

Abstract
Aims
Previous studies have shown that the antimalarial drug artemisinin is a potent inducer of its own metabolism in both patients and healthy subjects. The aim of this study was to characterize the time-dependent pharmacokinetics of artemisinin in healthy subjects.
Methods
Twenty-four healthy males were randomized to receive either a daily single dose of 500 mg oral artemisinin for 5 days, or single oral doses of 100/100/250/250/500 mg on each of the first 5 days. Two subjects from each group were administered a new dose of 500 mg on one of the following days after the beginning of the study: 7, 10, 13, 16, 20, or 24. Artemisinin concentrations in saliva samples collected on days 1, 3, 5, and on the final day were determined by HPLC. Data were analysed using a semiphysiological model incorporating (a) autoinduction of a precursor to the metabolizing enzymes, and (b) a two-compartment pharmacokinetic model with a separate hepatic compartment to mimic the processes of autoinduction and high hepatic extraction.
Results
Artemisinin was found to induce its own metabolism with a mean induction time of 1.9 h, whereas the enzyme elimination half-life was estimated to 37.9 h. The hepatic extraction ratio of artemisinin was estimated to be 0.93, increasing to about 0.99 after autoinduction of metabolism. The model indicated that autoinduction mainly affected bioavailability, but not systemic clearance. Non-linear increases in AUC with dose were explained by saturable hepatic elimination affecting the first-pass extraction.
Conclusion
Artemisinin produces a rapid onset of enzyme induction, resulting in a decrease in its own bioavailability over time. The proposed model successfully described the time-course of the onset and normalization of the autoinduction of metabolism in healthy subjects receiving two different dosage regimens of the compound.
Keywords: Artemisinin, antoinduction, pharmacokinetic model
Introduction
Artemisinin, a sesquiterpene peroxide extracted from Artemisia annua L., is the parent compound of a novel family of antimalarial drugs that clear parasites rapidly [1]. The compound is effective against strains of falciparum malaria otherwise resistant to conventional antimalarials. Plasma artemisinin concentrations reach a peak within 2–3 h after oral intake and decline with a short half-life of 1.5–2 h [2]. Owing to the lack of parenteral formulations, no information is available on the absolute bioavailability of the compound. Artemisinin is believed to pass through the gut membrane relatively easily [3, 4], although high oral clearance values are indicative of high first-pass metabolism of the compound, resulting in low bioavailability [5].
Artemisinin is a potent inducer of the enzymes responsible for its own metabolism in both healthy subjects [6, 7] and malaria patients [5, 8–10]. Autoinduction results in a 5–7-fold decrease in plasma artemisinin concentrations at the end of a 5-day treatment period in patients with uncomplicated falciparum malaria. The elimination half-life, and thus the system clearance of artemisinin, remains unchanged during the induction phase [5, 7–10]. Thus, it was suggested that artemisinin is highly extracted by the liver, with the elevated enzyme activity primarily affecting its bioavailability. It has been shown that a single oral dose of 500 mg or higher is enough to initiate the autoinduction, which may last for 5–7 days [11, 12]. Decreasing plasma concentrations during multiple dosing have also been reported for the analogues artemether [13, 14] and, less convincingly, artesunate [15]. Artemisinin metabolism in human liver microsomes is mediated primarily by CYP2B6 with probable secondary contribution from CYP3A4 and possibly CYP2A6 in individuals with low CYP2B6 expression [16]. Artemisinin has also been reported to induce CYP2C19 but not CYP3A4 [7]. However, it has not been established which enzyme(s) are affected in the autoinduction of artemisinin metabolism.
Artemisinin is a lipophilic, neutral compound and has been shown to distribute into saliva [17, 18]. Salivary concentrations of the drug are correlated with unbound plasma concentrations (correlation coefficient of 0.77) [18], making saliva an attractive alternative to blood in pharmacokinetic studies of this drug [10, 17, 18].
The aim of the present study was to characterize and model the pharmacokinetics of the autoinduction of artemisinin, both during the phase of falling artemisinin concentrations, and the phase when drug concentrations return to the same values as those observed after the first dose. The possibility of dose-dependency in the autoinduction of artemisinin metabolism was also examined.
