

Abstract
Aims
To evaluate the extent to which enterohepatic recycling circulation contributes to moxifloxacin bioavailability in healthy, males by administration of activated charcoal and to evaluate the efficacy of activated charcoal administration in decreasing systemic concentrations of moxifloxacin in the event of overdose.
Methods
Nine healthy males, mean age 34 years (range 23–45 years) participated in a single centre, randomized, nonplacebo-controlled, three way crossover study. The pharmacokinetics of moxifloxacin in plasma and urine were determined for up to 96 h following a 400 mg single dose randomly administered on three separate occasions with a minimum washout phase of 1 week. Treatment A was 400 mg moxifloxacin IV as a 1 h infusion, treatment B was 400 mg moxifloxacin IV as a 1 h infusion with oral activated charcoal (5 g directly before the start of the infusion, 5 g immediately after the end of the infusion, and 10 g at 2, 4 and 8 h after the start of the infusion), treatment C was 400 mg oral moxifloxacin with activated charcoal (10 g 15 min before and at 2, 4 and 8 h after drug administration). The subjects underwent a series of clinical and laboratory tests.
Results
Single 400 mg doses of moxifloxacin (PO and/or IV) were safe and well tolerated. The bioavailability of moxifloxacin was significantly decreased when given with charcoal (AUC = 35.5 (IV reference) vs 5.40 (PO) vs 28.5 (IV) mg l−1 h). Concurrently peak concentrations were lowered Cmax = 3.38 (IV reference) vs 0.62(PO) vs 2.97 (IV) mg l−1) by approximately 85% (P < 0.05) following oral administration and by 20% after IV treatment (P < 0.05). Bioavailability amounted to 15.4% (95% confidence interval 9.6, 25.0%) for treatment B while it was 80.4% (95% confidence interval 76.3.6, 84.6%) for treatment C. Terminal half-lives were not affected. The kinetics of urinary excretion corroborated these findings.
Conclusions
The results of this study show that moxifloxacin undergoes pronounced enteric recycling after systemic uptake. In addition, these findings confirm that activated charcoal may be useful in treating moxifloxacin overdose by preventing its absorption.
Keywords: activated charcoal, moxifloxacin, pharmacokinetics
Introduction
Activated charcoal is important in clinical practice as a tool for the management of drug or chemical overdose. Enterally administered compounds are effectively bound due to its large surface and high adsorption capacity thereby preventing systemic uptake from the gastrointestinal tract. A number of pharmacokinetic studies in healthy subjects have been conducted to evaluate the efficacy of antidotes in the management of drug overdose [1–9].
Activated charcoal has also been used to understand better the pharmacokinetics of fluoroquinolones. Based on animal and in vitro data these drugs have been found to undergo transepithelial transport processes leading to either transintestinal elimination or recirculation [10–13, 15, 16] It has been shown that transepithelial transport of these drugs occurs in humans in vivo [17–23]. Charcoal has been used extensively to elucidate the enteral transport of ciprofloxacin which is predominantly excreted via active renal elimination. Ideally, a comparison of the mass balance in healthy subjects with that of patients with impaired renal elimination suggested that the drug underwent transintestinal elimination [17]. Subsequently, oral administration of activated charcoal was given to block the (re) absorption of ciprofloxacin administered as a single intravenous infusion to healthy subjects. Eighteen percent of the drug was shown to undergo intestinal elimination [18, 19]. Charcoal has also been used as a probe to predict the pharmacokinetic behaviour of fluoroquinolone metabolites [24, 25].
Moxifloxacin is currently used worldwide in the treatment of respiratory tract infections and has received recent approval for the sequential intravenous and oral treatment of hospitalized patients. It is a fourth generation fluoroquinolone, and has improved activity against Gram-positive cocci, aerobic, anaerobic, intracellular bacteria, and ‘atypical’ organisms such as Mycoplasma and Chlamydia compared with third generation agents. Its pharmacokinetic properties are characterized by complete absorption, high bioavailability (approximately 91%) and rapid penetration into target tissues. Moxifloxacin is cleared both renally and by the liver both as unchanged drug and conjugated metabolites [26, 28, 30] This allows once daily oral dosing without the need to adjust the dose in renal impairment or liver insufficiency [26, 29].
