

Abstract
Aims
To study the population pharmacokinetics of piperaquine after co-administration with dihydroartemisinin in uncomplicated malaria.
Methods
The disposition of piperaquine was studied in 85 Cambodian patients with uncomplicated falciparum or vivax malaria treated with the piperaquine-dihydroartemisinin coformulation Artekin®. All patients were given Artekin® orally at 0, 6, 24 and 32 h with a total piperaquine dose of 32–35 mg base kg−1. Adults were given tablets while children received either tablets or a dispersible granule formulation. Patients underwent either intensive (17–19 samples) or sparse (2–5 samples) blood sampling schedules over 35 days and clinical/parasitological follow-up over >28 days. Piperaquine in plasma was quantified by high performance liquid chromatography.
Results
All patients achieved fever clearance within 24 h and parasite clearance within 72 h. The 28-day cure rate was 97% in adults and 98% in children. A covariate-free two-compartment population model with first-order absorption and elimination gave the most robust representation of the plasma concentration-time data in both adults and children. In adults (n = 38), the median (interquartile range) derived pharmacokinetic descriptors CL/F, Vss/F and t1/2,z were 0.9 l h−1 kg−1 (0.79–1.02 l h−1 kg−1), 574 l kg−1(371–711 l kg−1) and 23 days (19–28 days), respectively. In children (n = 47), corresponding values were 1.8 l h−1 kg−1 (1.29–2.3 l h−1 kg−1), 614 l kg−1 (332–1205 l kg−1) and 14 days (10–18 days), respectively.
Conclusions
Piperaquine is a highly lipid-soluble drug with a large Vss/F, long t1/2,z and a clearance that is markedly higher in children than in adults.
Keywords: combination therapy, dihydroartemisinin, malaria, piperaquine, population pharmacokinetics
Introduction
Piperaquine (1,3-bis[1-(7-chloro-4′-quinolyl)-4′-piperazinyl] phosphate (Figure 1); Rhone Poulenc 13228) is a bisquinoline antimalarial drug synthesized in the 1960s by both the Shanghai Pharmaceutical Industry Research Institute in China and Rhone Poulenc in France [1]. Initial studies showed piperaquine to be active against Plasmodium spp., including chloroquine-resistant P. falciparum. Therapeutic and prophylactic efficacy was documented in clinical trials in the 1970s. Piperaquine subsequently replaced chloroquine as first-line treatment for falciparum and vivax malaria in many areas of China during the 1970s and 1980s [2], a strategy that led to the emergence of piperaquine-resistant strains of P. falciparum[3–7].
Figure 1.
Piperaquine phosphate – 1,3-bis[1-(7-chloro-4′-quinolyl)-4′-piperazinyl] phosphate
Although some preclinical animal toxicity data have been published [8, 9], conventional Phase I-IV evaluation in humans has not been carried out. The efficacy studies reported in the Chinese medical literature were often not conducted and/or reported with the rigour of a modern-day clinical trial, and some aspects of drug development were not addressed at all [1, 2, 4, 10–18]. Specifically, there have been no studies of the pharmacokinetics of piperaquine in volunteers, or in the target population of patients with malaria.
At the time piperaquine usage was at its peak in southern China, the artemisinin derivatives were being developed. These potent antimalarial drugs are now used first-line in many tropical countries but short-course therapy is associated with recrudescence. Because of this and concerns that artemisinin-resistant strains of P. falciparum might emerge if these compounds were used as sole therapy, combination treatment regimens incorporating a second longer-acting drug such as mefloquine have been recommended [19]. Because of its relatively low cost and good tolerability, piperaquine has enjoyed a recent resurgence in clinical use outside China as a coformulation with dihydroartemisinin in the product Artekin® (Holleykin Pharmaceuticals Co. Ltd, Guangzhou, China) [20, 21]. Therefore, it is imperative that data on the disposition of piperaquine in humans are obtained. Such data will allow critical evaluation of the current dosing regimen, which has been developed solely from empirical use in a large number of Chinese patients. A recent clinical evaluation of Artekin® in 106 Cambodians (76 children and 30 adults) with uncomplicated falciparum malaria showed excellent efficacy, with 100% and 92.3% 28-day cure rates in children and adults, respectively [22]. Given the limitations of conducting research in remote areas of the tropics [23], we have used a population approach in a pharmacokinetic study involving a subset of these patients and other Cambodian subjects with uncomplicated malaria who were also treated with Artekin®.
Methods
Study sites and overview
Patients were recruited from two sites. The first was Anlong Veng district in Oddor Meanchey Province in north-western Cambodia, close to the border with Thailand. The second was Snoul district, Kratie Province in eastern Cambodia on the border with Viet Nam. All the Anlong Veng subjects in the present study were recruited in 2001 and, together with patients studied at the Snoul site in the same year, participated in a larger study investigating the clinical efficacy of Artekin® that has been published recently [22]. Recruitment for the present pharmacokinetic study continued in Snoul during 2002.
Eligibility criteria and recruitment procedures
Patients were recruited by both active and passive case detection. Active recruitment was by arranged visits to villages where screening (thick and thin blood films) of those with symptoms of malaria was undertaken. Passive recruitment was through patient referrals from village volunteer workers, and via symptomatic patients seeking treatment at the local health centre. Giemsa-stained thick film blood smears were examined by an experienced microscopist. Slides were only designated as negative after examination of over 100 fields at 1000× magnification. Speciation on positive slides was determined by thin film microscopy.
