

Abstract
Aims
To assess the effect of rosuvastatin on oestrogen and progestin pharmacokinetics in women taking a commonly prescribed combination oral contraceptive steroid (OCS); the effect on endogenous hormones and the lipid profile was also assessed.
Methods
This open-label, nonrandomised trial consisted of 2 sequential menstrual cycles. Eighteen healthy female volunteers received OCS (Ortho Tri-Cyclen®) on Days 1–21 and placebo OCS on Days 22–28 of Cycles A and B Rosuvastatin 40 mg was also given on Days 1–21 of Cycle B.
Results
Co-administration did not result in lower exposures to the exogenous oestrogen or progestin OCS components. Co-administration increased AUC[0–24] for ethinyl oestradiol (26%; 90% CI ratio 1.19–1.34), 17-desacetyl norgestimate (15%; 90% CI 1.10–1.20), and norgestrel (34%; 90% CI 1.25–1.43), and increased Cmax for ethinyl oestradiol (25%; 90% CI 1.17–1.33) and norgestrel (23%; 90% CI 1.14–1.33). The increases in exposure were attributed to a change in bioavailability rather than a decrease in clearance. Luteinizing and follicle-stimulating hormone concentrations were similar between cycles. There were no changes in the urinary excretion of cortisol and 6β-hydroxycortisol. Rosuvastatin significantly decreased low-density lipoprotein cholesterol [-55%], total cholesterol [-27%], and triglycerides [-12%], and significantly increased high-density lipoprotein cholesterol[11%]. Co-administration was well tolerated.
Conclusions
Rosuvastatin can be coadministered with OCS without decreasing OCS plasma concentrations, indicating that contraceptive efficacy should not be decreased. The results are consistent with an absence of induction of CYP3A4 by rosuvastatin. The expected substantial lipid-regulating effect was observed in this study, and there was no evidence of an altered lipid-regulating effect with OCS coadministration.
Keywords: oral contraceptive steroids, pharmacodynamics, pharmacokinetics, rosuvastatin
Introduction
Rosuvastatin* (Crestor®) – an effective effective inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase – has been developed by AstraZeneca for the treatment of patients with dyslipidaemia. In clinical trials, 1–80-mg doses of the drug produced decreases in low-density lipoprotein cholesterol (LDL-C) (up to 65%), total cholesterol (TC), and triglycerides (TG), and increases in high-density lipoprotein cholesterol (HDL-C) [1–3]. Data on the pharmacokinetic of rosuvastatin have also been reported [4–11].
Selective hepatic uptake of rosuvastatin by an active transport process has been demonstrated in rats [12, 13]. HMG-CoA reductase inhibitors (including rosuvastatin [14]) have been shown to be ligands for a liver-specific human organic anion transporting polypeptide present in the basolateral membranes of hepatic cells [15]. Elimination of rosuvastatin is primarily via the liver, and – by analogy with pravastatin [16], liver to bile transport of rosuvastatin may be an active process. Metabolism appears to be a minor route of clearance [6, 7, 9, 17, 18].
An estimated 60–70 million women worldwide use oral contraceptive steroids (OCS) [19]. This large population may include many women with dyslipidaemia. Therefore, it is important that a drug for the treatment of patients with dyslipidaemia does not alter OCS efficacy.
Amongst the most commonly used OCS’s, are the combination types that contain both synthetic oestrogen (ethinyl oestradiol) and a synthetic progestin (e.g. norgestimate). These OCS hormones are metabolized by the hepatic enzyme cytochrome P450 3A4 (CYP3A4) [19, 20]. Drugs that induce CYP3A4 are known to increase the metabolism of the synthetic oestrogen and progestin OCS components, which can lead to contraceptive failure [19, 20].
There is no evidence that rosuvastatin affects on CYP3A4 activity. Thus, rosuvastatin does not induce CYP3A4 actively in animals [AstraZeneca data on file], and has no significant inhibitory effect on the cytochrome P450 enzymes isoforms in human hepatic microsomes [17]. However, it is not known whether rosuvastatin alters the activity of CYP3A4 in humans in vivo. Thus, the aim of this study was to investigate the effect of rosuvastatin on oestrogen and progestin pharmacokinetics in women taking a commonly prescribed combination OCS. The effect on endogenous hormones and the lipid profile was also assessed.
Methods
This study was designed and monitored in accordance with the ethical principles of Good Clinical Practice and the Declaration of Helsinki. An Institutional Review Board (Christiana Care Corporation, 501 West 14th Street, PO Box 1668, Wilmington, DE 19899, USA) approved the protocol before the trial started, and all subjects gave written informed consent.
Study population
Subjects were healthy, nonpregnant, nonbreastfeeding, nonsmoking female volunteers between 18 and 40 years of age with intact ovarian function who had received Ortho Tri-Cyclen® (Ortho-McNeil Pharmaceutical, Raritan, NJ, USA) throughout the previous 3 menstrual cycles. Volunteers were excluded from the trial if they had values outside the normal range for total bilirubin, alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, or creatine kinase, or a Class III or IV Pap test result. Throughout the study period, subjects were required to use barrier methods of contraception during sexual intercourse.