This study aimed to determine the efficacy of activated charcoal to prevent absorption of moxifloxacin in the event of overdose. In addition, we investigated the hypothesis that the drug undergoes recirculation. Earlier studies suggested this and also that the absorption characteristics of moxifloxacin along the gastrointestinal tract may differ from other quinolones [13, 14, 16].
Methods
Subjects
After having received detailed information about the study, nine healthy males (mean age 34, range 23–45 years, body mass index mean ± s.d. 26.5 ± 2.9, range 21.7–30.8) were enrolled. A sample size of at least six subjects was considered sufficient for accurate statistical assessment of the study objectives with adequate accuracy in this exploratory trial, assuming a within-subject coefficient variation of the log-transformed AUC of approximately 10%, which corresponds to previous clinical pharmacokinetic experience [28, 30]. Each subject provided written informed consent, and the study protocol was approved by an independent ethics committee (North Rhine Medical Council, Düsseldorf, Germany). The study was conducted in accordance with the Declaration of Helsinki (1964) in the revised version of 1996 (Somerset West), the ICH GCP Guideline (Note for Guidance on Good Clinical Practice) and the German drug law (AMG).
Design
The study had a single-centre, single-dose, randomized, nonblinded, nonplacebo-controlled, crossover design. Subjects received the following dosing regimens on separate occasions after an overnight fast:
400 mg moxifloxacin IV as a 1 h infusion,
400 mg moxifloxacin IV as a 1 h infusion with oral administration of activated charcoal (5 g immediately before starting the infusion, 5 g at the end of infusion, and 10 g at 2, 4 and 8 h after the start of infusion),
400 mg moxifloxacin orally with oral administration of activated charcoal (10 g each 15 min before and at 2, 4 and 8 h after drug administration).
The three treatments were separated by a minimum washout period of at least 1 week. The timing of charcoal administration was optimized to extend the interaction with moxifloxacin over a period accounting for (a) more than 90% of systemic drug exposure based on the absorption characteristics of the drug [14, 16], (b) an expected transit time of charcoal in the gut of approximately 24 h [32], and (c) data showing that 1 g of charcoal is able to completely adsorb 400 mg of moxifloxacin.
Blood and urine sampling
Venous blood samples were taken before the administration of moxifloxacin (0 h) and then at 0.5, 0.75, 1, 1.25, 1.5, 2, 2.25, 2.5, 3, 4, 4.25, 4.5, 5, 6, 8, 8.25, 8.5, 12, 24, 48, 72 and 96 h after dosing to determine plasma moxifloxacin concentrations. Venous blood (3 ml) was collected into NH4-heparinate coated tubes, and the samples were centrifuged within 15 min of collection for 5 min at 1600 g at 5 °C. The plasma samples were then transferred to polypropylene tubes, frozen and stored at −20 °C until analysis.
Urine was collected before drug administration (blank) and then at 0–4, 4–8, 8–12, 12–24, 24–48, 48–72 and 72–96 h after dosing. The volume of each urine fraction was recorded and a 5 ml aliquot was frozen and stored at −20 °C until analysis.
Analysis of moxifloxacin
Plasma and urine concentrations of moxifloxacin were determined by a validated HPLC method with fluorescence detection [27]. The limit of quantification was 10 µg l−1. Quality control samples produced from blank plasma spiked with known concentrations of the drug (3 mg l−1, 0.5 mg l−1, 30 µg l−1) were stored and analyzed together with the study samples. In plasma and urine interday precision (specification ≤ 20% throughout the working range) ranged from 1.3 to 5.4%, and 3.0–5.1%, respectively. Accuracy (specification 90–110% throughout the working range) was always within the range 100.9–105.7% in plasma and 102.4–108.5% in urine.