Symptomatic patients with positive blood-films (parasite density >1000 µl−1 whole blood) in the age-groups 2–10 years and >16 years were considered for admission to the trial. In children, a lower age limit of 2 years was chosen based on ethical considerations related to blood sampling, while the upper age limit was set at 10 years to define an age range comparable with that used in most other paediatric studies of antimalarial therapeutic efficacy conducted in endemic areas. In children, only those with P. falciparum infections were eligible for entry. In adults, both P. vivax and P. falciparum infected patients could be enrolled. Parasitaemia was quantified by counting the number of asexual parasites per 1000 leucocytes and using the whole blood leucocyte count to calculate parasite density. Exclusion criteria included pregnancy, significant comorbidity, recent antimalarial treatment (specifically, quinine or an artemisinin drug within the previous 7 days, chloroquine within 14 days, or sulfadoxine-pyrimethamine or mefloquine within 28 days), or clinical manifestations of severe or complicated malaria. The study protocol was approved by the National Ethics Committee for Health Research, Ministry of Health, Cambodia and by the Human Rights Committee of the University of Western Australia. Written informed consent was obtained from all adult patients or from parents or guardians of the children.
Clinical procedures
Patients were informed that they were required to remain hospitalized for at least 4 days, and that there would be follow-up visits at predetermined times up to 35 days. At enrolment, a full physical examination was performed, with recording of vital signs (heart rate, respiratory rate, systolic and diastolic blood pressures, axillary temperature, conscious state), parasite count, white cell count, plasma glucose concentration, haematocrit and an electrocardiogram. Vital signs were assessed 6-hourly during hospitalization and blood smears were taken daily until two consecutive slides were negative. Clinical progress, reports of adverse effects, and other measurements and data were recorded daily on standard forms.
Dose regimens
Artekin® tablets (320 mg piperaquine phosphate and 40 mg dihydroartemisinin) or granules (120 mg piperaquine phosphate and 15 mg dihydroartemisinin) were administered orally according to the manufacturer's recommendations at time 0, 6, 24 and 32 h in all patients. In the absence of published piperaquine pharmacokinetic data, the timing of doses was based on assumptions that piperaquine would behave similarly to related 4-aminoquinolines, on practical considerations concerning convenience of administration, and on optimizing the therapeutic potential of the dihydroartemisinin component of the combination. A divided dosing regimen over 32 h was thought to offer the best prospect of combining efficacy (by achieving a high cumulative dose of piperaquine) with a low risk of toxicity (by limiting high peak concentrations as may occur with a single dose) in the same way as chloroquine dosing regimens have been developed [24]. All children recruited in 2001 were treated with sachets of Artekin® granules (mean weight adjusted dose = 33 mg base kg−1, range 28–41 mg base kg−1). In 2002, children were treated according to age; 2–3 years (4 × 1 sachets), 4–6 years (4 × 1.5 sachets), 7–10 years (4 × 1 tablets). All adults (>16 years) were treated with tablets (4 × 2 tablets). Doses were not administered with food. Treatments were supervised and patients were observed closely for at least 1 h after the first dose. Any patients vomiting during this period were retreated with artesunate and mefloquine, as recommended under the Cambodian National Treatment Guidelines, and excluded from the study.
Blood sampling
Due to socio-cultural concerns about the safety and effects of frequent blood sampling especially in young children, we restricted intensive sampling to five adults recruited from Anlong Veng in 2001. Venous blood (3 ml) was drawn into plain tubes at 0, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24, 32, 40, 48, 72, 96 h, and at 7, 14, 28 days. Children (n = 17) recruited from Snoul in 2001 were randomly assigned for venous blood collection at two of these time points. During 2002, adults (n = 33) and children (n = 30) from Snoul were randomized to have heparinized blood (4 ml) collected at 4 or 5 time points selected from the following times: 0, 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 20, 24, 32, 72, 96 h, and 7, 14, 21, 28 and 35 days. In all cases, blood samples were centrifuged within 30 min of collection and separated serum or plasma stored at −20 °C until transported on dry ice to Australia for piperaquine assay.
Discharge criteria and follow-up
Patients were discharged from hospital after 3 days provided they were asymptomatic and aparasitaemic. All patients were followed up on days 7, 14, 21 and 28 and were advised to return if they became symptomatic at any other time. Each follow-up visit included clinical assessment, with recording of axillary temperature, and documentation of any side-effects. Thick and thin blood films were also taken.
Outcome measures
The primary efficacy outcome measures were fever and parasite clearance times. Fever clearance time (FCT) was defined as the time from commencement of treatment to that at which the patient's temperature first fell to and remained below 37.3 °C. Parasite clearance time (PCT) was taken as the time to the first of two negative blood smears, with smears taken daily in some patients and every 6 h in others.
Based on World Health Organization (WHO) criteria [25], a sensitive (S) response was recorded if no asexual stages of P. falciparum were found on day 7 and parasites had not reappeared by day 28. If asexual parasites disappeared by day 7 but reappeared before day 28, the isolate was considered to have probable RI resistance. If the asexual parasitaemia had dropped by at least 75% at 48 h but not cleared, and if parasites were still present on day 7, the parasites were considered resistant at the RII level. If the asexual parasitaemia had dropped by less than 75% at 48 h and the patient remained slide-positive on day 7, the parasites were considered RIII resistant. The WHO has recently revised its standardized methodology and definitions for assessing in vivo clinical and parasitological response [26], and patients were also categorized as adequate clinical and parasitological response (ACPR), early treatment failure (ETF), late treatment failure-parasitological (LPF) or late treatment failure-clinical (LCF).