Methods
Study subjects and materials
Twenty-four healthy, male, Vietnamese adults with an average (±SD) age of 34 (±12) years and a bodyweight of 51 (±5) kg were included in this randomized, parallel group study, which took place at the Clinical Study Unit of the National Institute of Malariology, Parasitology and Entomology, Hanoi, Vietnam. Written, informed consent was obtained from each subject prior to inclusion. The study was approved by the Ministry of Health, Hanoi, Vietnam, the ethics committee of the Medical Faculty of Uppsala University, Uppsala, Sweden, and the Medical Products Agency, Uppsala, Sweden.
All subjects underwent a routine health screen. The following were inclusion criteria: male adults between 18 and 55 years of age, declared healthy after physical examination, with no history of antimalarial drug use within 3 weeks before the study, and written consent to participate in the study. Subjects taking any current medication with other drug(s), including vitamins, were excluded.
Artemisinin hard gelatin capsules (250 mg, containing no bulk material) were purchased from the Institute of Materia Medica (Hanoi, Vietnam). The manufacturer performed the batch quality control analysis of the capsules. According to the Vietnamese Medical Branch, the batch acceptance limits for the 250 mg capsules were a content of 237.5–262.5 mg artemisinin in each capsule. The 100-mg artemisinin hard gelatin capsules were prepared by the Pharmaceutical Unit at the National Institute of Malariology, Parasitology and Entomology, Hanoi, Vietnam. Artemisinin from the same batch as the 250 mg capsules was used. Empty capsules (containing no bulk material) were filled with 100 mg artemisinin with acceptance limits of 95–105 mg.
Study design
Subjects who enrolled in the standard-dose group (n = 12) received single doses of 500 mg artemisinin per day for 5 consecutive days (Figure 1), the recommended monotherapy regimen for treatment of uncomplicated malaria at the time of the study. Those enrolled in the escalating-dose group (n = 12) were administered 100 mg artemisinin for the first 2 days, increased to 250 mg on days 3 and 4, and given a final dose of 500 mg on day 5 (Figure 1). Two subjects from each group were randomized to return for intake of an additional dose of 500 mg artemisinin on one of the following days: 7, 10, 13, 16, 20, or 24 after the beginning of the study. Thus, each subject was administered a total of 6 doses of artemisinin, including 5 consecutive dosings and a single dose on a return day. All subjects received a total number of two capsules on each occasion under the supervision of the study team; each ingestion was followed by intake of 100 ml water and the mouth was rinsed with another 100 ml water. Subjects ate no solid food for 2 h before and 2 h after drug intake on drug administration and blood sampling days.
Figure 1.

Artemisinin dosing schedules in the two study arms. Subjects in group A (denoted A1 to A12) received single daily doses of 500 mg for five days, whereas group B subjects (denoted B1-B12) received escalating doses of 100 +100 +250 +250 +500 mg artemisinin on the first 5 days. Two subjects from each group received an additional single dose of 500 mg on either of the days 7, 10, 13, 16, 20, or 24. All doses were administered as a single dose in the morning
Saliva sampling
Two millilitres of unstimulated saliva was collected from each subject and transferred to 3.6-ml plastic cryo tubes (Nunc, Hereford, UK) at 5 min before and at 30, 60, 90, 120, 150, 180, 240, 300, 360, 420, and 480 min after drug administration on days 1, 3, 5, and on the returning day. All samples were immediately frozen and kept at −20 °C prior to transport to Sweden, where they were kept at −80 °C prior to artemisinin analysis.
Determination of artemisinin
All samples from each subject were analysed in the same run. Saliva artemisinin concentrations were determined using HPLC with a column switching system allowing direct injection of samples [19]. The limit of determination of the assay was 2 ng ml−1 and the intraday coefficient variation was 11%.
Pharmacokinetic analysis
Different structural models were fitted to the log-transformed saliva concentration-time data from all individuals simultaneously, using the first-order method (FO) without centring in the nonlinear mixed effects modelling program NONMEM, version VI [20]. Discriminations between hierarchical models were based on the objective function value (OFV) provided by NONMEM at a significance level of 0.05, equal to a decrease of 3.84 in the OFV, graphical analysis of residuals, and predictions in model diagnostics using Xpose, version 3.0 [21].
