

Abstract
Peripheral vascular disease (PVD) is generally accepted to result in the failure of skeletal muscle blood flow to increase adequately at the onset of muscular work. There are currently no routine pharmacological interventions towards the treatment of PVD, however, recent Phase III trials in the USA have demonstrated the clinical potential of the phosphodiesterase III inhibitor Cilostazol for pain-free and maximal walking distances in patients with intermittent claudication. PVD is characterized by a marked reliance on oxygen-independent routes of ATP regeneration (phosphocreatine hydrolysis and glycolysis) in skeletal muscle during contraction and the rapid onset of muscular pain and fatigue. The accumulation of metabolic by-products of oxygen-independent ATP production (hydrogen and lactate ions and inorganic phosphate) has long been associated with an inhibition in contractile function in both healthy volunteers and PVD patients. Therefore, any strategy that could reduce the reliance upon ATP re-synthesis from oxygen-independent routes, and increase the contribution of oxygen-dependent (mitochondrial) ATP re-synthesis, particularly at the onset of exercise, might be expected to improve functional capacity and be of considerable therapeutic value. Historically, the increased contribution of oxygen-independent ATP re-synthesis to total ATP generation at the onset of exercise has been attributed to a lag in muscle blood flow limiting oxygen delivery during this period. However, recent evidence suggests that limited inertia is present at the level of oxygen delivery, whilst considerable inertia exists at the level of mitochondrial enzyme activation and substrate supply. In support of this latter hypothesis, we have reported on a number of occasions that activation of the pyruvate dehydrogenase complex, using pharmacological interventions, can markedly reduce the dependence on ATP re-synthesis from oxygen-independent routes at the onset of muscle contraction. This review will focus on these findings and will highlight the pyruvate dehydrogenase complex as a novel therapeutic target towards the treatment of peripheral vascular disease, or any other disease state where premature muscular fatigue is prevalent due to metabolite accumulation.
Keywords: contractile function, pyruvate dehydrogenase complex, substrate utilization
Introduction
The increasing elderly population of the western world, coupled to the greater incidence of cigarette smoking and poor dietary habits, has led to an increase in the clinical manifestation of peripheral vascular disease (PVD). Affecting approximately 12% of the general population [1], with an increased frequency in the diabetic subpopulation [2], PVD is characterized as a failure of skeletal muscle blood flow to increase adequately at the onset of muscular work, such as walking [3, 4]. The absence of a ‘normal’ hyperaemic response of the cardiovascular system to exercise is associated with increased reliance upon ATP re-synthesis from oxygen-independent routes (namely ATP and phosphocreatine (PCr) hydrolysis and glycolysis to lactate) to meet the energy demands of contraction [5, 6], and the concomitant development of muscular pain and fatigue. The disease is progressive, impinging severely on the range of mobility of the patient and can ultimately jeopardize the integrity of the limb (critical leg ischaemia). Indeed, patients at this stage of the disease have a reported quality of life index similar to critical–terminal phase cancer patients [7]. The socio-economic impact of PVD upon the health service is immense, estimated in 1994 to be approximately £215 million in the UK, with approximately 60% of these costs arising from bypass and amputation surgery alone [8].
Clearly, any strategy capable of improving functional capacity and halting disease progression could be of considerable therapeutic and economic value. Current evidence, however, does not support the hypothesis that an improvement in peripheral blood flow results in an improvement in functional capacity [9–11], a view supported by the lack of correlation between lower limb blood flow and walking distances in PVD patients (for review see [12]). There are currently no routine pharmacological interventions towards the treatment of PVD. However, the phosphodiesterase III inhibitor Cilostazol has recently demonstrated clinical potential by increasing both pain-free and maximal walking distance of sufferers in Phase III trials in the USA, although the mechanism underpinning this functional improvement is yet to be determined [13]. At present, the single best treatment strategy for patients at all levels of disease progression is exercise training [1, 12, 14], where improvements in muscular function can occur independent of any measurable increase in limb blood flow [15]. Although the benefits of exercise training upon walking distances in PVD sufferers are well founded [9, 16, 17], many patients find adherence to a training regime difficult to maintain due to exercise-induced limb pain (claudication) and other disease-related complications, i.e. heart disease, diabetes, obesity, respiratory problems [1]. The physiological adaptations that occur in skeletal muscle of PVD patients as a result of an exercise rehabilitation programme have not been fully elucidated. This is due, at least in part, to studies to date not taking into account the habitual activity patterns of patients prior to entry into any research study, i.e. not taking into account any metabolic and vascular adaptations that might occur as a result of habitual muscle contraction [18]. In addition, it is not known if these exercise-induced improvements differ from the normal training-like adaptations that occur in healthy skeletal muscle with use [18].