Laboratory methods
Piperaquine concentrations in serum and plasma were assayed using a validated high-performance liquid chromatographic assay with ultraviolet detection at 340 nm [27]. Briefly, following solvent extraction, piperaquine was resolved on an XTerra™ RP18 column (RP18 3.5 mm, 4.6 mm i.d. × 100 mm; Waters Associates, Sydney, Australia) with mobile phase containing 7% acetonitrile in water (with 0.025% trifluoroacetic acid, 0.1% NaCl, and 0.008% triethylamine v/w/v) pumped at 1.2 ml min-1. Quality control samples were included with each assay batch. The assay was linear over the range of 5–1000 µg l−1 with intraday and interday coefficients of variation <10% and <21%, respectively. The limit of quantification was 5 µg l−1.
The piperaquine content of Artekin® tablets and granules was also assayed using the above chromatographic conditions. Each tablet/granule (n = 5; Lot20011201, expiry date 2003.12.14; Lot20020401, expiry date 2004.01.04, respectively) was first dissolved in 100 ml of 0.01 m KCl (buffered to pH 2.4 with 0.01 m HCl), then further diluted in the same buffer. Aliquots (50 µl) of this final dilution were injected directly onto the column. The mean peak area (n = 3) was then calibrated against a four-point standard curve (range 200–2000 ng) with standards diluted in the same buffer.
Statistical analysis
Data have been summarized as mean ± SD or median (interquartile range) as appropriate. The Mann–Whitney Rank Sum Test or Student's t-test (SigmaStat Ver2.0, SPSS Inc, Chicago, IL, USA) was used to compare the distribution of the admission characteristics and pharmacodynamic parameters between groups of patients. A two-tailed level of significance of P < 0.05 was used for all tests.
Pharmacokinetic modelling
Pharmacokinetic modelling of the piperaquine plasma concentration-time data was performed using Kinetica™ software package (Version 4.1, InnaPhase Corporation, Philadelphia, PA, USA).
A covariate-free model was used initially to analyse concentration-time data obtained from the groups of adults and children. The method ‘PopFitExtravascularCompMultiDose’ implemented in Kinetica™ was used, with one or two compartments and first order absorption and elimination from the central compartment. The model was parameterized in terms of an absorption rate constant (ka), lag time, volume of the central compartment relative to bioavailability (Vc/F), elimination rate constant (kel), and transfer rate constants k12, k21 in the two-compartment model. Clearance relative to bioavailability (CL/F) and volume of distribution at steady state relative to bioavailability (Vss/F) were calculated from the Bayesian estimates of Vc/F, kel, k12 and k21 for each patient [28], and median half-lives of absorption, distribution and elimination (t1/2,abs, t1/2,λ1 and t1/2,z, respectively) were obtained from the Kinetica™ output.
Kinetica™ applies the two-step EM algorithm to conduct nonlinear mixed-effect model analysis. Step E estimates individual parameter using the Maximum A-Posteriory Probability Bayesian fitting procedure from a prior knowledge of the mean and dispersion of the parameter in the population and individual available information, including drug concentration measurements and/or covariables. Step M computes the population maximum likelihood parameter estimate given the current estimates of individual parameters.
Interindividual variability of the population parameters was based on normal distributions for Vc/F, lag, kel, k12 and k21 and lognormal distribution for ka. Homoscedastic and heteroscedastic error variance models were also explored in the fitting process. These are defined according to Sigma and weighting function, where the homoscedastic model assumes that the error variance is unknown but constant for all data, and heteroscedastic model specifies that the error variance is proportional to Yobs.
Investigation of covariates was undertaken using the forward stepwise regression analysis option implemented in Kinetica™. Covariates were centred by subtracting the median covariate value and the centred value was used in the regression analysis. The significance of the addition of a second covariate is evaluated if it produces the largest increase in r2 in the presence of the previous covariate. The F-test is used and the P value evaluated to check whether the F change is significant. The candidate covariate found to be significant in this process is then incorporated into the model which is re-run with the defined covariable relationship.
The goodness of fit of the model was assessed with the following additional factors: (a) residuals for individual patients, (b) linearity of the plot of predicted vs observed values and (c) numerical statistics including the Akaike Information Criterion (AIC) and the objective function (OF). Increasing complexity in nested models (including covariates) was compared with the base model using the likelihood ratio test. The Logarithm of Likelihood function (LL) is χ2 distributed and a change (LLlarger-LLsmaller) of more than 3.84 units is required to confirm a statistically significant improvement in the structural model at P < 0.05 [29, 30].
Uncertainty in the parameter estimates was assessed as the coefficient of variation (CV) and by graphical evaluation of the probability distribution of parameter estimates. The final model also was evaluated graphically by simulation of a concentration-time dataset using the mean derived pharmacokinetic parameters and average dose administered.
Results
Admission characteristics
One adult with vivax malaria refused to have further blood taken after the first sample. This patient completed the full-course of Artekin® and cure was confirmed during follow-up. One child was lost to follow-up after discharge. The other 83 subjects completed all study procedures. Details of these subjects are summarized by age group in Table 1.
Table 1.
Admission characteristics of patients grouped according to age1
| Adults | Children | |
|---|---|---|
| Number of patients | 38 | 47 |
| Age (years) | 30 ± 13 | 7 ± 2 |
| Sex (% males) | 53 | 55 |
| Body weight (kg) | 47 ± 6 | 16 ± 4 |
| Pulse (beats min–1) | 89 ± 9 | 116 ± 18 |
| Systolic blood pressure (mmHg) | 109 ± 7 | 95 ± 17 |
| Diastolic blood pressure (mmHg) | 68 ± 6 | 60 ± 9 |
| Axillary temperature (°C) | 38.2 ± 0.6 | 38.4 ± 0.6 |
| Haematocrit (%) | 36 ± 5 | 30 ± 5 |
| Plasma glucose (mg dl–1) | 100 ± 17 | 98 ± 18 |
| Parasitaemia (µl–1) | 7700 (1,000–110 000) | 12 800 (3,100–33 342) |
| White blood cell count (l–1 | 5600 (4,850–6688) | 6 200 (5,700–7500) |
Results as mean ± SD. or median and interquartile range.