Pharmacokinetic analysis
Pharmacokinetic parameters were calculated using noncompartmental methods [31]. Peak concentrations (Cmax) and time to reach Cmax (tmax) were determined directly from individual plasma concentration vs time curves. AUC (area under the curve after single dose administration extrapolated to infinity) determination was based on the linear trapezoidal rule in the ascending part of the plasma concentration vs time curve, and the log-linear trapezoidal rule in the descending part. λz (the apparent terminal log-linear disposition rate constant) was derived from the terminal slope of the logarithmic plasma concentration vs time profile. t1/2 (apparent terminal half-life) was obtained by linear regression analysis after log-transformation of the data using not less than three data points. AUCnorm and Cmax,norm were calculated by normalizing the data to the dose and the body weight. The mean residence time (MRT) was determined as AUMC/AUC, where AUMC is the area under the first moment of the concentration-time curve determined by integrating the product of time and concentration from 0 to infinity. The volume of distribution at steady state was calculated according to Vss = CL × MRT (CL = Dose/AUC after intravenous administration). The amount of excreted unchanged drug into urine (Aeur) and the renal clearance (CLR calculated from the expression 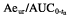 , where tn is the time of the last data point) were also determined.
, where tn is the time of the last data point) were also determined.
Statistical analysis
AUC and Cmax were analyzed assuming log-normally distributed data. To compare pharmacokinetics between treatments, the logarithms of these parameters were analyzed using analysis of variance (anova) including sequence, subject (sequence), period and treatment effects. Based on these analyses, point estimates (LS-Means) and exploratory two-sided 90% confidence intervals for the ratios ‘IV treatment with charcoal’ and ‘PO treatment with charcoal’, respectively, vs‘IV reference treatment’ were calculated by re-transformation of the logarithmic data using the intra-individual standard deviation of the anova.
Results
Of the nine subjects enrolled in this study, seven completed all of the treatment regimens and two subjects dropped out prior to moxifloxacin administration. The treatments were well tolerated, and only eight adverse events were reported by four subjects. Treatment related adverse events were reported by two subjects during each of the three treatments. All the adverse events were of mild severity, their relationship to the study medication was judged as ‘none’ (hypertonia, pharyngitis, maculopapular rash) or ‘unlikely’ (two cases of headache, ear pain/tinnitus). Treatment was given for headache (1 × 1000 mg paracetamol), rash (triamcinolone acetonide several times daily), and ear pain/tinnitus (Panotile® 3 × 2 drops a day). All adverse events resolved completely. A few mild and transient elevations of laboratory parameters were observed, but no clear pattern of onset or duration was apparent. The changes were either pre-existing or occurred during the follow up and therefore were not considered to be clinically relevant. ECG investigations showed a QTc prolonging effect of the order of magnitude observed in previous studies. There was no clinically relevant influence of the study drug on vital signs.
The plasma moxifloxacin concentration vs time curves for each treatment group are shown in Figure 1 and pharmacokinetic parameters are summarized in Table 1. Plasma concentrations following intravenous treatment were generally detectable for at least 72 h, and those after oral administration with charcoal up to 48 h in the majority of the cases. Co-administration of activated charcoal markedly decreased plasma concentrations (IV > IV + charcoal >> PO + charcoal). In all three treatments, a biphasic decline in plasma moxifloxacin concentrations was detectable in the terminal phase. This was less pronounced during oral treatment due to the absence of plasma concentration data beyond 48 h. No obvious differences in shape of these plots were detectable at the times of charcoal administration (10 g during dose administration and at 2, 4 and 8 h).
Figure 1.

Plasma-concentration vs time profiles for moxifloxacin following a single 400 mg dose given intravenously or orally with and without co-administration of charcoal (geometric mean, SD). A: 400 mg IV as 1 h infusion, B: 400 mg IV as a 1 h infusion with charcoal, C: 400 mg PO with charcoal. 400 mg i.v. (▴); 400 mg i.v. + charcoal (□); 400 mg p.o. + charcoal (•)
Table 1.