The heavy reliance upon oxygen-independent ATP production at the onset of muscular contraction is a symptom not solely associated with PVD muscle, but reflects an exaggeration of what occurs in healthy, normally perfused skeletal muscle during the transition from rest to muscular work. Indeed, at the onset of skeletal muscle contraction there is a marked increase in energy demand which must be matched by a rapid increase in ATP re-synthesis to enable the exercise workload to continue for longer than a few seconds. The re-adjustment of oxygen-dependent (mitochondrial) ATP re-synthesis to meet this demand is not immediate and follows an approximately exponential time course (for review see [19]). During this period, the shortfall in ATP supply is met by ATP re-synthesis from oxygen-independent routes. By way of example, Bangsbo et al.[20] observed in healthy human skeletal muscle that PCr hydrolysis and glycolysis contributed approximately 80% of the total ATP generated during the initial 30 s of high-intensity exercise. This value declined to approximately 45% during the subsequent 60–90 s, and to approximately 30% after 120 s of exercise; this decrease appeared to be accomplished by a parallel increase in oxygen-dependent ATP re-synthesis [20]. Although ATP production from oxygen-independent routes enables rapid rates of ATP turnover to be achieved, it has only a finite capacity and also results in the accumulation of metabolites that are deleterious to muscle function (hydrogen ions, lactate ions and inorganic phosphate; [21]). Indeed, without the progressive increase in mitochondrial ATP production at the onset of contraction, and thereby the reduction in oxygen-independent energy delivery, the onset of muscular fatigue would be markedly accelerated, as typified in sufferers from PVD.
Classically, the lag in oxygen-dependent ATP re-synthesis at the onset of contraction, and the resulting activation of oxygen-independent ATP regeneration, has been attributed to a finite rate of increase, or inertia, in skeletal muscle blood flow and thereby oxygen delivery to contracting muscle fibres [22–24 Richardson et al. 1995]. Indeed, the temporal changes in muscle oxygen utilization at the onset of exercise closely follow the increase in total limb blood flow during this period; hence the general acceptance of the phrase ‘oxygen deficit’ within the literature [25, 26]. Over the past decade, however, there has been a growing body of evidence indicating that neither muscle blood flow (bulk oxygen delivery) nor capillary diffusion limit oxygen utilization, and thereby oxygen-dependent ATP re-synthesis, at the onset of exercise [27–29]. For example, Grassi et al.[28], using a blood-perfused canine gastrocnemius muscle model, demonstrated that when the delay in blood flow (and thereby oxygen delivery) during the rest-to-steady state exercise transition was eliminated, there was no further acceleration in the rate of increase in muscle oxygen consumption over that observed under control conditions. Using the same model, the authors went on to present strong evidence to suggest that muscle oxygen diffusion also does not limit muscle oxygen consumption at the onset of exercise [29]. They concluded that the limitations to the rate of increase in oxygen consumption at the onset of exercise are probably attributable to heterogeneous microvascular oxygen delivery and/or an ‘intrinsic inertia’ within mitochondrial energy production of unspecified origin.
The pyruvate dehydrogenase complex: a site of metabolic inertia?
Work within our laboratory over the past decade has investigated the pyruvate dehydrogenase complex as a potential site of limitation to mitochondrial energy production at the onset of muscular contraction. The pyruvate dehydrogenase complex (PDC) is a multienzyme complex, located on the mitochondrial inner membrane, which regulates carbohydrate entry into the tricarboxylic acid (TCA) cycle. The PDC catalyses the physiologically irreversible reaction that commits carbohydrates to their oxidative fate inside the mitochondria through the conversion of the glycolytic product pyruvate into mitochondrial acetyl-CoA (involving NAD+ and free-coenzyme A; Figure 1). Regulation of the rate of formation of acetyl-CoA by the PDC (i.e. flux through the enzyme complex) is achieved by two strategies. The first of these is by altering the fraction of PDC that exists in its active form. This is achieved by covalent modification of PDC, either from its inactive (phosphorylated) to active (dephosphorylated) state by loosely associated pyruvate dehydrogenase phosphatases, or vice versa by a number of intrinsic and tissue-specific pyruvate dehydrogenase kinases (Figure 1) [30, 33]. These effectors of PDC activation are sensitive to pulsatile changes in calcium availability, cellular energetics and substrate/product accumulation [31, 32]. Second, the rate of pyruvate oxidation by PDC is regulated by end-product inhibition of flux through the enzyme complex by NADH and acetyl-CoA (Figure 1) [33]. The acetyl groups produced by PDC can be utilized by the TCA cycle or, alternatively, can be stockpiled in the form of acetylcarnitine, presumably when acetyl-CoA re-synthesis exceeds its rate of utilization by citrate synthase [34]. Buffering acetyl groups in this way has been proposed as a mechanism for the maintenance of a viable pool of free-coenzyme A, which is essential for sustained TCA cycle flux. This highlights an important metabolic role of carnitine, in addition to its function in mitochondrial long-chain acyl group translocation [34].