The induction model for artemisinin consisted of a PK component and an enzyme component, with the former influencing the concentrations of the enzyme and the latter influencing the concentration of artemisinin. During the early model-building phase several structural models were tested. With respect to the enzyme model, one-compartment models with hepatic drug concentrations stimulating the synthesis rate, or inhibiting the rate of elimination were tried, with stimulation/inhibition described by linear or nonlinear (Emax) functions. In addition, a model with hepatic drug concentrations stimulating the synthesis of a precursor to the enzyme was tried. The absorption of artemisinin from gut to hepatic compartment was assumed to be first-order, with or without a lag-time. Distribution between the hepatic and the sampling compartment was assumed to be first-order. Elimination was also assumed to take place from the hepatic compartment as linear, nonlinear (Michaelis-Menten) or mixed linear and nonlinear processes. Intrinsic CL was assumed to be proportional to the concentration of enzymes, and hepatic elimination was alternatively described by a well-stirred or parallel tube model taking into account hepatic plasma/blood flow and artemisinin plasma protein binding. The final model, which incorporated a nonlinear hepatic elimination with a linear stimulating effect of hepatic artemisinin concentrations on the production of enzyme precursors, is depicted in Figure 2 and described in further detail below.
Figure 2.

Schematic diagram of the induction model applied to saliva artemisinin concentration data. kENZ: zero-order production rate constants for the enzyme precursor or first order elimination rate of the metabolizing enzymes, kPRE: first-order production rate constant for the metabolizing enzymes, CLint: intrinsic clearance, fu: plasma unbound fraction, QH: hepatic plasma flow, EH: extraction ratio, FH: bioavailability from the liver compartment to the sampling compartment, ka: absorption constant rate, kSH: transfer rate constant of artemisinin from the sampling compartment to the hepatic compartment (set equal to QH/Vs, VS being the volume of distribution of the sampling compartment), CLH: hepatic clearance, VH: volume of the liver compartment (set equal to 1), SIND: slope of the inducing effect of artemisinin hepatic concentration on the production rate of enzyme precursor
The final model, applied to log-transformed concentration vs. time data from all subjects and all sampling days, consisted of one part describing the pharmacokinetics of artemisinin and another the time-variant concentrations of inducible enzyme(s), with the two compartments being linked in a circular fashion. In this model, artemisinin is introduced to the system in a gut compartment with subsequent absorption into a liver compartment, where elimination is described by a well-stirred model. The compound is further distributed into a sampling compartment, which constitutes the entire body, except for the liver. In this case sampling is from saliva, thus it is assumed that saliva concentrations are in instantaneous equilibrium with plasma as well as all other tissues except liver, to which distribution occurs. The rate of change of the amount of artemisinin in the liver compartment was described as follows:
| (1) |
Where AH and CH are the amounts and concentrations of the compound in the liver compartment, ka is the absorption rate constant, and AG is the amount of the compound in the gut compartment (equal to dose at time zero). QH was set to 0.63 × body weight (in kg) as a default to mimic hepatic plasma flow, but higher values of QH were also used to assess the influence of this value on the goodness-of-fit and on other parameter estimates. The transfer rate constant kSH of artemisinin from the sampling compartment to the hepatic compartment was estimated as QH/VS, VS being the volume of distribution of the sampling compartment, and AS the amount of the artemisinin in the sampling compartment. The hepatic volume (VH) was fixed to a default value of 1l, but this parameter was also given other values as part of a sensitivity analysis. The hepatic extraction ratio (EH), the hepatic CL (CLH), and the fraction of absorbed drug escaping hepatic first-pass extraction (FH) were defined according to a well-stirred model:
| (2) |
| (3) |
| (4) |
| (5) |
where CLint,0 and CLint,t represent the preinduced and time-variable intrinsic clearance, respectively. fu is the unbound plasma fraction of artemisinin, which was given the value 0.14, based on previous findings [18]. AENZ is the amount of the enzyme(s) in the enzyme pool compartment relative to the amounts in the preinduced state and Km is the parameter governing saturable hepatic elimination and represents the hepatic artemisinin concentration at which CLint is half-maximal.
The rate of drug change in the sampling compartment was described by:
| (6) |
Total body volume of distribution at steady state (Vss) and clearance (CL) of artemisinin are described by Equations. In the two-compartment model, the distribution phase will be rapid and of insignificant quantitative importance. The expression for the two rate constants is given by Equation and can be approximated to CL/VH and CL/Vss for the distribution (λ1) and the elimination (λ2) rate constant, respectively.