Eighteen volunteers entered and completed the trial. Their mean (range) age, height, and weight were 25 (20–33), years, 163 (150–173) cm, and 60 (51–70) kg, respectively.
Study design
This open-label, nonrandomised study was conducted at a single centre (Christiana Care Health Systems Inc., 4755 Ogletown-Stanton Road, Newark, DE 19718, USA). There were two 28-day treatment-assessment cycles (A and B) that corresponded to 2 full menstrual cycles. During Cycles A and B, all subjects were given a daily oral dose of oestrogen-progestin OCS on Days 1–21 and placebo OCS on Days 22–28. During Cycle B, subjects were also given a daily oral dose of rosuvastatin (4 × 10-mg capsules) on Days 1–21. Treatments were administered at 0800 h (following an 8- h fast) with 240 ml water. The OCS was Ortho Tri-Cyclen® (Ortho-McNeil), which consisted of 21 days of ethinyl oestradiol (EO) 0.035 mg (Weeks 1–3), and 7 days each of norgestimate (NGM) 0.180 mg (Week 1), 0.215 mg (Week 2), and 0.250 mg (Week 3).
Determination of plasma concentration of exogenous hormones
Venous blood samples (5 ml) for the assay of exogenous hormones (EO, NGM and its active metabolites 17-desacetyl norgestimate [DesAc-NGM] and norgestrel [NG]) were collected before dosing and at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 18, 24, 48, and 72 h after dosing on Days A-21 and B-21. These analyses were to confirm that hormones were maintained at concentrations adequate for contraceptive efficacy. Samples were collected into tubes containing lithium heparin anticoagulant and cooled to 5 °C before centrifugation. Plasma was then harvested and stored at −20 °C until assay.
Plasma samples were analysed for EO at Phoenix International (Quebec, Canada) using high-resolution gas chromatography with negative chemical ionization mass spectrometric detection. Mean accuracy values for quality control samples were 95–100%, and the imprecision (coefficient of variation) was 6–16.0%. Plasma samples were analysed for NGM, DesAc-NGM, and NG (also at Phoenix International) using high-performance liquid chromatography with mass spectrometric detection. Mean accuracy values for quality control samples were 98–103%, 98–103%, and 96–105%, respectively, and the imprecision (coefficient of variation) was 4–15%, 6–7%, and 8–13%, respectively. Lower limits of quantification for the assays were: EO 2 pg ml−1, NGM 0.02 ng ml−1, DesAc-NGM 0.02 ng ml−1, and NG 0.08 ng ml−1.
Determination of the urinary concentrations of cortisol and 6β-hydroxycortisol
Urine samples for the analysis of cortisol and 6β-hydroxycortisol (markers of CYP3A4 induction [21] were collected over a 24-h period on Days A-21 and B-21. Samples were collected into containers and stored at −20 °C until assay.
Urine samples were analysed at BAS Analytics (Warwickshire, UK) using high-performance liquid chromatography with ultraviolet detection. The lower limits of quantification of the assays were 10 ng ml−1for cor-tisol and 20 ng ml−1for 6β-hydroxycortisol. Accuracy ranged from 100 to 106% and imprecision ranged from 1.2 to 3.6% for cortisol, and 95–99% and 1.1–2.5% for 6β-hydroxycortisol.
Determination of the plasma concentrations of rosuvastatin
Blood samples (5 ml venous blood) for rosuvastatin were collected before dosing and at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 18, 24, 48, and 72 h after dosing on Day B-21. Samples were collected into tubes containing lithium heparin anticoagulant and cooled to 5 °C before centrifugation. Plasma was then harvested, mixed with an equal volume of acetate buffer 0.1 m (pH 4.0), and stored at −70 °C until assay.
Plasma samples were analysed at Quintiles Scotland Ltd (Edinburgh, UK) using high-performance liquid chromatography with mass spectrometric detection [22]. Briefly, samples were subjected to automated solid-phase extraction on 96-well plates containing a hydrophobic-lipophilic balanced copolymer sorbent. The extract was chromatographed on a high-performance liquid chromatography column, and rosuvastatin detected using a triple quadrupole mass spectrometer fitted with a turbo-ionspray source. Correlation coefficients for rosuvastatin calibration curves were 0.998–1.00 Mean inaccuracy levels and imprecision values for quality control samples (at all concentrations) were <13% and <7%, respectively. The lower limit of quantification of the assay was 0.1 ng ml−1. However, because all samples were diluted 2-fold prior to analysis, the effective limit was 0.2 ng ml−1.