Over the two recruitment periods, a total of 216 samples were collected from 38 adults. The first five patients recruited from Anlong Veng in 2001 were subjected to an intensive sampling schedule (median = 18; total 88 samples) while the other 33 adults recruited in 2002 underwent sparse sampling (median = 4; total 128 samples). Twenty-eight of the patients were blood slide positive for falciparum malaria and 10 for vivax malaria. There were no significant differences in the mean doses of piperaquine (P. falciparum 31.9 ± 4.1 mg base kg−1 (mean ± SD), P. vivax 32.1 ± 4.2 mg base kg−1 (mean ± SD); t = 0.08, P = 0.938) or initial parasitaemias (P. falciparum, median = 9300; P. vivax, median = 4600; U = 138, P = 0.059) between the two groups.
A total of 47 children with uncomplicated falciparum malaria aged between 2 and 10 years were also recruited. In 2001, one or two samples were collected from each of the 17 children (4–10 years old) for a total of 30 samples. A further 30 children were recruited in 2002 and those aged between 2 and 6 years were given sachets of granules (n = 10) and those aged 7 to 10 years were given tablets (n = 20). Up to four samples (median = 4) were collected from each child with a total of 38 samples for the granule formulation and 77 samples for the tablet formulation.
Clinical course
These data are summarized in Table 2. All patients achieved parasite clearance within 72 h of commencing treatment. In both adults and children, at least 96% of patients had cleared their parasite load by 48 h and 100% by 72 h. Median FCTs ranged from 9 to 12 h and were similar in both adults and children. Overall 28-day cure rates were >97% in each of the three subject groups.
Table 2.
28-day cure results and pharmacodynamic parameters grouped according to age1 and parasite species
| Formulation | Adults (P. falciparum) | Adults (P. vivax) | Children (P. falciparum) |
|---|---|---|---|
| Number of patients | 28 | 10 | 47 |
| Total dose of dihydroartemisinin (mg kg–1) | 6.9 ± 0.9 | 6.9 ± 0.9 | 7.6 ± 1.6 |
| Total dose of piperaquine base (mg kg–1) | 31.9 ± 4.1 | 32.1 ± 4.2 | 34.9 ± 7.2 |
| Fever clearance times (h) | 12 (6–18) | 9 (6–12) | 12 (12–24) |
| Parasite clearance time (h) | NA2 | 12 (12–18) | NA2 |
| % patients with PCT ≤ 24 h | 71 | 100 | 85 |
| % patients PCT ≤ 48 h | 96 | 100 | 96 |
| % patients PCT ≤ 72 h | 100 | 100 | 100 |
| % of S/RI/RII/RIII | 96 (S) | 100 (S) | 98 (S) |
| % of ACPR/ETF/LCF/LPF3 | 96 (ACPR) | 100 (ACPR) | 98 (ACPR) |
Results as mean ± SD, or median and interquartile range
NA = not available as some patients had smears taken only every 24 h, and others 6 hourly
Adequate clinical parasitological response (ACPR), Early treatment failure (ETF), Late treatment failure-clinical (LCF), Late treatment failure- parasitological (LPF).
Safety and tolerability
Full adverse effect profiles over 28-days were obtained from 80 of the 85 patients recruited. Overall, 46% of patients reported one or more of the symptoms shown in Table 3 between day 1 and 28. Eighteen percent of patients reported symptoms during the 32-h dosing period that were not reported on day 0. Headache, dizziness and nausea were prevalent on the first day of treatment.
Table 3.
Adverse events reported by both adult and paediatric patients (percent with symptom) on admission (day 0) and during the whole of the 28-day follow-up period
| Patients with symptom (%) | ||
|---|---|---|
| Symptom | Day 0 (n = 85) | Post-treatment (n = 83)1 |
| Headache | 55% | 36% |
| Dizziness | 24% | 14% |
| Abdominal pain | 12% | 11% |
| Anorexia | 15% | 1% |
| Nausea | 14% | 4% |
| Tinnitus | 8% | 6% |
| Deafness | 5% | 2% |
| Palpitations | 0% | 1% |
| Insomnia | 2% | 1% |
Two patients lost to follow-up.
Piperaquine content of Artekin® tablets and granules
Piperaquine phosphate content was 339 ± 9 mg for tablets (n = 5) and 112 ± 4 mg for granules (n = 5). This corresponds to 5.9% ± 2.8% above and 6.7% ± 3.3% below the stated piperaquine phosphate content of tablets (320 mg) and granules (120 mg), respectively.
Pharmacokinetic model development
The data were considered in two groups, according to age. Model development included numerical comparisons of AIC and OF, together with visual inspection of correlation plots of observed vs predicted piperaquine concentrations, plots of weighted residuals vs ycalc for individual patients, and of probability distributions of parameter estimates. A mix of sparse and rich data from all 38 adults was best represented by a two-compartment model with a lag time and first-order absorption and elimination. Both homoscedastic and heteroscedastic error models were analysed but the latter did not improve the overall fit. Initially, we attempted to analyse the data from children given tablets or granule formulations separately. However, since there were only 20 and 27 subjects, respectively, we chose to combine these two datasets to improve the validity of analysis. In children, a two-compartment structural model with an heteroscedastic (1/Yobs) error model was most appropriate, but inclusion of an absorption lag time could not be justified. Using the stepwise inclusion approach, the variables screened for possible correlation with Vc/F, ka, kel, k12 and k21 were age and body weight, both centred using the population median. At P = 0.05, no correlations were found between either of these covariables and the pharmacokinetic parameters in both adults and children.