| (7) |
| (8) |
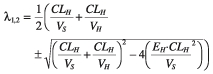 |
The enzyme part of the model consisted of two compartments: the precursor and the enzyme pool. The amount of enzyme was set to 1 for the preinduced state. The change in the precursor pool with time was modelled by letting the amount of drug in the liver to increase the precursor formation rate linearly:
| (10) |
where APRE is the amount in the precursor compartment, kENZ is the first-order enzyme elimination rate constant, which, through the normalization of AENZ to 1 in the preinduced state, is also equal to the zero-order precursor production rate constant. SIND is a slope describing the linear effect of artemisinin concentrations in the liver on the rate of production of enzyme precursor with units of l ng−1, and kPRE is the first-order rate constant of the transformation of precursor to enzyme, the reciprocal of which was defined as mean induction time (MIT). The amount of precursor in the preinduced state was set to kENZ/kPRE. Incorporation of the precursor compartment into the model enabled capture of the time delay between artemisinin administration and elevated amounts of enzyme. Changes in the enzyme pool were defined as follows:
| (11) |
The model was parameterized such that the elimination half-life of the enzymes, t½12,ENZ, rather than kENZ, was estimated.
The absorption lag-time for artemisinin was estimated using log-transformed concentration data from the standard-dose group (group A) on day 1, described by a one-compartment model with oral absorption. The population estimate of the absorption lag-time was used as fixed parameter in the final model.
An exponential variance model was used to describe the interindividual variability (IIV) in the intrinsic clearance and the volume of distribution. Inter-occasional variability (IOV) in the absorption rate and lag-time of artemisinin, expressed in exponential terms, was also incorporated into the model. A residual error model with a proportional component was applied in the final model with the log-transformed data.
The effects of the covariates age, body weight, and smoking habits on different parameters were analysed using general additive models (GAM) [22], implemented in Xpose.
Results
Artemisinin was well tolerated by all subjects with no reported adverse effects. Every subject completed the protocol. Owing to mechanical problems during the HPLC analysis of the samples, data from one subject from the standard dosing group could not be included in the final analysis. This subject had received the final dose on day 24 after the beginning of study. Furthermore, artemisinin concentrations in several samples collected at 30 min after drug intake were abnormally high in several subjects. Similar observations have been reported in other studies involving saliva sampling [10, 18] and are believed to be due to residual contamination of the drug after oral intake. All concentration data obtained at this time point were considered unreliable and were not used in the data analysis.
Saliva artemisinin concentrations in the first period of the study (including 5 consecutive days of dosing) decreased with each new dose (Figure 3). Consequently, dose-normalized artemisinin AUCs decreased with time during the continuous administration of the compound (Figure 4). A large variability in the pharmacokinetics was observed between the different occasions that artemisinin was administered.
Figure 3.

Individual artemisinin saliva concentrations vs. time in the two study arms (Group A: upper figure, Group B: lower figure)
Figure 4.

Dose-normalized salivary artemisinin AUCs in the two study arms (Group A: thick lines, Group B: thin lines)
The concentration-time profiles of artemisinin were best described by the proposed final model (Figures 2 and 5) with both the nonlinear elimination and the enzyme precursor compartments contributing significantly to improving the goodness-of-fit. A model with drug influencing the elimination, rather than production of enzymes did not describe the data accurately. Furthermore, models with mixed linear and nonlinear elimination did not further improve the fit to the data and neither did a model, where the influence of hepatic drug concentration on the rate of synthesis of the precursor was described by an Emax, rather than a linear function. The population estimates of mean time of enzyme induction and enzyme half-life were 1.85 and 37.9 h, respectively (Table 1). The intrinsic clearance of artemisinin at low concentrations (CH < < Km) was estimated to be 2880 l h−1 before the onset of induction, resulting in a preinduced hepatic extraction rate of 0.93. The volume of distribution, based on saliva sampling, was estimated to be 48.8 l. The contribution of the drug in the liver compartment to Vss is negligible (about 2%) and therefore Vss is almost equal to Vs. The model-predicted saliva elimination half-life for artemisinin was 1.0 h in the uninduced state. High interoccasional variability was found for both the absorption lag times and absorption rate constants.
Figure 5.

Logarithm of the population prediction (PRED) and individual predictions (IPRED) vs. observations (DV)
Table 1.