Pharmacokinetic analysis
For the exogenous hormones, the following pharmacokinetic parameters were estimated in the absence (Cycle A) and presence (Cycle B) of rosuvastatin: area under the plasma concentration-time curve from time zero to 24 h (AUC[0–24]); maximum observed plasma drug concentration (Cmax); time of Cmax (tmax); and terminal elimination half-life (t1/2).
For cortisol and 6β-hydroxycortisol, daily urinary excretion was estimated in the absence (Cycle A) and presence (Cycle B) of rosuvastatin.
For rosuvastatin, the following steady-state pharmacokinetic parameters were estimated when coadministered with OCS (Cycle B): AUC[0–24]; Cmax; tmax; and t1/2.
AUC[0–24] was determined using the linear trapezoidal rule. Cmax and tmax were determined by visual inspection of the plasma concentration-time curves. t1/2 was calculated as 0.693/λz (where λz was the terminal elimination rate constant derived from log-linear regression of the terminal portion of the plasma concentration-time curves).
Determination of plasma concentration of endogenous hormones
Blood samples (15 ml venous blood) for the assay of (luteinizing hormone [LH], follicle-stimulating hormone [FSH], sex hormone binding globulin [SHBG], and progesterone) were taken before dosing on Days 7, 14, 20, and 21 of Cycles A and B. Samples were collected into vacutainers (containing no anticoagulant). Serum was then harvested and stored at −20 °C until assay.
Samples were analysed at the Medical Research Laboratories (Kentucky, USA) using radioimmunoassay techniques.
Determination of the plasma concentrations of lipids and lipoproteins
Blood samples (10 ml venous blood) for LDL-C, TC, TG, and HDL-C and apolipoprotein B [ApoB] and ApoA-I were taken before dosing on Days 1 and 21 of Cycles A and B (subjects fasted for at least 12 h before collection). Samples were collected into tubes containing powdered EDTA and centrifuged within 30 min Plasma was harvested and stored at −20 °C until assay.
Samples were analysed at the Medical Research Laboratories, which is certified for lipid analysis as specified by the Standardization Program of the Center for Disease Control and Prevention and the National Heart, Lung and Blood Institute. Concentrations of LDL-C were calculated using the Friedewald formula for volunteers who had TG concentrations ≤0.53.38 mmol l−1 (300 mg dl−1), and using the beta-quantification method for volunteers who had TG concentrations >3.38 mmol l−1 (300 mg dl−1).
Pharmacodynamic evaluation
For the endogenous hormones, plasma concentrations were compared in the absence (Cycle A) and presence (Cycle B) of rosuvastatin.
For the lipids and lipoproteins, changes from baseline (defined as the mean of available values from Days A-1, A-21, and B-1) to Day B-21 were evaluated.
Statistical methods
A sample size of 16 would have had 90% power to detect a 30% difference in the AUC [0–24] of EO between Cycle A (OCS alone) and Cycle B (OCS + rosuvastatin).
AUC [0–24] and Cmax of EO and NGM (primary parameters), DesAc-NGM, and NG were compared between Cycles A and B. Differences were assessed using the geometric mean (gmean) ratio ([OCS + rosuvastatin]/OCS) and corresponding 90% confidence interval (CI). The criterion for the absence of a clinically relevant difference was a CI within the range 0.7–1.43 for the ration of the pharmacokinetic parameters. Differences between cycles in the t1/2 of EO, NGM, DesAc-NGM, and NG were assessed using the arithmetic mean difference ([OCS + rosuvastatin]–OCS) and corresponding 90% CI.
Differences between cycles in the urinary excretion of cortisol and 6β-hydroxycortisol, the 6β-hydroxycortisol/cortisol ratio, and the concentrations of LH, FSH, SHBG, and progesterone were assessed using the arithmetic mean difference ([Day B-21]–[Day A-21]) and corresponding 95% CI. The mean percent change from baseline to Day B-21 in LDL-C, TC, TG, HDL-C, ApoB, and ApoA-I was assessed using the 95% CI and P-value based on a paired t-test.
Tolerability evaluation
Tolerability was assessed from adverse event reports, clinical laboratory tests (clinical chemistry, haematology, and urinalysis), physical examination and vital signs, and from 12-lead electrocardiograms (ECGs).
Results
Pharmacokinetics of exogenous hormones
The gmean plasma concentrations of EO before, and for 72 h following, the final dose of trial medication in Cycle A (OCS alone) and Cycle B (OCS + rosuvastatin) are presented in Figure 1.
Figure 1.
