increase of saquinavir bioavailability by the pharmaceutic aid cremophor EL. Br J Clin Pharmacol. 2002;53:576–81. doi: 10.1046/j.1365-2125.2002.01595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noble S, Faulds D. Saquinavir. A review of its pharmacology and clinical potential in the management of HIV infection. Drugs. 1996;52:93–112. doi: 10.2165/00003495-199652010-00007. [DOI] [PubMed] [Google Scholar]
- 33.Kitchen VS, Skinner C, Ariyoshi K, et al. Safety and activity of saquinavir in HIV infection. Lancet. 1995;345:952–5. doi: 10.1016/s0140-6736(95)90699-1. [DOI] [PubMed] [Google Scholar]
- 34.Mitsuyasu RT, Skolnik PR, Cohen SR, et al. Activity of the soft gelatin formulation of saquinavir in combination therapy in antiretroviral-naive patients. AIDS. 1998;12:F103–F109. doi: 10.1097/00002030-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Malaty LI, Kuper JJ. Drug interactions of HIV protease inhibitors. Drug Saf. 1999;20:147–69. doi: 10.2165/00002018-199920020-00005. [DOI] [PubMed] [Google Scholar]
- 36.Kupferschmidt HH, Fattinger KE, Ha HR, Follath F, Krähenbühl S. Grapefruit juice enhances the bioavailability of the HIV protease inhibitor saquinavir in man. Br J Clin Pharmacol. 1998;45:355–9. doi: 10.1046/j.1365-2125.1998.t01-1-00687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yasuda K, Lan L-B, Sanglard D, Furuya K, Schuetz JD, Schuetz EG. Interaction of cytochrome P450 2002, 3A inhibitors with P–glycoprotein. J Pharmacol Exp Ther. 303:323–32. doi: 10.1124/jpet.102.037549. [DOI] [PubMed] [Google Scholar]
- 38.Woodcock DM, Jefferson S, Linsenmeyer ME, et al. Reversal of the multidrug resistance phenotype with Cremophor EL, a common vehicle for water-insoluble vitamins and drugs. Cancer Res. 1990;50:4199–203. [PubMed] [Google Scholar]
- 39.Goodwin B, Ridinbo MR, Kliewer SA. Regulation of CYP3A gene transcription by the pregnane X receptor. Annu Rev Pharmacol Tocicol. 2002;42:1–23. doi: 10.1146/annurev.pharmtox.42.111901.111051. [DOI] [PubMed] [Google Scholar]
- 40.Backman JT, Kivisto KT, Olkkola KT, Neuvonen PJ. The area under the plasma concentration-time curve for oral midazolam is 400-fold larger during treatment with itraconazole than with rifampicin. Eur J Clin Pharmacol. 1998;54:53–8. doi: 10.1007/s002280050420. [DOI] [PubMed] [Google Scholar]
- 41.Wacher VJ, Silverman JA, Zhang Y, Benet LZ. Role of P- glycoprotein and cytochrome P450 1998, 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci. 87:1322–8. doi: 10.1021/js980082d. [DOI] [PubMed] [Google Scholar]
- 42.Vella S, Floridia M. Saquinavir. Clinical pharmacology and efficacy. Clin Pharmacokinet. 1998;34:189–201. doi: 10.2165/00003088-199834030-00002. [DOI] [PubMed] [Google Scholar]
- 43.Figgitt DP, Plosker GL. Saquinavir soft-gel capsule. An updated review of its use in the management of HIV infection. Drugs 2000. 2002;60:481–516. doi: 10.2165/00003495-200060020-00016. [DOI] [PubMed] [Google Scholar]
- 44.Kurowski M, Sternfeld T, Hill A, Sawyer AW, Moecklinghoff C. Comparative pharmacokinetics and short-term safety of Fortovase®/ritonavir and Invirase®/ritonavir 1000mg/100mg BID. 9th Conference on Retroviruses and Opportunistic Infections; Abstract 432-W. [Google Scholar]
- 45.Buss N, Snell P, Bock J, Hsu A, Jorga K. Saquinavir and ritonavir pharmacokinetics following combined ritonavir and saquinavir (soft gelatin capsules) administration. Br J Clin Pharmacol. 2001;52:255–64. doi: 10.1046/j.0306-5251.2001.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purba HS, Maggs JL, Orme MLE, Back DJ, Park BK. The metabolism of 17a-ethinyloestradiol by human liver microsomes: formation of catechol and chemically reactive metabolites. Br J Clin Pharmac. 1987;23:447–53. doi: 10.1111/j.1365-2125.1987.tb03074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Böcker R, Kleingeist B, Eichhorn M, Lepper H. In vitro interaction of contraceptive steroids with human liver cytochrome P-450 enzymes. Adv Contracept. 1991;7(Suppl 3):140–8. [Google Scholar]
- 48.Back DJ, Houlgrave R, Tjia JF, Ward S, Orme MLE. Effect of the progestogens, gestodene, 3-ketodesogestrel, levonorgestrel, norethisterone and norgestimate on the oxidation of ethinyloestradiol and other substrates by human liver microsomes. J Steroid Biochem Molec Biol. 1991;38:219–25. doi: 10.1016/0960-0760(91)90129-s. [DOI] [PubMed] [Google Scholar]
- 49.Stoehr GP, Kroboth PD, Juhl RP, Wender DB, Phillips JP, Smith RB. Effect of oral contraceptives on triazolam, temazepam, alprazolam, and lorazepam kinetics. Clin Pharmacol Ther. 1984;36:683–90. doi: 10.1038/clpt.1984.240. [DOI] [PubMed] [Google Scholar]
- 50.Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]