Typical pharmacokinetic parameter values for artemisinin and associated interoccasional (IOV) and interindividual (IIV) variability in 23 healthy Vietnamese subjects
| Parameter (unit) | Estimate (RSE%) | IOV (RSE%) | IIV (RSE%) |
|---|---|---|---|
| t½,ENZ (h) | 37.9 (22) | NE | NE |
| SIND (l ng−1) | 0.018 (27) | NE | NE |
| CLint,0 (L h−1) | 2880 (27) | NE | 0.32 (44) |
| Vs (L) | 48.8 (20) | NE | NE |
| Lag-time (h) | 0.50 (FIXED) | 2.5 (39) | NE |
| ka (h−1) | 0.18 (30) | 0.55 (32) | NE |
| fu | 0.14 (FIXED) | NE | NE |
| Km (ng ml−1) | 1370 (70) | NE | NE |
| MIT (h) | 1.85 (40) | NE | NE |
| Residual error | 0.5 (13) | NE | NE |
t½12,ENZ: enzyme elimination half-life; SIND: slope of the inducing effect of artemisinin hepatic concentration on the production rate of enzyme precursor; CLint: intrinsic clearance; Vs: volume of sampling compartment; Lag-time: absorption lag-time; ka: absorption constant rate; fu: plasma unbound fraction; Km: hepatic artemisinin concentration resulting in 50% of maximum intrinsic clearance; MIT: Mean induction time; RSE%: Relative standard error in per cent.
Sensitivity analyses were performed with respect to the parameters VH, QH and fu, where each was varied one at a time and changes in OFV and parameter estimates were noted. Changing fu from 0.14 to 0.10 or 0.20 only resulted in marginal changes in OFV (about 1 unit) and small changes in the parameter values. The default value of QH (0.63 l h−1 kg−1) corresponds approximately to hepatic plasma flow. Changing QH to 1.2 l h−1 kg−1, corresponding approximately to hepatic blood flow, which is the other possible extreme value QH could reasonably take, resulted in a nonsignificant change in OFV but an approximate doubling of the values for SIND and VS. Increasing VH from 1l to values of 5 and 10l, increased the OFV by 10 and 12 units and was associated with a decrease in Vs. None of the tested covariates had any significant effect on any of the parameters of the pharmacokinetic or enzyme part of the model.
The typical values for the estimated model parameters (Table 1) were used to simulate the artemisinin concentration-time profile for two typical subjects (body weight = 51 kg), one receiving 500 mg artemisinin for 5 consecutive days, and the other receiving the alternative daily doses of 100 +100 +250 +250 +500 mg for 5 consecutive days. Both subjects received a further dose of 500 mg on day 20 (Figure 6). Model-estimated relative changes in the amounts of enzyme for all individual subjects are depicted. The approximately 10-fold increase in enzyme that occurs in the high dose group is predicted to lead to an increase in EH from 0.93 to almost 0.99.
Figure 6.

Model-simulated concentration-time data for two typical individuals receiving oral artemisinin doses of: A) 500 mg daily on days 1-5 and day 20, and B) 100 +100 +250 +250 +500 +500 mg on days 1–5 and day 20. Artemisinin concentrations on days 1, 3, 5, and 20 are shown
Since NONMEM VI is a beta version, the final model was re-run using NONMEM V. The two versions gave the same objective function values, and the parameter estimates differed only on the third significant figure.
Discussion
In the present study we monitored artemisinin concentrations in saliva samples collected from healthy subjects during 5 consecutive days of administration of two different regimens of the compound followed by a further single dose given after various washout periods. The ease of collecting and handling saliva samples enabled us to obtain frequent measurements during 4 out of the 6 days of administration. This allowed for the characterization of the pharmacokinetics of artemisinin during both the induction period and the period of the return of the enzyme activity to its preinduced values.
In accordance with previous studies, a decrease in dose-normalized saliva artemisinin concentrations during the 5 days of drug administration was observed, indicating auto-induction of metabolism [10]. An increase in the rate of enzyme synthesis is the most common mechanism for many inducing agents [23, 24]. In the present study, a model assuming an increase in the amount of the enzymes in an enzyme pool was applied to describe the autoinduction of artemisinin metabolism. In our model the intrinsic clearance of artemisinin was proportional to the amount of enzymes in the enzyme pool compartment. The increase in the amount of enzyme resulted from an increase in the rate of production of precursor, driven by the amount of artemisinin in the liver. Thus, elevated amounts of enzyme due to induction resulted in increased hepatic extraction of artemisinin, accelerating its elimination from the liver. This model described the data better than the other models tested (see Methods).