The AIC and LL values during pharmacokinetic model development are shown in Table 4. In the final analysis of both datasets, a two-compartment model (extravascular administration) with first-order absorption and elimination was used. Since inclusion of covariates did not improve the models, a covariate-free model was used to derive the final pharmacokinetic parameters for both adults and children (Table 5). Plots of weighted residuals against predicted piperaquine concentrations for datasets from adults and children are shown in Figure 2. While the residuals were generally evenly distributed about zero, there were some deviations particularly at high concentrations in the adult dataset and at low concentrations in the child dataset. The mean absorption rate constant was very small in both age groups, resulting in long absorption half-lives of 8.3 h and 9.2 h for adults and children, respectively. Mean CL/F was approximately two-times larger in children than adults and was also associated with a shorter elimination half-life. Figure 3 shows piperaquine concentration-time plots, and simulations based on mean population parameters for adults (A) and children (B). Using mean data shown in the simulated plasma concentration-time curves (Figure 3), the λz-phase AUC was approximately 79% and 71% of the total AUC for adults and children, respectively, indicating that the λz half-life is a key determinant of the disposition of piperaquine.
Table 4.
Development of the structural pharmacokinetic model for piperaquine; changes in Akaike Information Criteria (AIC) and Logarithm of Likelihood function (LL) with change in model complexity
| Adults | Children | |||
|---|---|---|---|---|
| Model | AIC | LL | AIC | LL |
| One compartment | 5.76 | −1154 | 43.76 | −5641 |
| Two compartments, no lag time1 | 5.62 | −1126 | 4.68 | −599 |
| Two compartments with lag time1 | 5.59 | −1117 | 4.82 | −616 |
Homoscedastic error model for adults and heteroscedastic (1/Yobs) error model for children.
Table 5.
Population estimates of structural and derived pharmacokinetic parameters for piperaquine in 85 patients with uncomplicated falciparum and vivax malaria, grouped according to age1
| Adults | Children | |
|---|---|---|
| Vc/F (l kg–1) | 14.5 (46%) | 19.7 (51%) |
| ka (h–1) | 0.083 (108%) | 0.075 (44%)2 |
| lag (h) | 0.49 (179%) | – |
| kel (h–1) | 0.058 (78%) | 0.090 (52%) |
| k12 (h–1) | 0.19 (57%) | 0.33 (75%) |
| k21 (h–1) | 0.0056 (131%) | 0.0099 (85%) |
| t1/2,λ1 (h) | 2.6 (2.4–3.0) | 1.7 (1.3–1.9) |
| t1/2,z (h) | 543 (457–663) | 324 (245–438) |
| t1/2,abs (h) | 9.09 (7.67–15.4) | 9.26 (9.25–9.27) |
| CL/F (l h–1 kg–1) | 0.90 (0.79–1.02) | 1.85 (1.29–2.30) |
| Vdss/F (l kg–1) | 574 (371–711) | 614 (332–1205) |
Results as population mean and coefficient of variation (CV%) or, because of the presence of outlying values, median and (interquartile range)
Parameter estimated with a log normal distribution.
Figure 2.

Plots of weighted residuals against predicted piperaquine concentrations for datasets from (A) adults and (B) children. Residuals were approximately evenly distributed above and below zero (solid line). Homoscedastic and heteroscedastic weighted error models were used for the adult and children datasets, respectively
Figure 3.
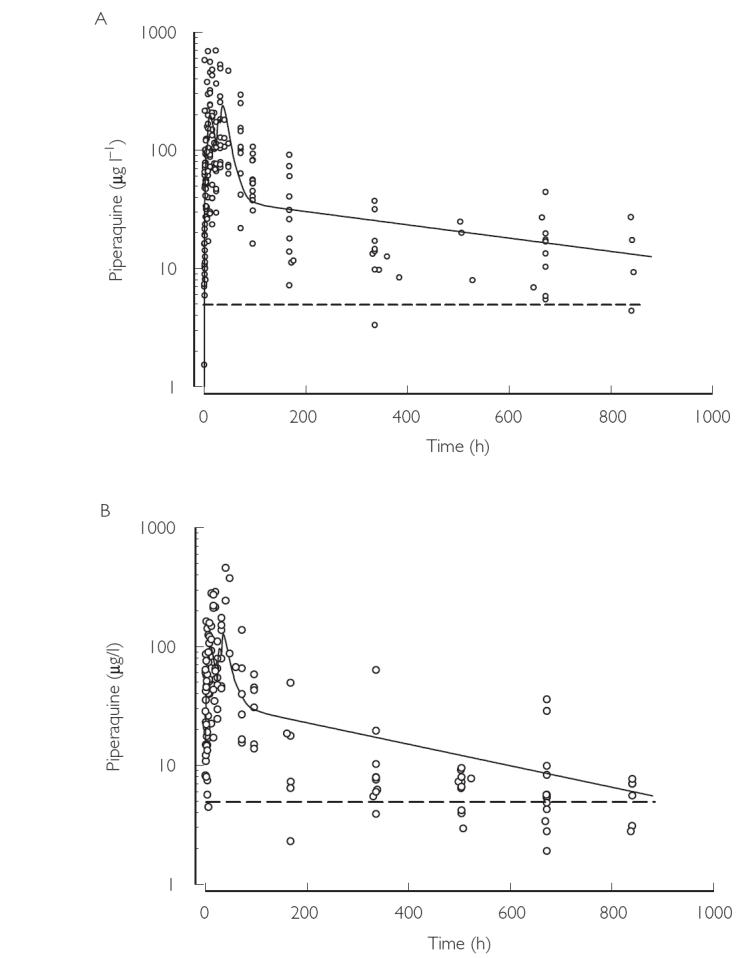
Observed plasma piperaquine concentration-time data for adults (A) and children (B). The solid lines show simulated concentration-time data (solid line) derived from the mean pharmacokinetic parameters (Table 5) and the average doses (Table 2). The dotted line indicates the assay limit of quantification (LOQ; 5 µg l−1). Note that concentrations below the LOQ do not contribute to the regression analysis. The biphasic appearance of the simulated curves within the first 3 days results from the close proximity of doses 1 and 2, and 3 and 4 (see Methods)
Discussion
This is the first report describing the pharmacokinetics of piperaquine in adults and children with malaria. Because the content of piperaquine in the tablet and granule formulations was within a range that is acceptable for similar pharmaceuticals (92.5%−107.5% of stated content) [31], the nominal content of piperaquine base was used in deriving the pharmacokinetic parameters. In addition, we chose to model the pharmacokinetics of the adults and children separately as they represented discrete population subgroups. The population pharmacokinetic parameters derived from a mix of rich and sparse data from 38 adults treated with Artekin® included a long median t1/2,z (23 days), a large Vdss/F (574 l kg−1) and slow CL/F (0.90 l h−1 kg−1). Similar pharmacokinetic characteristics are seen for other very lipid-soluble drugs. For example, chloroquine has a large Vd (115 ± 61 l kg−1) [32], mefloquine a long t1/2,z (20 ± 4 days) [33] and clofazamine a long t1/2,z (70 days) [34]. The long mean t1/2,z and the large molecular weight (535.51) suggest that piperaquine may undergo enterohepatic recycling. However, at this stage we have no data to show that piperaquine is a substrate for biliary transport.