Figure 1.
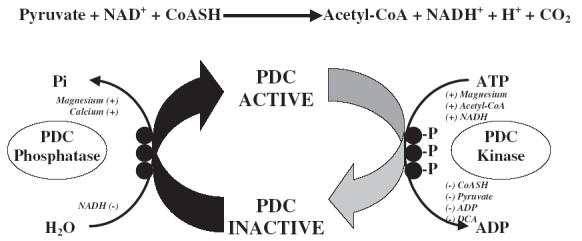
The pyruvate dehydrogenase complex reaction and covalent regulation of activation status by the intrinsic pyruvate dehydrogenase phosphatase and kinase system. CoASH, Free-coenzyme A; Pi, inorganic phosphate; (–), an inhibitor of the enzyme it is beside; (+), an activator of the enzyme it is beside; P, phosphorylation of the three specific serine residues upon the haloenzyme core of the pyruvate dehydrogenase complex; DCA, the systemic pyruvate dehydrogenase kinase inhibitor sodium dichloroacetate
In 1996, we were the first to demonstrate that pharmacological activation of the PDC, using the systemic PDC kinase (PDK) inhibitor dichloroacetate (Figure 1) [35, 36], markedly increased acetylcarnitine availability in resting skeletal muscle and appreciably reduced PCr hydrolysis and lactate accumulation during subsequent intense contraction, and under conditions where muscle blood flow and oxygen delivery were fixed at close to resting levels [37]. Subsequent to this, we demonstrated in both canine and human skeletal muscle that the rapid hydrolysis of PCr and accumulation of lactate that occur at the onset of exercise were at least partly due to an inherent lag in the activation of oxygen-dependent (mitochondrial) ATP regeneration [38, 39]. In particular, we were able to show that activation of the PDC at rest, using dichloroacetate, was accompanied by an approximately 30% reduction in ATP re-synthesis from oxygen-independent routes after 1 min of contraction, even though muscle force production was identical to the saline (control) group. Following 6 min of contraction, the contribution from oxygen-independent routes to ATP re-synthesis had fallen to approximately 50% of that observed in the control group, while tension development was greater [38]. It also appeared from these studies that some of the acetyl groups that were stockpiled at rest after PDC activation were utilized during contraction, indicating that the mitochondria were able to utilize more acetyl groups at the onset of exercise when provision was increased by dichloroacetate administration [37, 38]. From these investigations, it was concluded that the activation, and thereby flux, through PDC must limit acetyl-CoA availability and consequently mitochondrial ATP re-synthesis at the onset of exercise. Moreover, that the activation of PDC and ‘priming’ of mitochondria with acetyl groups prior to exercise, by administering dichloroacetate, could significantly increase the overall contribution of oxidative pathways to total ATP production at the onset of exercise. Another important finding from this series of studies was that the decline in muscle tension development during contraction (i.e. fatigue) was substantially reduced following dichloroacetate administration, probably due to PCr hydrolysis and lactate accumulation being reduced at the immediate onset of contraction [37, 38]. Furthermore, this effect was sustainable throughout contraction, at least until the exercise workload was increased to a near maximal intensity [39].
The ‘acetyl group deficit’
If inertia in the rate of increase in oxygen-dependent ATP regeneration at the onset of exercise does indeed reside at the level of PDC, which our previous work certainly seems to indicate, then it stands to reason that a period of time must exist at the onset of exercise when acetyl-CoA supply via PDC is insufficient to match the demands of the TCA cycle, and the concentration of acetyl-CoA should therefore decline. However, studies to date have shown that acetyl groups appear to accumulate throughout moderate-to-intense muscular contraction [34, 40–42], with this accumulation being greater in skeletal muscle contracting under ischaemic conditions [41]. From these findings, it has been inferred that acetyl-CoA production is probably in excess of TCA cycle demands throughout contraction, which contrasts with our hypothesis that metabolic inertia resides at the level of PDC. Closer scrutiny of the relevant literature reveals, however, that studies to date have failed to investigate the metabolic events occurring within the initial seconds of contraction, or indeed, at any time point during contraction prior to significant PDC activation. We therefore decided to test our contention that early in the rest-to-work transition period there is a lag in mitochondrial ATP re-synthesis, which is in part due to an inadequate supply of acetyl-CoA via PDC [43]. Using a canine hind-limb perfusion model [41], five muscle biopsy samples were obtained from the gracilis muscle during the first minute (rest, 10, 20, 40 and 60 s) of ischaemic muscle contraction, which we envisaged would give us sufficient resolution to elucidate the temporal relationship between PDC activation, acetyl group accumulation, and PCr hydrolysis and lactate accumulation at the onset of contraction [43]. The results demonstrated that a lag in acetyl group provision (in the form of acetyl-CoA and acetylcarnitine) occurred during the initial 20 s of contraction, which resulted from, and was mirrored by, a lag in PDC activation (Figure 2). This unequivocally demonstrated the existence of a period of metabolic inertia (the so called ‘acetyl group deficit’) in skeletal muscle at the onset of contraction, and was directly in line with our earlier observations that the supply of acetyl groups to the TCA cycle was limited during the rest-to-work transition [43].