In our model we have separated the sampling compartment from the liver compartment. Multiple compartments are normally used to explain multiexponential disposition. However, in this case the division into two compartments was not made for describing bi-exponential decay but to account for the effect of artemisinin on hepatic enzymes during its absorption phase. Our model assumes that hepatic enzymes are affected by the hepatic concentration of drug and that a significant contribution derives from presystemic exposure. Thus, each consecutive oral dose of artemisinin will affect the enzymes to a similar degree, in contrast to a successively diminished systemic exposure, driven by the autoinduction. Furthermore, the model allows the effects of enzyme induction on clearance and bioavailability to be accounted for in a way that would not be possible in a one-compartment disposition model. The use of two disposition compartments, despite a mono-exponential elimination phase, was made possible by the introduction of physiologically based information into the model. It was assumed that VH was considerably smaller than VS, as larger values of VH gave rise to poorer prediction of the observed data. The intercompartmental clearance was selected based on knowledge about the hepatic plasma/blood flow. Under normal circumstances the most appropriate assumption would be to equate QH with hepatic blood flow. However, for artemisinin, little drug can be expected to be released from erythrocytes to be available for hepatic elimination, and thus hepatic plasma flow is probably more appropriate. There was no noticeable difference in the goodness-of-fit between QH fixed to hepatic plasma flow compared with hepatic blood flow.
According to the well-stirred model, the preinduced intrinsic hepatic clearance of artemisinin was estimated to be 2880 l h−1 at hepatic concentrations below the Km value (1370 ng ml−1). The hepatic extraction ratio of the compound was 93%, resulting in a bioavailability of 7%. The high extraction ratio of artemisinin is in accordance with previous reports [5, 6, 8]. Thus, increased intrinsic clearance due to auto-induction would not result in any major change in the systemic clearance of artemisinin, but would decrease its bioavailability.
Artemisinin exhibited a 2-fold higher oral clearance values in malaria patients receiving a first dose of 100 mg of the compound compared with those administered 500 mg [10]. Our final model incorporated a concentration-dependent intrinsic clearance term for artemisinin to account for this observation as well as the dose-dependency of the data from the present study.
Our model estimated a mean induction time of 1.9 h, indicating the phenomenon to be occurring after the first dose. However, this rapid initiation of induction would not have an effect on the first dose, since the induction is mostly manifested as an increased first pass metabolism of the compound and most of the dose will have been absorbed prior to its onset. In a previous report a single oral dose of artemisinin was found sufficient to affect the pharmacokinetics of a second dose administered 1 week later [11]. In another study, induction of artemisinin metabolism in healthy subjects during a 7-day period was investigated. Artemisinin AUCs on days 4 and 7 were observed to decrease to 43% and 25% of those on the first day [6].
The model estimated an enzyme elimination half-life of 37.9 h. Thus, after a last dose of artemisinin, it would take 6–8 days for the enzyme activity to return to its preinduction values. This is in accordance with published studies, where 2–3 weeks of wash-out period were found to result in normal enzyme activities compared with day 1 [6,7]. There is not much known about the metabolizing enzymes responsible for the elimination of artemisinin that are induced upon its administration. Therefore, the model-estimated half-life could account for a mix of several enzymes. Since the information on the kinetics of the enzyme induction is based on artemisinin concentrations, there is a theoretical risk that a long half-life of artemisinin would mask a shorter enzyme half-life. However, artemisinin's short half-life of 1 h for artemisinin, does not rate-limit the degradation kinetics of the induced enzyme(s) whereas the enzyme half-life is estimated to be 37.9 h. A less likely scenario would be the presence of artemisinin metabolite(s) with longer half-lives, affecting the elimination half-life of the enzymes.
Induction of drug metabolism has been modelled in several ways. Carbamazepine autoinduction was explained by a model assuming an exponential increase in its metabolic clearance [25], whereas cyclophosphamide elimination was described by a noninducible and an inducible route, the latter being mediated by a hypothetical enzyme compartment [26]. A third approach has been to model the changes in clearance over the induction period, as applied to methadone [27] and isofosfamide [28]. Our model offers two advantages over these models for enzyme induction. Whereas previous approaches predict a change in the clearance of the drug, our model describes the induction in terms of an increase in the intrinsic clearance of the compound, theoretically allowing for the prediction of the time course of drugs with various degrees of extraction. A second feature of our model is the incorporation of the mean induction time, which allows the capture of the lag-time of the induction process, which is often observed experimentally.
Use of saliva sampling in pharmacokinetic studies of artemisinin offers a simple and convenient way of monitoring the time course of artemisinin. Saliva samples are easier to handle and their collection is more acceptable to study subjects than blood sampling. Moreover, changes in saliva concentrations reflect those in plasma. However, care must be taken to rinse the mouth thoroughly after oral intake of the drug. In this study, a few samples at 30 min after the dose were found to be contaminated by artemisinin present in the mouth. One disadvantage is that saliva artemisinin concentrations are generally low and thus require more powerful analytical methods for their measurement.