We first attempted to analyse separately the data from children who were administered either tablets or granules. However, since there were 30 or fewer subjects, respectively, in these two groups, and minimal differences in pharmacodynamic descriptors between the formulations (data not shown, e.g. ka was similar), we chose to combine all the paediatric data in a single pharmacokinetic analysis. This approach produced a robust description of the data but has the disadvantage that it ignores possible differences in bioavailability that might arise from the two formulations. While the median value for Vdss/F in the children (614 l kg−1) was broadly similar to that in the adults, the shorter half-life (14 days) in children resulted in a much more rapid CL/F (1.85 l h−1 kg). The latter most probably reflects higher rates of hepatic metabolism and/or biliary excretion in children.
A short absorption lag time was an appropriate parameter in the analysis of the adult dataset. In addition, the absorption half-lives for piperaquine were long in both adults and children. Slow absorption is likely to be a function of the high lipophilicity of piperaquine (log10 P = 6.157) [35]. Similar absorption profiles are seen with drugs such as halofantrine and tafenoquine which have a low bioavailability when given in the fasting state and improved bioavailability when administered with food [36, 37]. Further work is necessary to define the absorption profile for piperaquine in its various formulations. Nevertheless, the Artekin® formulations used in our study achieved plasma concentrations that were generally above reported in vitro IC50 values for piperaquine (4.4 µg l−1 and 8.6 µg l−1, respectively, in sensitive and in resistant P. falciparum strains) [38].
Artekin® proved a highly efficacious combination in the treatment of uncomplicated falciparum and vivax malaria. Our 28-day test data are consistent with the recently published larger Cambodian efficacy study which included 22 of the present patients out of a total of 106 adults and children [22]. It should be noted that the standard 28-day test may over-estimate efficacy when evaluating a long half-life drug such as piperaquine. However the previous Cambodian efficacy study was conducted in a similar population and under similar conditions, employed follow up to 56 days at which time cure rates were 98%, and found no confirmed recrudescences between 28 and 56 days in any of 72 patients completing 56 days of follow-up [22]. Median FCTs in adults and children ranged from 8 to 12 h. Parasite clearance occurred in >96% of patients by 48 h and in 100% by 72 h.
Although primarily concerned with pharmacokinetic analysis, some assessment of the tolerability and safety of piperaquine was made in this study. All adverse events reported were mild or moderate in severity and none precipitated withdrawal from the study or necessitated additional interventions for symptom relief. Among the 46% of patients who reported one or more symptoms, headache, dizziness, abdominal pain, anorexia and nausea were most commonly reported. However, this finding was confounded by the large number of patients (83%) reporting the same symptoms when first presenting with untreated malaria. An accurate assessment of tolerability and safety cannot be made reliably without appropriate comparison arms such as artesunate-mefloquine combination therapy. A detailed analysis of the laboratory investigations performed during follow-up will be reported elsewhere.
It has been recommended that partner drugs for artemisinin-based combination therapy have a t1/2,z of >4 days (at least two parasite life-cycles) [19]. Piperaquine has a median t1/2,z that is between 3 and 6 times this figure. Combination regimens such as artesunate-mefloquine may ensure mutual protection against the development of drug resistance in low transmission areas [39]. However, it is possible that the persistence of a drug such as piperaquine at subtherapeutic plasma concentration may select resistant parasite strains following re-infection in hyperendemic areas.