Pharmacokinetic parameters for moxifloxacin in plasma and urine following a single oral dose of 400 mg drug with oral activated charcoal (combi PO), and given intravenously with (combi IV) and without (mono IV) oral activated charcoal, respectively (n = 7, geometric mean/SD, range)
| Parameter | Mono IV | Combi IV | Combi PO |
|---|---|---|---|
| AUC (mg l−1 h) | 35.5/1.15 (31.1–46.8) | 28.5/1.13 (25.3–35.8) | 5.40/1.92 (2.98–18.2) |
| AUC(0,12 h) (mg l−1 h) | 19.4/1.10 (17.5–23.1) | 16.9/1.08 (15.92–19.2) | 3.18/1.96 (1.51–10.7) |
| Cmax (mg l−1) | 3.38/1.22 (2.71–4.13) | 2.97/1.13 (2.37–3.44) | 0.618/2.07 (0.211–1.84) |
| tmax† (h) | 1.00 (0.75–1.50) | 1.00 (0.50–1.50) | 0.75 (0.50–1.25) |
| t1/2,24 h (h) | 10.7/1.10 (9.0–11.7) | 8.8/1.11 (7.6–10.1) | 9.6/1.09 (8.9–11.5) |
| t1/2, 48 h (h) | 11.6/1.11 (10.0–14.0) | 11.8/1.06 (10.9–12.8) | 10.8/1.12 (9.5–13.2) |
| t1/2, 72 h (h) | 12.6/1.10 (11.7–15.5) | 13.0/1.07 (11.6–13.9) | 11.2/1.16 (9.6–13.8) |
| t1/2 (h) | 14.3/1.18 (11.7–17.5) | 14.3/1.13 (12.6–17.0) | 11.2/1.16 (9.58–13.8) |
| Vss (l kg−1) | 2.01/1.14 (1.55–2.32) | 2.28/1.15 (1.78–2.74) | |
| CL, CL/F (l h−1) | 11.3/1.15 (8.54–12.9) | 14.0/1.13 (11.2–15.8) | 74.1/1.92 (21.9–134) |
| CLR (l h−1) | 3.07/1.19 (2.43–3.84) | 3.34/1.23 (2.43–4.34) | 3.48/1.16 (2.78–4.15) |
| Aeur* (%) | 27.5 ± 5.5 (18.8–34.6) | 23.9 ± 3.7 (19.1–28.2) | 6.1 ± 5.7 (2.27–18.5) |
arithmetic mean ± SD.
Median (range)
The absolute bioavailability of intravenously administered moxifloxacin was significantly decreased by approximately 20% (P < 0.05) when charcoal was given concomitantly with the drug, whereas it decreased by approximately 85% (P < 0.05) after concomitant oral administration (Table 2).
Table 2.
The influence of activated charcoal on the absolute bioavailability of intravenous and oral moxifloxacin in healthy males (LS-means of ratios (see legend) with two-sided 90% confidence intervals on the primary pharmacokinetic parameters AUC and Cmax, n = 7)
| Analyte | Ratio | Parameter | n | Estimated ratio (LS-mean, %) | 95% confidence interval |
|---|---|---|---|---|---|
| Moxifloxacin | Combi PO/Mono IV | AUC | 7 | 15.4 | 9.6, 25.0* |
| Cmax | 7 | 18.5 | 11.9, 28.7* | ||
| Combi IV/Mono IV1 | AUC | 7 | 80.4 | 76.3, 84.6* | |
| Cmax | 7 | 86.6 | 73.0, 102.8 |
Combi PO = 400 mg oral moxifloxacin together with 40 g of activated charcoal given in 4 portions 15 min before, 2, 4 and 8 h after administration, Combi IV = 400 mg intravenous moxifloxacin (1 h infusion) together with 40 g activated charcoal given in 4 portions immediately before, 2, 4 and 8 h start of infusion, Mono IV = reference treatment with 400 mg IV moxifloxacin infused at a constant rate over 1 h.
P < 0.05.
estimates for Combi IV/Mono IV based only on the data of the two IV periods.
Discussion
In this study the effect of charcoal co-administration on the pharmacokinetics of moxifloxacin following oral administration and intravenous infusion of a single 400 mg moxifloxacin doses was investigated in a three arm cross over design using intravenous administration as the reference. In vitro data showing complete binding of moxifloxacin to 1 g of activated charcoal suggested that the 40 mg dose recommended for the management of overdose should be sufficient to quantify its effect on the pharmacokinetics of moxifloxacin using the current study design. An additional fourth oral control treatment was not deemed necessary, since moxifloxacin is known to have high oral bioavailability (91%) and all relevant reference data can be obtained from the IV control.