In conclusion, a semiphysiological model has been developed to describe the autoinduction of metabolism, through increased production of an enzyme precursor for a compound with high hepatic extraction. The proposed model was successfully applied to saliva concentration-time data from healthy subjects, receiving two different dosage regimens of artemisinin. Our model provides estimates of the parameters describing the kinetics of both drug and inducible enzymes.
Acknowledgments
This work was supported in part by the Swedish International Development Cooperation Agency.
References
- 1.van Agtmael MA, Eggelte TA, van Boxtel CJ. Artemisinin drugs in the treatment of malaria: from medicinal herb to registered medication. Trends Pharmacol Sci. 1999;20:199–205. doi: 10.1016/s0165-6147(99)01302-4. [DOI] [PubMed] [Google Scholar]
- 2.Alin MH, Ashton M, Kihamia CM, Mtey GJ, Björkman A. Clinical efficacy and pharmacokinetics of artemisinin monotherapy and in combination with mefloquine in patients with falciparum malaria. Br J Clin Pharmacol. 1996;41:587–92. doi: 10.1046/j.1365-2125.1996.35116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Augustijns P, D'Hulst A, Van Daele J, Kinget R. Transport of artemisinin and sodium artesunate in Caco-2 intestinal epithelial cells. J Pharm Sci. 1996;85:577–9. doi: 10.1021/js960001i. [DOI] [PubMed] [Google Scholar]
- 4.Svensson USH, Sandstrom R, Carlborg O, Lennernas H, Ashton M. High in situ rat intestinal permeability of artemisinin unaffected by multiple dosing and with no evidence of P-glycoprotein involvement. Drug Metab Dispos. 1999;27:227–32. [PubMed] [Google Scholar]
- 5.Ashton M, Sy ND, Gordi T, Hai TN, Thach DC, Farah MH, et al. Evidence for time-dependent artemisinin kinetics in adults with uncomplicated malaria. Pharm Pharmacol Lett. 1996;6:127–30. [Google Scholar]
- 6.Ashton M, Hai TN, Sy ND, Huong DX, Huong NV, Nieu NT, et al. Artemisinin pharmacokinetics is time-dependent during repeated oral administration in healthy male adults. Drug Metab Dispos. 1998;26:25–7. [PubMed] [Google Scholar]
- 7.Svensson US, Ashton M, Hai TN, Bertilsson L, Huong DX, Huong NV, et al. Artemisinin induces omeprazole metabolism in human beings. Clin Pharmacol Ther. 1998;64:160–7. doi: 10.1016/S0009-9236(98)90149-7. [DOI] [PubMed] [Google Scholar]
- 8.Alin MH, Ashton M, Kihamia CM, Mtey GJ, Björkman A. Multiple dose pharmacokinetics of oral artemisinin and comparison of its efficacy with that of oral artesunate in falciparum malaria patients. Trans R Soc Trop Med Hyg. 1996;90:61–5. doi: 10.1016/s0035-9203(96)90480-0. [DOI] [PubMed] [Google Scholar]
- 9.Ashton M, Sy ND, Huong NV, Gordi T, Hai TN, Huong DX, et al. Artemisinin kinetics and dynamics during oral and rectal treatment of uncomplicated malaria. Clin Pharmacol Ther. 1998;63:482–93. doi: 10.1016/S0009-9236(98)90044-3. [DOI] [PubMed] [Google Scholar]
- 10.Gordi T, Huong DX, Hai TN, Nieu NT, Ashton M. Artemisinin pharmacokinetics and efficacy in uncomplicated-malaria patients treated with two different dosage regimens. Antimicrob Agent Chemother. 2002;46:1026–31. doi: 10.1128/AAC.46.4.1026-1031.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashton M, Gordi T, Hai TN, Huong NV, Sy ND, Nieu NT, et al. Artemisinin pharmacokinetics in healthy adults after 250, 500 and 1000 mg single oral doses. Biopharm Drug Dispos. 1998;19:245–50. doi: 10.1002/(sici)1099-081x(199805)19:4<245::aid-bdd99>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 12.Zhang SQ, Hai TN, Ilett KF, Huong DX, Davis TM, Ashton M. Multiple dose study of interactions between artesunate and artemisinin in healthy volunteers. Br J Clin Pharmacol. 2001;52:377–85. doi: 10.1046/j.0306-5251.2001.01461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Agtmael MA, Cheng-Qi S, Qing JX, Mull R, van Boxtel JC. Multiple dose pharmacokinetics of artemether in Chinese patients with uncomplicated falciparum malaria. Int J Antimicrob Agents. 1999;12:151–8. doi: 10.1016/s0924-8579(99)00063-1. [DOI] [PubMed] [Google Scholar]