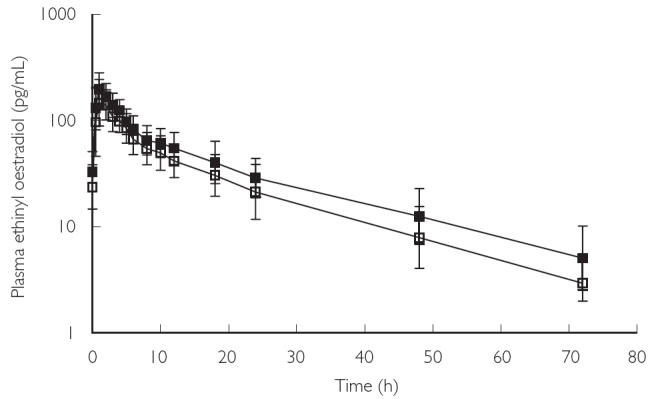
Geometric mean plasma concentrations of ethinyl oestradiol (± standard deviation) over time in Cycles A and B. Volunteers received OCS alone in Cycle A (□) and OCS + rosuvastatin in Cycle B (▪)
NGM is metabolized to 2 active metabolites: DesAc-NGM and NG. The rapidity and completeness of these reactions meant that circulating concentrations of NGM were extremely low, and probably contributed little to activity. Thus NGM concentrations have not been reported here. The gmean plasma concentrations of DesAc-NGM and NG before, and for 72 h following the final dose of trial medication in Cycle A (OCS alone) and Cycle B (OCS + rosuvastatin) are presented in Figure 2.
Figure 2.
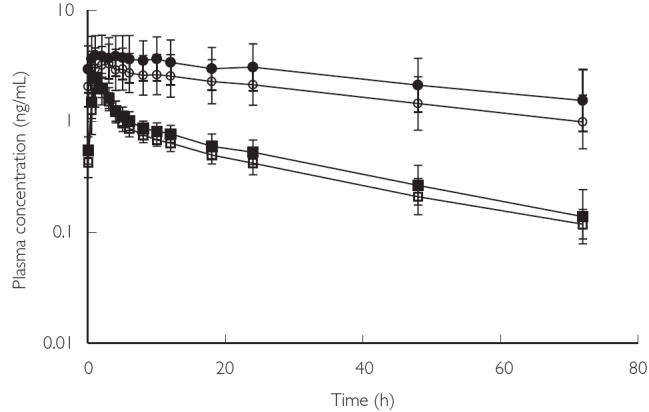
Geometric mean plasma concentrations of 17-desacetyl norgestimate (DesAc-NGM) and norgestrel (NG) (± standard deviation) over time in Cycles A and B. Volunteers received OCS alone in Cycle A and OCS + rosuvastatin in Cycle B. DesAc-NGM (Cycle A) (□), DesAc-NGM (Cycle B) (▪), NG (Cycle A) (○), NG (Cycle B) (•)
The effect of rosuvastatin administration on the pharmacokinetic parameters of EO, DesAc-NGM, and NG is summarized in Table 1.
Table 1.
Pharmacokinetic parameters for ethinyl oestradiol, 17-desacetyl norgestimate, and norgestrel in Cycles A and B
| Hormone Parameter (units) | Summary statistic | Cycle A OCS alone n = 18 | Cycle B OCS + rosuvastatin n = 18 | Treatment effecta | 90% confidence interval of the treatment effect |
|---|---|---|---|---|---|
| Ethinyl oestradiol (EO) | gmean (CV%) | 1339 (35) | 1690 (31) | 1.26 | 1.19–1.34 |
| AUC(0–24) (pg·h/ml) | gmean (CV%) | 1339 (35) | 1690 (31) | 1.26 | 1.19–1.34 |
| Cmax (pg/ml) | gmean (CV%) | 159 (41) | 198 (36) | 1.25 | 1.17–1.33 |
| tmax (h) | Median (range) | 1.0 (1.0–4.0) | 1.0 (1.0–4.0) | NA | NA |
| t1/2 (h) | Mean (s.d.) | 13.8 (3.31)b | 16.6 (3.68)b | 2.37 | 0.89–3.85 |
| 17-desacetyl norgestimate(DesAc-NGM) | gmean (CV%) | 19.1 (16) | 22.0 (16) | 1.15 | 1.10–1.20 |
| AUC(0–24) (ng·h/ml) | gmean (CV%) | 19.1 (16) | 22.0 (16) | 1.15 | 1.10–1.20 |
| Cmax (ng/ml) | gmean (CV%) | 2.47 (33.9) | 2.53 (19.3) | 1.03 | 0.91–1.16 |
| tmax (h) | Median (range) | 1.0 (0.5–4.0) | 1.0 (1.0–3.0) | NA | NA |
| t1/2 (h) | Mean (s.d.) | 23.7 (4.22) | 24.0 (4.71)c | 0.71 | −0.50–1.91 |
| Norgestrel (NG) | gmean (CV%) | 62.0 (44) | 82.8 (48) | 1.34 | 1.25–1.43 |
| AUC(0–24) (ng·h/ml) | gmean (CV%) | 62.0 (44) | 82.8 (48) | 1.34 | 1.25–1.43 |
| Cmax (ng/ml) | gmean (CV%) | 3.46 (41.0) | 4.27 (44.8) | 1.23 | 1.14–1.33 |
| tmax (h) | Median (range) | 2.0 (0.5–8.0) | 3.0 (0.5–10.0) | NA | NA |
| t1/2 (h) | Mean (s.d.) | 31.2 (3.31)d | 29.5 (NC)d | 1.20 | NC |
Geometric mean of the ratio (OCS + rosuvastatin)/OCS for AUC(0–24) and Cmax; arithmetic mean of the difference (OCS + rosuvastatin)-OCS for t1/2
t1/2 data in both Cycles A and B were available for 16 volunteers
t1/2 data in Cycle B were available for 15 volunteers
t1/2 data in Cycle A were available for 3 volunteers; data in Cycle B were available for 1 volunteer (because half-life could not be characterized over 2 full half-lives with the sampling schedule used).