Figure 2.
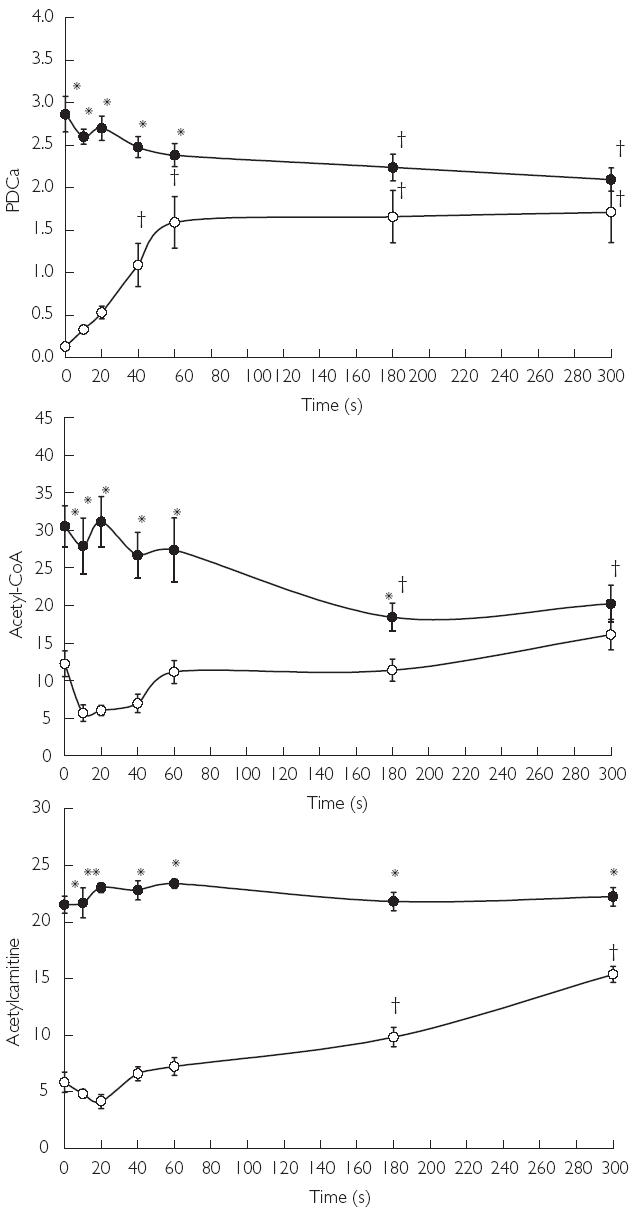
Active form of the pyruvate dehydrogenase complex (PDCa) and acetyl-CoA and acetylcarnitine concentrations at rest and during 5 min of ischaemic contraction following pretreatment with saline (CON (○)) or sodium dichloroacetate (DCA (•)). Units are as follows: PDCa, mmol of acetyl-CoA min−1 kg−1 dry muscle (at 37 °C); acetyl-CoA, µmol kg−1 dry muscle; acetylcarnitine, mmol kg−1 dry muscle. Results are expressed as means ± SEM. Significant differences: *P < 0.05 compared with corresponding CON value; ‡P < 0.05 compared with value at rest within the same treatment group
As dichloroacetate activates the PDC and near maximally acetylates the free-coenzyme A and carnitine pools at rest (Figure 2), it was not possible to determine in any of our previous studies whether the reduction in oxygen-independent ATP re-synthesis at the onset of contraction following dichloroacetate (Figure 3) was attributable to acetyl-CoA delivery via the PDC being increased at the immediate onset of contraction and/or was due to the readily available pool of acetyl groups being sequestered by the TCA cycle. With this question in mind, we have recently investigated whether pharmacologically increasing the availability of acetyl-CoA and acetylcarnitine, independent of PDC activation, could overcome the acetyl group deficit at the onset of exercise [44]. We were able to show that administration of sodium acetate increased the availability of acetyl-CoA and acetylcarnitine in resting skeletal muscle, but did not increase PDC activation. Furthermore, during the first minute of ischaemic muscle contraction, when the PDC was largely inactive, treatment with sodium acetate increased the contribution of oxygen-dependent ATP regeneration towards the energy demands of the muscle when compared with the saline-treated (control) group [44]. However, following this first minute, when near maximal activation of PDC had been achieved in both control and acetate groups, it appeared that PDC-derived acetyl-CoA, rather than stockpiled acetyl groups per se, was the principal route of substrate delivery to the TCA cycle. Collectively these investigations have established the activation of the pyruvate dehydrogenase complex as a rate-limiting step in the rate of rise in oxygen-dependent ATP production in skeletal muscle at the onset of exercise, which in turn will dictate the magnitude of oxygen-independent ATP delivery and thereby the rate of fatigue development.