When activated charcoal was co-administered during a 1 h intravenous infusion of 400 mg moxifloxacin, an absolute bioavailability of approximately 80% was obtained. This decrease in exposure can be explained by the adsorption of part of the moxifloxacin dose undergoing gastrointestinal recycling to the activated charcoal, thus preventing subsequent reabsorption. Previous work has shown that activated charcoal also blocks the reabsorption of other quinolones [18–25]. The results from the intravenous treatment arms of this study further support previous preclinical findings suggesting the occurrence of gastrointestinal circulation for moxifloxacin and/or its metabolites [13].
The latter was blocked to the same extent as that for ciprofloxacin reported previously [19]. In view of the longer terminal half-life of moxifloxacin (approximately 12 h vs 4 h for ciprofloxacin) the schedule for the charcoal administration in the present study was designed to maximize its effect on the bioavailability of moxifloxacin. The charcoal administration was timed to cover the period of maximum gastrointestinal turnover of moxifloxacin [28]. In addition, taking into account that moxifloxacin is also secreted into and absorbed from the upper and lower parts of the gastrointestinal tract, the transit time of charcoal should prevent adsorption of moxifloxacin excreted into the gastrointestinal tract for more than 1 day. Since 90% of systemic exposure to the drug occurs within 36 h, only a small portion of the moxifloxacin dose still being excreted into the gastrointestinal tract would not be adsorbed onto activated charcoal. This can be considered as a minor limitation of the present study design. Administration of higher charcoal doses was not considered because of the associated increased risk of gastro-intestinal side-effects (e.g. obstipation). A comparison of the AUC and AUC(0,12 h) ratios in this study underlines this conclusion (see Table 1).
Activated charcoal is a well documented standardized method for the clinical management of overdose with drugs or chemical agents [6]. It can prevent the absorption of almost all drugs present in the gastrointestinal tract independent of their physico-chemical properties with a few exceptions (e.g. ethanol, cyanides). Since its antidotal efficacy is dependent upon on when and for how long it is given after the overdose, it is recommended that activated charcoal is administered as early as possible. Our results illustrate that charcoal may be considered as an option for the management of moxifloxacin overdose. Thus, concomitant administration of 40 g of activated charcoal with 400 mg oral moxifloxacin leads to the almost total prevention of drug absorption.
In humans, oral moxifloxacin is absorbed within 4 h [30], and there is no indication of delayed absorption for doses of up to 800 mg. The present data suggest that doses of charcoal normally used in the management of overdose are adequate for moxifloxacin.
In summary, moxifloxacin was safe and well tolerated when administered as a single intravenous or oral dose alone or in combination with activated charcoal. Our findings indicate that charcoal can be used to manage moxifloxacin overdose, and suggested the occurrence of gastrointestinal circulation of the drug.
Acknowledgments
This study was conducted by Bayer AG, Wuppertal, Germany. The authors thank Mr Peter Hopfe and Guido Schwarz for their excellent technical support.
The authors would like to dedicate this manuscript to Professor Jochen Kuhlmann on the occasion of his 60th birthday.