AUC(0–24), Area under the plasma concentration-time curve from time zero to 24 h; Cmax, Maximum observed plasma drug concentration; CV%, Coefficient of variation expressed as a percentage of the gmean (geometric mean); NA, Not applicable; NC, Not calculable; OCS, Oral contraceptive steroid; s.d, Standard deviation; t1/2, terminal elimination half-life; tmax, time of Cmax
Co-administration of rosuvastatin and OCS did not result in lower exposures to this oestrogen or progestin components. Co-administration increased AUC[0–24] for EO, DesAc-NGM, and NG, and increased Cmax for EO and NG. The 90% CIs for all but 1 of these parameters (AUC[0–24] for (NG) were within the prespecified range for the absence of a clinically relevant difference[i.e. 0.7–1.43].
There was a small increase in the mean t1/2 for EO with rosuvastatin treatment. However, the CIs for this parameter were wide[0.89–3.85], indicating considerable variation, and passed through unity. The increased EO exposure (AUC[0–24] and Cmax) was not explained by this small increase in t1/2 and was therefore attributed to an increase in bioavailability. Elimination rates for DesAc-NGM and NG did not appear to differ between treatment with OCS alone and OCS + rosuvastatin.
Urinary excretion of cortisol and 6β-hydroxycortisol
No significant change in the urinary excretion of cortisol or 6β-hydroxycortisol occurred with coadministration of rosuvastatin and OCS [Table 2]. The 6β-hydroxycortisol/cortisol ratio was also unchanged, although the study was not powered to determine differences in this ratio.
Table 2.
Urinary excretion of cortisol and 6β-hydroxycortisol, and cortisol/6β-hydroxycortisol ratio, in Cycles A and B
| Variable (units) | Day A-21a Mean (s.d.)n = 18 | Day B-21a Mean (s.d.)n = 18 | Mean differenceb (s.d.) | 95% confidence interval of the mean difference |
|---|---|---|---|---|
| Cortisol (µg)c | 57.1 (46.1) | 41.9 (24.4) | −28.4 (61.8) | −80.0 to −23.3 |
| 6β-hydroxycortisol (µg)d | 161 (92.6) | 174 (121) | 21.8 (133) | −46.8 to −90.3 |
| 6β-hydroxycortisol/cortisole | 4.26 (2.65) | 4.70 (3.08) | −0.98 (2.00) | −2.83 to −0.86 |
Volunteers received OCS alone in Cycle A and OCS + rosuvastatin in Cycle B
(Day B-21)–(Day A-21)
Data for Day A-21 were available for 15 volunteers; data for Day B-21 were available for 11 volunteers; paired values were available for 8 volunteers
Data for Day B-21 were available for 17 volunteers
Data for Day A-21 were available for 15 volunteers; data for Day B-21 were available for 10 volunteers; paired values were available for 7 volunteers.
Abbreviations are: s.d.: Standard deviation.
Pharmacokinetics of rosuvastatin
The gmean AUC[0–24] of rosuvastatin on Day B-21 was 290 ng ml−1. The gmean Cmax of 35.2 ng ml−1 was achieved at a median tmax of 3.0 h. The arithmetic mean t1/2 (determined in 14 volunteers) was 15.3 h.
Plasma concentrations of endogenous hormones
The effect of rosuvastatin administration on the concentrations of endogenous hormones is summarized in Table 3. There were no relevant changes in LH (mean difference −0.56 mIU/ml; 95% CI −1.66–0.55), FSH (−0.68 mIU/ml; −1.68–0.33), SHBG (28.1 nmol l−1; −1.93–58.0), or progesterone (−0.05 ng ml−1; −0.12–0.02) concentrations with coadministration of rosuvastatin and OCS (although LH, FSH, and progesterone were slightly decreased and SHBG was slightly increased). In addition, the concentrations of progesterone were low at Day 21 (luteal phase) in both cycles in all volunteers.
Table 3.