Figure 3.
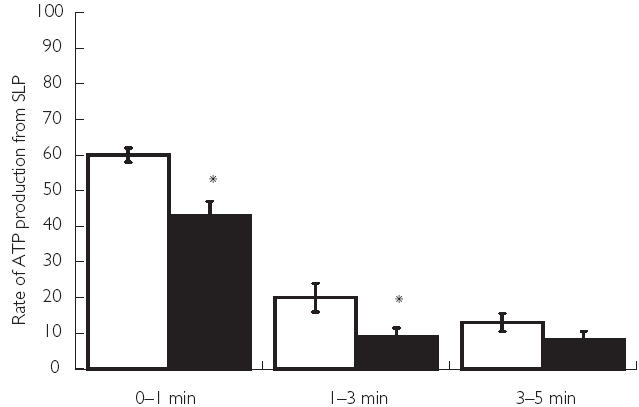
Rates of ATP re-synthesis from phosphocreatine hydrolysis and glycolysis between rest and 1 min, 1 min and 3 min and 3 and 5 min of ischaemic contraction following pretreatment with saline (CON (□)) or sodium dichloroacetate (DCA (▪)). Results are expressed as means ± SEM, with units of mmol of ATP equivalents min−1 kg−1 dry muscle. Significant differences: *P < 0.05 compared with corresponding CON value
Conclusion and future perspectives
In conclusion, in the present review we have provided convincing evidence to support the contention that PDC activation and acetyl-CoA availability limit oxygen-dependent (mitochondrial) ATP re-synthesis at the onset of skeletal muscle contraction (the so called ‘acetyl group deficit’). Increasing the provision of acetyl groups, through the pharmacological activation of the PDC, can overcome this period of metabolic inertia, accelerate the rate of mitochondrial ATP re-synthesis and concomitantly improve the maintenance of contractile function throughout the rest-to-work transition under both ischaemic and non-ischaemic conditions. We here highlight the tissue-specific activation of the pyruvate dehydrogenase complex as a potentially new and novel therapeutic target towards the treatment of peripheral vascular disease or any other disease state where premature muscular fatigue is prevalent due to metabolite accumulation, particularly as a relatively muscle-specific PDK isoform is now known to exist [30].
The systemic PDK inhibitor, and thereby PDC activator, dichloroacetate has been used clinically for many years, most notably in the treatment of congenital lactic acidosis (for review see [45]). However, the chronic administration of dichloroacetate is not known to be without adverse side-effects. Indeed, Cicmanec et al.[46] failed to establish a ‘no-adverse-effect level’ of dichloroacetate during a 90-day toxicity study in beagle dogs. Not surprisingly, safety concerns have curtailed the use of dichloroacetate as a therapeutic agent in clinical settings, with dichloroacetate regarded today more as a probe with which to investigate intermediary metabolism. Hopefully, structurally distinct [47], less toxic and tissue-specific PDK inhibitors [30] will become available in the near future that can subsequently be evaluated for their clinical potential.
It is of note that a period of low-intensity exercise (commonly referred to as ‘warm-up’ exercise) has been shown to result in the acceleration of oxygen uptake kinetics and produce a range of positive biochemical and ergogenic effects during a second, more strenuous, bout of exercise [48–51]. These effects of warm-up exercise have classically been attributed to an exercise-induced elevation of muscle temperature and/or the augmentation of local muscle blood flow which remain elevated at the onset of the second bout of exercise. However, in light of our investigations outlined above, we have recently demonstrated that low-intensity exercise can result in muscular acetyl group accumulation, and that the stockpiling of these acetyl groups is associated with the acceleration of oxygen-consumption kinetics and mitochondrial ATP re-synthesis during a subsequent bout of more intense exercise [52]. It is likely that such exercise-induced metabolic responses will have important clinical implications in the treatment and amelioration of symptoms associated with peripheral vascular disease; however, this remains to be established.
Acknowledgments
We acknowledge AstraZeneca Pharmaceuticals, the British Heart Foundation and the Medical Research Council for their support of this work. We also acknowledge the contributions of our coworkers, as detailed in the original publications.