- 14.Ezzet F, Mull R, Karbwang J. Population pharmacokinetics and therapeutic response of CGP 56697 (artemether + benflumetol) in malaria patients. Br J Clin Pharmacol. 1998;46:553–61. doi: 10.1046/j.1365-2125.1998.00830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khanh NX, de Vries PJ, Ha LD, van Boxtel CJ, Koopmans R, Kager PA. Declining concentrations of dihydroartemisinin in plasma during 5-day oral treatment with artesunate for falciparum malaria. Antimicrob Agents Chemother. 1999;43:690–2. doi: 10.1128/aac.43.3.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svensson US, Ashton M. Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br J Clin Pharmacol. 1999;48:528–35. doi: 10.1046/j.1365-2125.1999.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sidhu JS, Ashton M. Single-dose, comparative study of venous, capillary and salivary artemisinin concentrations in healthy, male adults. Am J Trop Med Hyg. 1997;56:13–6. doi: 10.4269/ajtmh.1997.56.13. [DOI] [PubMed] [Google Scholar]
- 18.Gordi T, Hai TN, Hoai NM, Thyberg M, Ashton M. Use of saliva and capillary blood samples as substitutes for venous blood sampling in pharmacokinetic investigations of artemisinin. Eur J Clin Pharmacol. 2000;56:561–6. doi: 10.1007/s002280000179. [DOI] [PubMed] [Google Scholar]
- 19.Gordi T, Nielsen EYuZ, Westerlund D, Ashton M. Direct analysis of artemisinin in plasma and saliva using coupled- column high-performance liquid chromatography with a restricted-access material pre-column. J Chromatogr B Biomed Sci Appl. 2000;742:155–62. doi: 10.1016/s0378-4347(00)00156-0. [DOI] [PubMed] [Google Scholar]
- 20.Beal SL, Sheiner LB. Introductory guide. San Francisco: NONMEM Project Group; 1998. NONMEM users guide. [Google Scholar]
- 21.Jonsson EN, Karlsson MO. Xpose- an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comp Meth Prog Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 22.Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic/pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm. 1992;20:511–28. doi: 10.1007/BF01061469. [DOI] [PubMed] [Google Scholar]
- 23.Bock KW, Lipp HP, Bock-Hennig BS. Induction of drug-metabolizing enzymes by xenobiotics. Xenobiotica. 1990;20:1101–11. doi: 10.3109/00498259009046831. [DOI] [PubMed] [Google Scholar]
- 24.Whitlock JP. The regulation of cytochrome P-450 gene expression. Ann Rev Pharmacol Toxicol. 1986;26:333–69. doi: 10.1146/annurev.pa.26.040186.002001. [DOI] [PubMed] [Google Scholar]
- 25.Levy RH, Dumain MS, Cook JL. Time-dependent kinetics. V. Time course of drug levels during enzyme induction (one-compartment model) J Pharmacokinet Biopharm. 1979;7:557–78. doi: 10.1007/BF01061209. [DOI] [PubMed] [Google Scholar]
- 26.Hassan M, Svensson US, Ljungman P, Bjorkstrand B, Olsson H, Bielenstein M, et al. A mechanism-based pharmacokinetic-enzyme model for cyclophosphamide autoinduction in breast cancer patients. Br J Clin Pharmacol. 1999;48:669–77. doi: 10.1046/j.1365-2125.1999.00090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rostami-Hodjegan A, Wolff K, Hay AW, Raistrick D, Calvert RR, Tucker GT. Population pharmacokinetics of methadone in opiate users: characterization of time-dependent changes. Br J Clin Pharmacol. 1999;48:43–52. doi: 10.1046/j.1365-2125.1999.00974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Body AV, Cole M, Pearson AD, Idle JR. The kinetics of the auto-induction of ifosfamide metabolism during continuous infusion. Cancer Chemother Pharmacol. 1995;36:53–60. doi: 10.1007/BF00685732. [DOI] [PubMed] [Google Scholar]