References
- 1.Olkolla KT, Neuvonen PJ. Effect of gastric pH on antidotal efficacy of activated charcoal in man. Int J Clin Pharmacol Ther Toxicol. 1984;22:565–9. [PubMed] [Google Scholar]
- 2.Neuvonen PJ, Olkolla KT, Alanen T. Effect of ethanol and pH on the absorption of drugs to activated charcoal: studies in vitro and in man. Acta Pharmacol Toxicol. 1984;54:1–7. doi: 10.1111/j.1600-0773.1984.tb01888.x. [DOI] [PubMed] [Google Scholar]
- 3.Olkolla KT, Neuvonen PJ. Do gastric contents modify antidotal efficacy of oral activated charcoal? Br J Clin Pharmacol. 1985;18:663–9. doi: 10.1111/j.1365-2125.1984.tb02527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olkolla KT, Neuvonen PJ. Effect of cathartics on antidotal efficacy of oral charcoal. Acta Pharmacol Toxicol. 1986;59(Suppl 5: Part 1):223. [Google Scholar]
- 5.Davies RL, Koup JR, Roon RA, Opheim KE, Smith AN. Effect of oral activated charcoal on tobramycin clearance. Antimicrob Agents Chemother. 1988;32:274–5. doi: 10.1128/aac.32.2.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuvonen PJ, Olkkolla KT. Oral activated charcoal in the treatment of intoxications: Role of single and repeated doses; Med Toxicol. 1988;3:33–58. doi: 10.1007/BF03259930. [DOI] [PubMed] [Google Scholar]
- 7.Danel V, Henry JA. Activated charcoal, emesis, and gastric lavage in aspirin overdose. Br Med J. 1988;296:1507. doi: 10.1136/bmj.296.6635.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lapatto-Reinimluoto O, Kivistö KT, Neuvonen PJ. Effect of activated charcoal alone or given after gastric lavage in reducing the absorption of diazepam, ibuprofen and citalopram. Br J Clin Pharmacol. 1999;48:148–153. doi: 10.1046/j.1365-2125.1999.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lapatto-Reinimluoto O, Kivistö KT, Neuvonen PJ. Efficacy of activated charcoal versus gastric lavage half an hour after ingestion of moclobemide, temazepam, and verapamil. Eur J Clin Pharmacol. 2000;56:285–8. doi: 10.1007/s002280000139. [DOI] [PubMed] [Google Scholar]
- 10.Torre D, Sampietro C, Quadrelli C, Bianchi W, Maggiolo F. Effects of orally administered activated charcoal on ciprofloxacin pharmacokinetics in healthy volunteers. Chemotherapia. 1988;7:382–6. [PubMed] [Google Scholar]
- 11.Singh GN, Gupta RP. Effect of absorbents on permeation of norfloxacin. Pharmazie. 1988;43:699–700. [PubMed] [Google Scholar]
- 12.Rubinstein E, Dautrey S, St. Farinoti R, Julien L, Ramon J, Carbon C. Intestinal elimination of sparfloxacin, fleroxacin and ciprofloxacin in rats. Antimicrob Agents Chemother. 1995;39:99–102. doi: 10.1128/aac.39.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siefert HM, Kohlsdorfer C, Steinke W, Witt A. Pharmacokinetics of the 8-methoxyquinolone, moxifloxacin: tissue distribution in male rats. J Antimicrob Chemother 1999. 1999;43(Suppl B):61–7. doi: 10.1093/jac/43.suppl_2.61. [DOI] [PubMed] [Google Scholar]
- 14.Siefert HM, Hucke F, Kohlsdorfer C, Zimmer D. Proceedings of the 39th Interscience Conference on Antimicrobial Agents and Chemotherapy. San Francisco: 1999. Absorption of moxifloxacin, trovafloxacin and ciprofloxacin from rat ascending colon; p. Abstract 1198. [Google Scholar]
- 15.Griffiths N, Hirst BH, Simmons NL. Active intestinal secretion of the fluoroquinolone antibacterials ciprofloxacin, norfloxacin and pefloxacin; a common secretory pathway? J Pharmacol Exp Ther. 1994;269:496–502. [PubMed] [Google Scholar]
- 16.Musafija A, Ramon J, Shtelman Y, Yoseph G, Rubinovitz B, Segev S, Rubinstein E. Trans-epithelial intestinal elimination of moxifloxacin in rabbits. J Antimicrob Chemother. 2000;45:803–5. doi: 10.1093/jac/45.6.803. [DOI] [PubMed] [Google Scholar]