References
- 1.Regensteiner JG, Hiatt WR. Exercise rehabilitation for patients with peripheral arterial disease. Exercise Sport Sci Rev. 1995;23:1–24. [PubMed] [Google Scholar]
- 2.Brand FN, Abbott RD, Kannel WB. Diabetes intermittent claudication, and the risk of cardiovascular events. The Framingham Study. Diabetes. 1989;38:504–509. doi: 10.2337/diab.38.4.504. [DOI] [PubMed] [Google Scholar]
- 3.Lewis T. Pain in muscular ischemia (its relation to anginal pain) Arch Intern Med. 1932;49:713–729. [Google Scholar]
- 4.Holm J, Björntorp P, Scherstén T. Metabolic activity in human skeletal muscle; effect of peripheral arterial insufficiency. Eur J Clin Invest. 1972;2:321–325. doi: 10.1111/j.1365-2362.1972.tb00657.x. [DOI] [PubMed] [Google Scholar]
- 5.Jansson E, Johansson J, Sylven C, Kaijser L. Calf muscle adaption in intermittent claudication. Side-differences in muscle metabolic characteristics in patients with unilateral arterial disease. Clin Physiol. 1988;8:17–29. doi: 10.1111/j.1475-097x.1988.tb00258.x. [DOI] [PubMed] [Google Scholar]
- 6.Lundgren F, Bennegard K, Elander A, Lundholm K, Schersten T, Bylund-Fellenius A. Substrate exchange in human limb muscle during exercise at reduced blood flow. Am J Physiol. 1988;255:H1156–H1164. doi: 10.1152/ajpheart.1988.255.5.H1156. [DOI] [PubMed] [Google Scholar]
- 7.Albers M, Fratezi AC, Deluccia N. Assessment of quality of life of patients with severe ischemia as a result of infrainguinal arterial occlusive disease. J Vasc Surg. 1992;16:54–59. [PubMed] [Google Scholar]
- 8.Drummond M. Socio-economic impact of peripheral vascular disease. Atherosclerosis. 1997;131:S33–S34. doi: 10.1016/s0021-9150(97)06123-6. [DOI] [PubMed] [Google Scholar]
- 9.Gardner AW, Poehlman ET. Exercise treatment programs for the treatment of claudication pain: a meta-analysis. J Am Med Assoc. 1995;274:975–980. [PubMed] [Google Scholar]
- 10.Perkins JMT, Collin J, Creasy TS, Fletcher EWL, Morris PJ. Exercise training versus angioplasty for stable claudication. Long term and medium term results of a prospective randomised trial. Eur J Vasc Endovasc Surg. 1996;11:409–413. doi: 10.1016/s1078-5884(96)80171-7. [DOI] [PubMed] [Google Scholar]
- 11.Whyman MR, Fowkes FGR, Kerracher EM, et al. Is intermittent claudication improved by percutaneous transluminal angioplasty? A randomised controlled trial. J Vasc Surg. 1996;26:551–557. doi: 10.1016/s0741-5214(97)70052-1. [DOI] [PubMed] [Google Scholar]
- 12.Tan KH, de Cossart L, Edwards PR. Exercise training and peripheral vascular disease. Br J Surg. 2000;87:553–562. doi: 10.1046/j.1365-2168.2000.01445.x. [DOI] [PubMed] [Google Scholar]
- 13.Strandness DE, Jr, Dalman RL, Panian S, et al. Effect of cilostazol in patients with intermittent claudication: a randomized, double-blind, placebo-controlled study. Vasc Endovasc Surg. 2002;36:83–91. doi: 10.1177/153857440203600202. [DOI] [PubMed] [Google Scholar]
- 14.Housley E. Treating claudication in five words. Br Med J. 1988;296:1483–1484. doi: 10.1136/bmj.296.6635.1483. (Editorial). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahloff A, Bjorntorp P, Holm J, Schersten T. Metabolic activity of skeletal muscle in patients with peripheral arterial insufficiency; effect of physical training. Eur J Clin Invest. 1974;4:9–15. doi: 10.1111/j.1365-2362.1974.tb00365.x. [DOI] [PubMed] [Google Scholar]
- 16.Ernst E, Fialka V. A review of the clinical effectiveness of exercise therapy for intermittent claudication. Arch Intern Med. 1993;153:2357–2360. [PubMed] [Google Scholar]
- 17.Robeer GG, Brandsma JW, Van Den Heuvel SP, Smit B, Oostendorp RAB, Wittens CHA. Exercise therapy for intermittent claudication: a review of the quality of randomised clinical trials and evaluation of predictive factors. Eur J Vasc Endovasc Surg. 1998;15:36–43. doi: 10.1016/s1078-5884(98)80070-1. [DOI] [PubMed] [Google Scholar]