Mean concentrations of endogenous hormones in Cycles A and B
| Mean (standard deviation) | ||||
|---|---|---|---|---|
| Hormone (units) Cyclea | Day 7 n = 18 | Day 14 n = 18 | Day 20 n = 18 | Day 21 n = 18 |
| Luteinizing hormone (mIU/ml) | ||||
| Cycle A | 9.94 (8.30) | NC | NC | NC |
| Cycle B | 8.00 (7.04) | NC | NC | NC |
| Follicle-stimulating hormone (mIU/ml) | ||||
| Cycle A | 6.88 (4.31) | NC | 3.74 (3.03) | 3.41 (2.88) |
| Cycle B | 6.83 (4.92) | 4.10 (3.46) | NC | NC |
| Sex hormone binding globulin (nmol/l)Cycle A | 497.0 (167.5) | 578.9 (192.0) | 580.3 (176.5) | 585.9 (191.1) |
| Cycle B | 526.1 (170.1) | 613.3 (187.7) | 623.2 (205.9) | 614.0 (196.3) |
| Progesterone (ng/ml) | ||||
| Cycle A | NA | NA | NA | 0.614 (0.164) |
| Cycle B | NA | NA | NA | 0.562 (0.163) |
Volunteers received OCS alone in Cycle A and OCS + rosuvastatin in Cycle B. Abbreviations are: NA: Not applicable, NC: Not calculable (> 50% of individual assessments were below the limit of quantification for the assay – the lower limits of quantification for luteinizing and follicle-stimulating hormones were 2.0 mIU/ml and 1.6 mIU/ml, respectively).
Effect on lipids and lipoproteins
Co-administration of rosuvastatin and OCS resulted in decreases in plasma LDL-C (mean percent change from baseline −55%; 95% CI −59 to −51), TC (−27%; −31 to −24), and TG (−12%; −22 to −3), and a significant increase in HDL-C (11% 5–17). A significant decrease in ApoB (−38%; −44 to −33) was also seen, along with a significant increase in ApoA-I (11% 6–15).
Tolerability
Co-administration of rosuvastatin and OCS was well tolerated over the 21-day treatment period. There were no serious adverse events, no withdrawals due to adverse events, and no clinically significant changes in clinical laboratory parameters, vital signs, ECGs, or physical examinations during the trial. Dysmenorrhoea and nausea were each reported by 1 subject in each treatment cycle. Breast tenderness and bloating were each reported by 2 subjects during Cycle B.
Discussion
This trial evaluated the potential interaction between rosuvastatin and an oestrogen-progestin OCS (Ortho Tri-Cyclen®) in order to determine the effect of coadministration on oestrogen and progestin pharmacokinetics and endogenous hormones. The results show that coadministration did not result in lower exposures to the exogenous oestrogen or progestin OCS components. In contrast, coadministration increased AUC[0–24] for EO[26%], DesAc-NGM[15%], and NG[34%], and increased Cmax for EO[25%] and NG[23%]. The increases in exposure were attributed to a change in bioavailability rather than a decrease in clearance. LH and FSH concentrations were similar between Cycles A and B. Together these results indicate that coadministration of rosuvastatin and OCS should not decrease contraceptive efficacy.
OCS hormones are metabolised by the hepatic enzyme CYP3A4 [19, 20]. Drugs that induce this enzyme are known to increase the metabolism of the oestrogen and progestin OCS components resulting in lower OCS hormone exposures, which can lead to contraceptive failure. In-vivo observations in animals indicate that rosuvastatin does not induce CYP3A4 (AstraZeneca data on file). This is supported by the present findings, namely the lack of a significant change in the urinary excretion of cortisol and 6β-hydroxycortisol, and the observation that OCS hormone exposures did not decrease, with coadministration of rosuvastatin.
The mechanism by which rosuvastatin increased exposure to EO and the NGM active metabolites DesAc-NGM and NG is unknown. As mentioned above, CYP3A4 is an important enzyme involved in the clearance of these steroids. However, in vitro studies with human hepatic microsomes [17] have demonstrated a lack of inhibitory activity of rosuvastatin on CYP3A4. This suggests that inhibition of CYP3A4 was not the mechanism by which rosuvastatin increased EO, DesAc-NGM, and NG exposure. An interaction via a transport protein may be an alternative explanation. The absorption, distribution, and elimination of rosuvastatin are known to involve transporters though these have not been well characterized.
The effects of the increases in exposure to EO and the active metabolites of NGM seen with coadministration of rosuvastatin and OCS in this trial are unlikely to be of clinical relevance. The 26% increase in EO exposure is modest and unlikely to increase the risk of adverse events associated with administration of low-dose OCS preparations containing EO 20–35 µg. The small increases in exposure to DesAc-NGM and NG, are also unlikely to change the progestin activity of the triphasic OCS used in the present work. The concentrations of EO during treatment with rosuvastatin would be expected to be below the concentrations resulting from administration of OCS preparations containing EO 50 µg. Another HMG-CoA reductase inhibitor, atorvastatin, also increases exposure to EO following coadministration with OCS [23, 24].