- 17.Rohwedder RW, Bergan T, Thorsteinsson SB, Scholl H. Transintestinal elimination of ciprofloxacin. Chemother. 1990;36:77–84. doi: 10.1159/000238751. [DOI] [PubMed] [Google Scholar]
- 18.Sörgel F, Naber KG, Jaehde U, Mahr G, Muth P, Reiter A. Die gastrointestinale Sektretion von Antibiotika am Beispiel von Ciprofloxacin. Fortschritte Antimikrob Antineoplast Chemother. 1991;10:319–26. [Google Scholar]
- 19.Sörgel F, Naber KG, Jaehde U, Reiter A, Seelmann R, Sigl G. Gastrointestinal secretion of ciprofloxacin, Evaluation of the charcoal model for investigations in healthy volunteers. Am J Med. 1989;87(Suppl 5A):5A–62S. doi: 10.1016/0002-9343(89)90025-9. [DOI] [PubMed] [Google Scholar]
- 20.Kamidono S, Fujii A. Clinical studies of DNA gyrase inhibitors on renal dysfunction. Pharmacokinetics of enoxacin and its elimination effects by haemodialysis and adsorption with charcoal. Nishinihon J Urol. 1985;47:135–9. [Google Scholar]
- 21.Sörgel F, Naber KG, Martinkova J, Kinzig M, Chladek J, Müller C. Effect of charcoal on rufloxacin pharmacokinetics to assess intestinal secretion in humans. Pharm Res. 1994;11(Suppl):447. [Google Scholar]
- 22.Kinzig M, Sörgel F, Naber KG, Schugg C, Weiss G, Weinz C, Grannemann R, Drajsek JF. Gastrointestinal dialysis of temafloxacin in man. Proceedings of the Interscience conference on antimicrobial agents and chemotherapy. 1992;32:355. [Google Scholar]
- 23.Kinzig M, Seelmann R, Mahr G, Sörgel F, Naber KG. Significant gastrointestinal secretion of fleroxacin in man. In: Ehrlich Paul., editor. Proceedings of the 17th International Congress on Chemotherapy, Berlin. ed. Munich: Gesellschaft für Chemotherapie, Futuramed; 1991. p. Abstract 388. [Google Scholar]
- 24.Seelmann R, Jähde U, Muth P, Mahr G, Reiter A. Comparison of the effect of charcoal and probenecid on the pharmacokinetics of ciprofloxacin's major metabolite, CIP-M1. Eur J Clin Microbiol Infect Dis. 1991:379–80. (Special issue) [Google Scholar]
- 25.Sörgel F, Kinzig M, Naber KG, Jähde U, Birner B, Schunack W. Absorption of metabolites from the gut may be predicted from charcoal interaction studies with iv administration of parent drug. Pharm Res. 1993;10(Suppl 10):355. [Google Scholar]
- 26.Product information. Avelox® 400 mg infusion solution (Zul. no. 45263.00.01) Bayer Vital GmbH. 2002.
- 27.Stass H, Dalhoff A. Determination of BAY 12–8039, a new 8-methoxy-quinolone, in human body fluids by HPLC with fluorescence detection using on-column focusing. J Chromatogr B. 1997;702:163–74. doi: 10.1016/s0378-4347(97)00371-x. [DOI] [PubMed] [Google Scholar]
- 28.Stass H, Kubitza D. Pharmacokinetics and elimination of moxifloxacin after oral and intravenous administration in man. J Antimicrob Chemother. 1999;43(Suppl B):83–90. doi: 10.1093/jac/43.suppl_2.83. [DOI] [PubMed] [Google Scholar]
- 29.Stass H, Kubitza D, Halabi A, Delesen H. Pharmacokinetics of moxifloxacin, a novel 8- methoxy-quinolone, in patients with renal dysfunction; Br J Clin Pharmacol. 2002;53:232–7. doi: 10.1046/j.0306-5251.2001.01557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wise R. A review of the clinical pharmacology of moxifloxacin, a new 8-methoxy-quinolone and its potential relation to therapeutic efficacy. Clin Drug Invest. 1999;17:365–87. [Google Scholar]
- 31.Gibaldi M, Perrier D. Pharmacokinetics. New York: Marcel Dekker Inc; 1982. [Google Scholar]
- 32.Rowland M, Tozer TN. 3rd edn. Williams & Wilkins; 1995. Clinical Pharmacokinetics Concepts and Applications; p. 131. chapter 9. [Google Scholar]