- 18.Saltin B, Gollnick PD. Skeletal muscle adaptability: significance for metabolism and performance. In: Peachy LD, Adrian RH, Geiger SR, editors. Handbook of physiology, skeletal muscle. Bethesda, MD: American Physiological Society; 1983. pp. 555–631. Sect. 10. [Google Scholar]
- 19.Tschakovsky ME, Hughson RL. Interaction of factors determining oxygen uptake at the onset of exercise. J Appl Physiol. 1999;86:1101–1113. doi: 10.1152/jappl.1999.86.4.1101. [DOI] [PubMed] [Google Scholar]
- 20.Bangsbo J, Gollnick PD, Graham TE, et al. Anaerobic energy production and O2 deficit–debt relationship during exhaustive exercise in humans. J Physiol. 1990;422:539–559. doi: 10.1113/jphysiol.1990.sp018000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitts RH. Cellular mechanisms of fatigue. Physiol Rev. 1994;74:49–94. doi: 10.1152/physrev.1994.74.1.49. [DOI] [PubMed] [Google Scholar]
- 22.Knight DR, Schaffartzik W, Poole DC, Hogan MC, Bebout DE, Wagner PD. Effects of hyperoxia on maximal leg O2 supply and utilization in men. J Appl Physiol. 1993;75:2586–2594. doi: 10.1152/jappl.1993.75.6.2586. [DOI] [PubMed] [Google Scholar]
- 23.Richardson RS, Knight DR, Poole DC, et al. Determinants of maximal exercise VO2 during single leg knee-extensor exercise in humans. Am J Physiol. 1995;268:H1453–H1461. doi: 10.1152/ajpheart.1995.268.4.H1453. [DOI] [PubMed] [Google Scholar]
- 24.MacDonald M, Pederson PK, Hughson RL. Acceleration of VO2 kinetics in heavy submaximal exercise by hyperoxia and prior high-intensity exercise. J Appl Physiol. 1997;83:1318–1325. doi: 10.1152/jappl.1997.83.4.1318. [DOI] [PubMed] [Google Scholar]
- 25.Margaria R, Edwards HT, Hill DB. The possible mechanisms of contracting and paying the oxygen debt and the role of lactic acid in muscle contraction. Am J Physiol. 1933;106:689–715. [Google Scholar]
- 26.Saltin B. Anaerobic capacity: past, present, and prospective. In: Taylor AW, Gollnick PD, Green HJ, et al., editors. Biochemistry of exercise VII. Champaign, IL: Human Kinetics; 1990. pp. 387–412. [Google Scholar]
- 27.Yoshida T, Kamiya J, Hishimoto K. Are oxygen uptake kinetics at the onset of exercise speeded up by local metabolic status in active muscles? Eur J Appl Physiol Occup Physiol. 1995;70:482–486. doi: 10.1007/BF00634376. [DOI] [PubMed] [Google Scholar]
- 28.Grassi B, Gladden LB, Stary CM, Wagner PD, Hogan MC. Faster adjustment of O2 delivery does not affect V(O2) on-kinetics in isolated in situ canine muscle. J Appl Physiol. 1998;85:1394–1403. doi: 10.1152/jappl.1998.85.4.1394. [DOI] [PubMed] [Google Scholar]
- 29.Grassi B, Gladden LB, Stary CM, Wagner PD, Hogan MC. Peripheral O2 diffusion does not affect V(O2) on-kinetics in isolated in situ canine muscle. J Appl Physiol. 1998;85:1404–1412. doi: 10.1152/jappl.1998.85.4.1404. [DOI] [PubMed] [Google Scholar]
- 30.Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329:191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cooper RH, Randle PJ, Denton RM. Stimulation of phosphorylation and inactivation of pyruvate dehydrogenase by physiological inhibitors of the pyruvate dehydrogenase reaction. Nature. 1975;257:808–809. doi: 10.1038/257808a0. [DOI] [PubMed] [Google Scholar]
- 32.Constantin-Teodosiu D, Cederblad G, Hultman E. PDC activity and acetyl group accumulation in skeletal muscle during isometric contraction. J Appl Physiol. 1993;74:1712–1718. doi: 10.1152/jappl.1993.74.4.1712. [DOI] [PubMed] [Google Scholar]
- 33.Wieland OH. The mammalian pyruvate dehydrogenase complex: structure and regulation. Rev Physiol Biochem Pharmacol. 1983;96:123–127. doi: 10.1007/BFb0031008. [DOI] [PubMed] [Google Scholar]
- 34.Childress CC, Sacktor B, Traynor DR. Function of carnitine in the fatty acid oxidase-deficient insect flight muscle. J Biol Chem. 1966;242:754–760. [PubMed] [Google Scholar]
- 35.Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974;141:761–781. doi: 10.1042/bj1410761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pratt ML, Roche TE. Mechanisms of pyruvate inhibition of kidney pyruvate dehydrogenasea kinase and synergistic inhibition by pyruvate and ADP. J Biol Chem. 1979;254:7191–7196. [PubMed] [Google Scholar]