In summary, piperaquine and dihydroartemisinin form an effective combination in the treatment of uncomplicated falciparum and vivax malaria in adults and children. Using population pharmacokinetic modelling of rich and sparse data from 38 adults and 47 children, disposition of piperaquine was characterized by a large Vdss/F, a long t1/2,z and a CL/F that was markedly higher in children than in adults. Of the parameter estimates generated in the present study, those relating to absorption were not defined optimally, particularly in the children. We would recommend that further studies target the absorption profile of piperaquine using a more intensive initial sampling protocol. In addition, collection of samples beyond 28 days would assist in improved characterization of the terminal elimination phase, especially in adults. Although we do not anticipate a significant interaction between the two components of Artekin®, studies of possible drug interactions between piperaquine and dihydroartemisinin and/or other likely comedications are needed. Studies in healthy volunteers could also be useful to assess bioavailability of new formulations, and to examine whether malaria itself alters piperaquine pharmacokinetic properties.
Acknowledgments
We thank the National Centre for Parasitology, Entomology and Malaria Control, Phnom Penh and the European Commission Cambodia Malaria Control Project, Phnom Penh for invaluable support. We also thank staff at the Anlong Veng and Snoul health centres in Cambodia for their assistance with the conduct of the field studies. T-Y. Hung was the recipient of a Raine Medical Research Foundation Bachelor of Medical Science Scholarship. Valuable financial support also came from the University of Western Australia and the Ramaciotti Research Foundation.
References
- 1.Chen L, Qu FY, Zhou YC. Field observations on the antimalarial piperaquine. Chin Med J (Engl) 1982;95:281–6. [PubMed] [Google Scholar]
- 2.Chen L. Recent studies on antimalarial efficacy of piperaquine and hydroxypiperaquine. Chin Med J (Engl) 1991;104:161–3. [PubMed] [Google Scholar]
- 3.Fan B, Zhao W, Ma X, et al. In vitro sensitivity of Plasmodium falciparum to chloroquine, piperaquine, pyronaridine and artesunate in Yuxi prefecture of Yunnan province. Chin J Parasitol Parasit Dis. 1998;16:460–2. [PubMed] [Google Scholar]
- 4.Lan CX, Lin X, Huang ZS, Chen YS, Guo RN. In vivo sensitivity of Plasmodium falciparum to piperaquine phosphate assayed in Linshui and Baisha counties, Hainan Province. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 1989;7:163–5. [PubMed] [Google Scholar]
- 5.Yang H, Liu D, Huang K, et al. Assay of sensitivity of Plasmodium falciparum to chloroquine, amodiaquine, piperaquine, mefloquine and quinine in Yunnan province. Chin J Parasitol Parasit Dis. 1999;17:43–5. [PubMed] [Google Scholar]
- 6.Yang HL, Yang PF, Liu DQ, et al. Sensitivity in vitro of Plasmodium falciparum to chloroquine, pyronaridine, artesunate and piperaquine in south Yunnan. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 1992;10:198–200. [PubMed] [Google Scholar]
- 7.Zhang KY, Zhou JX, Wu Z, Huang QL. Susceptibility of Plasmodium falciparum to chloroquine, piperaquine, amodiaquine, mefloquine and quinine with in vitro microtechnique in Hainan Island. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 1987;5:165–9. [PubMed] [Google Scholar]
- 8.Sheng N, Jiang W, Tang HL. Pre-clinical toxical study of new antimalarial agents. II. Piperaquine phosphate and its compound ‘Preventive no. 3′. Acad J Second Mil Med College. 1981;1:40–6. [Google Scholar]
- 9.Zhao HJ, Xia YY, Zheng Z. Pre-clinical toxicity study of new antimalarial agents. IV. Liver ultrastructure changes affected by antimalarial agent, compound tablet of piperaquine phosphate and sulfadoxine. Acad J Second Mil Med College. 1981;1:47–8. [Google Scholar]
- 10.Qu FY. The antimalarial effects of piperaquine phosphate and sulphadoxine composite as tested in Hainan Island (author's transl) Zhonghua Yi Xue Za Zhi. 1981;61:388–91. [PubMed] [Google Scholar]
- 11.Qu FY, Li CJ, Chen ZD. Prophylactic efficacy on malaria of piperaquine phosphate combined with sulfadoxine. Chin Med J (Engl) 1981;61:388–91. [Google Scholar]
- 12.Chen L, Qu FY, Zhou YC. Field observation of prophylactic effect of the new antimalarial piperaquine in Hainan province. Med J PLA. 1979;4:104–8. [Google Scholar]
- 13.Guo XB. Randomised comparison on the treatment of falciparum malaria with dihydroartemisinin and piperaquine. Zhonghua Yi Xue Za Zhi. 1993;73:602–4. [PubMed] [Google Scholar]
- 14.Guo XB, Fu LC. Comparative study of artemisinin suppositories and piperaquine phosphate in the treatment of falciparum malaria. Zhong Xi Yi Jie He Za Zhi. 1989;9:475–3. [PubMed] [Google Scholar]
- 15.Huang JZ, Lan XH, Xu WZ. Sensitivity of Plasmodium falciparum to piperaquine in Baoting County, Hainan Island. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 1985;3:276–7. [PubMed] [Google Scholar]
- 16.Wang G. Curing three patients of chloroquine-resistant falciparum malaria with resistant level III by piperaquine. Chin J Inf Dis. 1985:3–78. [Google Scholar]
- 17.Chen L, Dai ZR, Qian YL, et al. Observation on the efficacy of combined use of some new antimalarials for the treatment of falciparum malaria in Hainan Province. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 1989;7:81–4. [PubMed] [Google Scholar]
- 18.Anonymous. A brief report of piperaquine in the treatment of 280 vivax malaria patients. Geneva: World Health Organisation; 1973. [Google Scholar]
- 19.Nosten F, Brasseur P. Combination therapy for malaria: the way forward? Drugs. 2002;62:1315–29. doi: 10.2165/00003495-200262090-00003. [DOI] [PubMed] [Google Scholar]
- 20.Anonymous. Meeting on Antimalarial Drug Development. Manila, Philippines: World Health Organisation Regional Office for the Western Pacific; 2002. http://www.wpro.who.int/malaria/docs/shanghai.pdf, Report series number RS//GE/33 (CHN) [Google Scholar]
- 21.World Health Organisation. Western Pacific Regional Office Country Profile, Vietnam. http://www.wpro.who.int/malaria/t1f2vietnam.asp.