Concentrations of the endogenous hormones LH, FSH, and progesterone appeared similar between Cycles A and B. In particular, no increase in progesterone during the luteal phase was observed, which suggests that ovulation did not occur in any volunteer. Although changes in hormone concentrations can be missed if not measured at the appropriate time, it is unlikely that rosuvastatin would cause a reduction in OCS effi-cacy without decreasing exposure to the exogenous hormones.
The pharmacokinetics of rosuvastatin were consistent with those seen in other populations in which rosuvastatin was administered alone [4–7, 9, 10]. In addition, when coadministered with OCS, rosuvastatin produced significant decreases in LDL-C [−55%], TC [−27%], and TG [−12%], and a significant increase in HDL-C [11%]. None of the subjects had dyslipidaemia. The mean baseline LDL-C and TG values were both less than 2.5 mmol l−1 (100 mg dl−1) and the mean baseline HDL-C-value was high at 1.6 mmol l−1 (63 mg dl−1). Nonetheless, the magnitudes and directions of the changes in plasma lipids are considered clinically relevant. There was no evidence of an altered lipid-regulating effect with OCS coadministration [1–3].
It should be noted that this study was performed in selected healthy subjects over a single menstrual cycle under carefully controlled conditions. The effects of rosuvastatin on oestradiol pharmacokinetics using other OCS preparations would be expected to be similar, but the use of a single OCS in the present work makes that inference speculative.
In conclusion, rosuvastatin can be coadministered with OCS without decreasing OCS plasma levels, indicating that contraceptive efficacy should not be decreased. The results are consistent with an absence of induction of CYP3A4 by rosuvastatin. The expected substantial lipid-regulating effect was observed, and there was no evidence of an altered lipid-regulating effect with OCS coadministration.
Acknowledgments
This trial was supported by AstraZeneca and has been included in the marketing applications for rosuvastatin. The authors thank Phoenix International (Montreal, Quebec, Canada) for performing the exogenous hormone assays, Quintiles Scotland Ltd (Edinburgh, Scotland, UK) for performing the rosuvastatin assay, BAS Analytics (Kenilworth, Warwickshire, UK) for performing the cortisol and 6b.5-hydroxycortisol assays, Medical Research Laboratories (Highland Heights, Kentucky, USA) for performing the endogenous hormone and lipid lipoprotein-1 assays, and Elizabeth Eaton PhD for manuscript preparation.
References
- 1.Olsson AG, Istad H, Luurila O, et al. Effects of rosuvastatin and atorvastatin compared over 52 weeks of treatment in patients with hypercholesterolemia. Am Heart J. 2002;144:1044–51. doi: 10.1067/mhj.2002.128049. [DOI] [PubMed] [Google Scholar]
- 2.Brown WV, Bays HE, Hassman DR, et al. Efficacy and safety of rosuvastatin compared with pravastatin and simvastatin in patients with hypercholesterolemia: a randomized, double-blind, 52-week trial. Am Heart J. 2002;144:1036–43. doi: 10.1067/mhj.2002.129312. [DOI] [PubMed] [Google Scholar]
- 3.Blasetto JW, Stein EA, Brown WV, Chitra R, Raza A. Efficacy of rosuvastatin compared with other statins at selected starting doses in hypercholesterolemic patients and in special population groups. Am J Cardiol. 2003;91:3C–10C. doi: 10.1016/s0002-9149(03)00003-1. [DOI] [PubMed] [Google Scholar]
- 4.Martin PD, Dane AL, Nwose OM, Schneck DW, Warwick MJ. No effect of age or gender on the pharmacokinetics of rosuvastatin: a new HMG-CoA reductase inhibitor. J Clin Pharmacol. 2002;42:1116–21. doi: 10.1177/009127002401382722. [DOI] [PubMed] [Google Scholar]
- 5.Martin PD, Mitchell PD, Schneck DW. Pharmacodynamic effects and pharmacokinetics of a new HMG-CoA reductase inhibitor, rosuvastatin, after morning or evening administration in healthy volunteers. Br J Clin Pharmacol. 2002;54:472–7. doi: 10.1046/j.1365-2125.2002.01688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper KJ, Martin PD, Dane AL, Warwick MJ, Raza A, Schneck DW. Lack of effect of ketoconazole on the pharmacokinetics of rosuvastatin in healthy subjects. Br J Clin Pharmacol. 2003;55:94–9. doi: 10.1046/j.1365-2125.2003.01720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper KJ, Martin PD, Dane AL, Warwick MJ, Schneck DW, Cantarini MV. The effect of fluconazole on the pharmacokinetics of rosuvastatin. Eur J Clin Pharmacol. 2002;58:527–31. doi: 10.1007/s00228-002-0508-8. [DOI] [PubMed] [Google Scholar]