- 37.Timmons JA, Poucher SM, Constantin-Teodosiu D, Worrall V, Macdonald IA, Greenhaff PL. Increased acetyl group availability enhances contractile function of canine skeletal muscle during ischemia. J Clin Invest. 1996;97:879–883. doi: 10.1172/JCI118490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Timmons JA, Poucher SM, Constantin-Teodosiu D, Macdonald IA, Greenhaff PL. Metabolic responses from rest to steady state determine contractile function in ischemic skeletal muscle. Am J Physiol. 1997;273:E233–E238. doi: 10.1152/ajpendo.1997.273.2.E233. [DOI] [PubMed] [Google Scholar]
- 39.Timmons JA, Gustafsson T, Sundberg CJ, et al. Substrate availability limits human skeletal muscle oxidative ATP regeneration at the onset of ischemic exercise. J Clin Invest. 1998;101:79–85. doi: 10.1172/JCI1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris RC, Foster CVL, Hultman E. Acetylcarnitine formation during intense muscular contraction in humans. J Appl Physiol. 1987;63:440–442. doi: 10.1152/jappl.1987.63.1.440. [DOI] [PubMed] [Google Scholar]
- 41.Timmons JA, Poucher SM, Constantin-Teodosiu D, Worrall V, Macdonald IA, Greenhaff PL. Metabolic responses of canine gracilis muscle during contraction with partial ischemia. Am J Physiol. 1996;270:E400–E406. doi: 10.1152/ajpendo.1996.270.3.E400. [DOI] [PubMed] [Google Scholar]
- 42.Howlett RA, Heigenhauser GJF, Hultman E, Hollidge-Horvat MG, Spriet LL. Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. Am J Physiol. 1999;277:E18–E25. doi: 10.1152/ajpendo.1999.277.1.E18. [DOI] [PubMed] [Google Scholar]
- 43.Roberts PA, Loxham SJG, Poucher SM, Constantin-Teodosiu D, Greenhaff PL. The acetyl group deficit at the onset of contraction in ischaemic canine skeletal muscle. J Physiol. 2002;544:591–602. doi: 10.1113/jphysiol.2002.021097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts PA, Loxham SJG, Poucher SM, Constantin-Teodosiu D, Greenhaff PL. Skeletal muscle pyruvate dehydrogenase complex flux dictates the magnitude of the acetyl group deficit at the onset of contraction. J Physiol. 2001;531P:57P. doi: 10.1113/jphysiol.2002.021097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stacpoole PW. The pharmacology of dichloroacetate. Metabolism: Clin Exp. 1989;38:1124–1144. doi: 10.1016/0026-0495(89)90051-6. [DOI] [PubMed] [Google Scholar]
- 46.Cicmanec JL, Condie LW, Olson GR, Wang S-R. 90-day toxicity study of dichloroacetate in dogs. Fund Appl Toxicol. 1991;17:376–389. doi: 10.1016/0272-0590(91)90227-u. [DOI] [PubMed] [Google Scholar]
- 47.Mann WR, Dragland CJ, Vinluan CC, Vedananda TR, Bell PA, Aicher TD. Diverse mechanisms of inhibition of pyruvate dehydrogenase kinase by structurally distinct inhibitors. Biochimica Biophysica Acta. 2000;1480:283–292. doi: 10.1016/s0167-4838(00)00079-0. [DOI] [PubMed] [Google Scholar]
- 48.Martin BJ, Robinson S, Wiegman DL, Aulick LH. Effect of warm-up on metabolic responses to strenuous exercise. Med Sci Sports. 1975;7:146–149. [PubMed] [Google Scholar]
- 49.Essen B, Kaijser L. Regulation of glycolysis in intermittent exercise in man. J Physiol. 1978;281:499–511. doi: 10.1113/jphysiol.1978.sp012436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Genovely H, Stamford BA. Effects of prolonged warm-up exercise above and below anaerobic threshold on maximal performance. Eur J Appl Physiol Occup Physiol. 1982;48:323–330. doi: 10.1007/BF00430222. [DOI] [PubMed] [Google Scholar]
- 51.Robergs RA, Pascoe DD, Costill DL, et al. Effect of warm-up on muscle glycogenoloysis during intense exercise. Med Exercise Sport Sci. 1991;23:37–43. [PubMed] [Google Scholar]
- 52.Campbell-O’Sullivan SP, Constantin-Teodosiu D, Peirce N, Greenhaff PL. Low intensity exercise in humans accelerates mitochondrial ATP production and pulmonary oxygen kinetics during subsequent more intense contraction. J Physiol. 2002;538:931–939. doi: 10.1113/jphysiol.2001.013238. [DOI] [PMC free article] [PubMed] [Google Scholar]