- 22.Denis MB, Davis TME, Hewitt S, et al. Efficacy and safety of dihydroartemisinin-piperaquine (Artekin) in Cambodian children and adults with uncomplicated falciparum malaria. Clin Infect Dis. 2002;35:1469–76. doi: 10.1086/344647. [DOI] [PubMed] [Google Scholar]
- 23.Simpson JA, Aarons L, White NJ. How can we do pharmacokinetic studies in the tropics? Trans R Soc Trop Med Hyg. 2001;95:347–51. doi: 10.1016/s0035-9203(01)90178-6. [DOI] [PubMed] [Google Scholar]
- 24.Krishna S, White NJ. Pharmacokinetics of quinine, chloroquine and amodiaquine. Clinical implications. Clin Pharmacokinet. 1996;30:263–99. doi: 10.2165/00003088-199630040-00002. [DOI] [PubMed] [Google Scholar]
- 25.World Health Organization. Chemotherapy of malaria and resistance to antimalarials. Geneva: World Health Organization; 1972. Technical Report Series no. 529. [PubMed] [Google Scholar]
- 26.World Health Organization. Monitoring Antimalarial Drug Resistance. Geneva: Switzerland; 2001. WHO/CDS/CSR/EPH/2002.17. http://www.who.int/csr/resources/publications/drugresist/whocdscsreph200217.pdf. Report of a WHO consultation. 3–5 December. [Google Scholar]
- 27.Hung TY, Davis TME, Ilett KF. Measurement of piperaquine in plasma by high-performance liquid chromatography with ultraviolet absorbance detection. J Chromatogr B Biomed Sci Appl. 2003;791:93–101. doi: 10.1016/s1570-0232(03)00209-5. [DOI] [PubMed] [Google Scholar]
- 28.Heinzel G. Compartmental analysis formulas. In: Heinzel G, Woloszczak R, Thomann P, editors. Topfit, Version 2.0 Pharmacokinetic, pharmacodynamic data analysis for the PC. Stuttgart: Gustav Fischer; 1993. pp. 4–158. Chapter 4. [Google Scholar]
- 29.Kinetica™. Version 4.1. User Manual R1. Philadelphia PA: Innaphase Corporation; 2002. [Google Scholar]
- 30.Davidian M, Giltinan DM. Nonlinear Models for Repeated Measurement Data. 1. London: Chapman & Hall; 1995. Inference based on linearization; pp. 151–90. [Google Scholar]
- 31.British Pharmacopoeia Commission. British Pharmacopoeia. London: The Stationery Office; 2000. p. 1807. [Google Scholar]
- 32.White NJ. Clinical pharmacokinetics of antimalarial drugs. Clin Pharmacokinet. 1985;10:187–215. doi: 10.2165/00003088-198510030-00001. [DOI] [PubMed] [Google Scholar]
- 33.Benet L, Oie S, Schwartz JB. Design and Optimization of Dosage regimens; Pharmacokinetic Data. In: Hardman JG, Limbird LE, editors. Goodman and Gilman's Pharmacological Basis of Therapeutics. 9. New York: McGraw-Hill; 1996. pp. 1707–1792. [Google Scholar]
- 34.Holdiness MR. Clinical pharmacokinetics of clofazimine: A review. Clin Pharmacokinet. 1989;16:74–85. doi: 10.2165/00003088-198916020-00002. [DOI] [PubMed] [Google Scholar]
- 35.Chemical Abstracts Service. Columbus, Ohio: American Chemical Society; 2002. Piperaquine. SciFinder Scholar, 1999. [Google Scholar]
- 36.Edstein MD, Kocisko DA, Brewer TG, Walsh DS, Eamsila C, Charles BG. Population pharmacokinetics of the new antimalarial agent tafenoquine in Thai soldiers. Br J Clin Pharmacol. 2001;52:663–70. doi: 10.1046/j.1365-2125.2001.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watkins WM, Winstanley PA, Mberu EK, et al. Halofantrine pharmacokinetics in Kenyan children with non-severe and severe malaria. Br J Clin Pharmacol. 1995;39:283–7. doi: 10.1111/j.1365-2125.1995.tb04450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vennerstrom JL, Ellis WY, Ager AL, Jr, Andersen SL, Gerena L, Milhous WK. Bisquinolines, 1. N,N-bis (7-chloroquinolin-4-yl) alkanediamines with potential against chloroquine-resistant malaria. J Med Chem. 1992;35:2129–34. doi: 10.1021/jm00089a025. [DOI] [PubMed] [Google Scholar]
- 39.Nosten F, van Vugt M, Price R, et al. Effects of artesunate-mefloquine combination on incidence of Plasmodium falciparum malaria and mefloquine resistance in western Thailand: a prospective study. Lancet. 2000;356:297–302. doi: 10.1016/s0140-6736(00)02505-8. [DOI] [PubMed] [Google Scholar]