- 8.Martin PD, Kemp J, Dane AL, Warwick MJ, Schneck DW. No effect of rosuvastatin on the pharmacokinetics of digoxin in healthy volunteers. J Clin Pharmacol. 2002;42:1352–7. doi: 10.1177/0091270002042012008. [DOI] [PubMed] [Google Scholar]
- 9.Cooper KJ, Martin PD, Dane AL, Warwick MJ, Schneck DW, Cantarini MV. Effect of itraconazole on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther. 2003;73:322–9. doi: 10.1016/s0009-9236(02)17633-8. [DOI] [PubMed] [Google Scholar]
- 10.Martin PD, Dane AL, Schneck DW, Warwick MJ. An open-label, randomized, three-way crossover trial of the effects of coadministration of rosuvastatin and fenofibrate on the pharmacokinetic properties of rosuvastatin and fenofibric acid in healthy male volunteers. Clin Ther. 2003;25:459–71. doi: 10.1016/s0149-2918(03)80089-9. [DOI] [PubMed] [Google Scholar]
- 11.Simonson SG, Martin PD, Mitchell P, Schneck DW, Lasseter KC, Warwick MJ. Pharmacokinetics and pharmacodynamics of rosuvastatin in subjects with hepatic impairment. Eur J Clin Pharmacol. 2003;58:669–75. doi: 10.1007/s00228-002-0541-7. [DOI] [PubMed] [Google Scholar]
- 12.Nezasa K, Higaki K, Takeuchi M, Nakano M, Koike M. Uptake of rosuvastatin by isolated rat hepatocytes: comparison with pravastatin. Xenobiotica. 2003;33:379–88. doi: 10.1080/0049825031000066259 . [DOI] [PubMed] [Google Scholar]
- 13.Nezasa K, Higaki K, Matsumura T, et al. Liver-specific distribution of rosuvastatin in rats: comparison with pravastatin and simvastatin. Drug Metab Dispos. 2002;30:1158–63. doi: 10.1124/dmd.30.11.1158. [DOI] [PubMed] [Google Scholar]
- 14.Brown CDA, Windass A, Bleasby K, Lauffart B. Rosuvastatin is a high affinity substrate of hepatic organic anion transporter OATP-C. Atheroscler Supple. 2001;2:90. (abstract P174) [Google Scholar]
- 15.Hsiang B, Zhu Y, Wang Z, et al. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J Biol Chem. 1999;274:37161–8. doi: 10.1074/jbc.274.52.37161. [DOI] [PubMed] [Google Scholar]
- 16.Yamazaki M, Kobayashi K, Sugiyama Y. Primary active transport of pravastatin across the liver canalicular membrane in normal and mutant Eisai hyperbilirubinaemic rats. Biopharm Drug Dispos. 1996;17:645–59. doi: 10.1002/(SICI)1099-081X(199611)17:8<645::AID-BDD986>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 17.McCormick AD, McKillop D, Butters CJ, et al. ZD4522 – an HMG-CoA reductase inhibitor free of metabolically mediated drug interactions: metabolic studies in human in vitro systems. J Clin Pharmacol. 2000;40:1055. (abstract 46) [Google Scholar]
- 18.Martin PD, Dane AL, Schneck DW, Warwick MJ. Disposition of new HMG-CoA reductase inhibitor ZD4522 following dosing in healthy subjects. J Clin Pharmacol. 2000;40:1056. (abstract 48) [Google Scholar]
- 19.Back DJ, Orme ML. Pharmacokinetic drug interactions with oral contraceptives. Clin Pharmacokinet. 1990;18:472–84. doi: 10.2165/00003088-199018060-00004. [DOI] [PubMed] [Google Scholar]
- 20.Wilbur K, Ensom MH. Pharmacokinetic drug interactions between oral contraceptives and second-generation anticonvulsants. Clin Pharmacokinet. 2000;38:355–65. doi: 10.2165/00003088-200038040-00004. [DOI] [PubMed] [Google Scholar]
- 21.Tran JQ, Kovacs SJ, McIntosh TS, Davis HM, Martin DE. Morning spot and 24-hour urinary 6 beta-hydroxycortisol to cortisol ratios: intraindividual variability and correlation under basal conditions of CYP3A4 induction. J Clin Pharmacol. 1999;39:487–94. [PubMed] [Google Scholar]
- 22.Hull CK, Penman AD, Smith CK, Martin PD. Quantification of rosuvastatin in human plasma by automated solid-phase extraction using tandem mass-spectrometric detection. J Chromatogr B Anal Technol Biomed Life Sci. 2002;772:219–28. doi: 10.1016/s1570-0232(02)00088-0. [DOI] [PubMed] [Google Scholar]
- 23.Parke-Davis. Physicians’ Desk Reference ®. 54. Montvale, NJ: Medical Economics Company Inc.; 2000. Lipitor ® (atorvastatin calcium) tablets product insert; pp. 2254–7. [Google Scholar]
- 24.Yang B-B, Siedlik PH, Smithers JA, Sedman AJ, Stern RH. Atorvastatin pharmacokinetic interactions with other CYP3A4 substrates: erythromycin and ethinyl estradiol. Pharm Res. 1996. p. S-437. (abstract PPDM 8179)