

Abstract
Purpose
To characterize artemisinin pharmacokinetics (PK) and its antimalarial activity in vivo.
Methods
Artemisinin salivary concentration and parasite count data were obtained from Vietnamese malaria patients receiving two different dosage regimens. PK data were analysed using a previously developed semiphysiological model incorporating autoinduction of eliminating enzymes. A pharmacodynamic (PD) model reflecting different stages of the parasite life-cycle was developed and fitted to the data. The model included visible and invisible compartments as well as sensitive, insensitive, and injured parasite stages. Salivary artemisinin concentrations functioned as the driving force for the observed decrease in the number of parasites.
Results
Large interindividual variability was observed in both PK and PD data. The PK model described reasonably well the observed decrease in salivary concentrations after repeated drug administration. The preinduction hepatic extraction ratio of artemisinin was estimated to be 0.87 with a volume of distribution of 27 L. Artemisinin half-life averaged 0.7 h. Incorporation of a saturable hepatic elimination affecting the first-pass extraction as well as a higher intrinsic clearance in female patients resulted in the best fit of the model to the data. The PD model described the decrease in the number of parasites during the course of treatment well. The longest mean transit time of parasites from sensitive, visible to invisible to insensitive visible stages was found to be 34.5 h through one life-cycle. The half-life of injured parasites was 2.7 h.
Conclusions
The proposed semimechanistic PK/PD model successfully described the time course of both salivary artemisinin concentrations after repeated dosing and the number of parasites in patients treated with the drug.
Keywords: artemisinin, mechanism-based model, pharmacokinetics, pharmacodynamics
Introduction
Artemisinin is a potent antimalarial compound, extracted from Artemisia annua. Artemisinin and its derivative are now widely used in several countries in South-east Asia as the first-line medication against falciparum malaria infections. Artemisinin therapy is safe, effective and results in a rapid decline of parasite numbers [1]. The drug is a potent inducer of cytochrome P450 enzymes, including those responsible for its own elimination. Such autoinduction has been observed in both healthy subjects [2] and in malaria patients [3], and results in decreased concentrations of the compound in both plasma [2–4] and saliva [5, 6]. The time-course of autoinduction has recently been characterized using a semiphysiological model based on saliva samples from healthy subjects [5].
The efficacy of artemisinin and its derivatives in the treatment of uncomplicated malaria cases has been reviewed [1, 7, 8]. In general, combination therapy involving an artemisinin compound with another antimalarial drug is recommended. There is, however, no proper rationale for the choice of dose or duration of therapy of artemisinins. Few attempts have been made to derive a pharmacokinetic/pharmacodynamic (PK/PD) model of artemisinin or its derivatives in vivo[9, 10]. In this paper we describe a semimechanistic approach to modelling of the time course of parasitaemia after administration of two different dosage regimens of artemisinin, and relate salivary drug concentrations to the observed decline in parasite numbers over time. The model is based on data from a previously published study which offered a descriptive presentation of the observations [6].
Methods
Study design
This double-blind, randomized study involved 77 patients with uncomplicated malaria, receiving either 500 mg artemisinin daily for 5 days, or an escalating dosage regimen of 100 + 100 + 250 + 250 + 500 mg (Table 1). Doses were taken with 100 mL water and the mouth was rinsed with the same volume of water. Artemisinin on the first and last day was administered as a single dose, whereas on days 2–4, it was divided and given as morning and evening doses. The study took place in Phu Rieng Rubber Plantation Hospital, Binh Phuoc, Vietnam and was carried out in accordance with the Helsinki Declaration. Informed consent was obtained from each patient prior to inclusion in the study, the protocol for which was approved by the Ministry of Health, Hanoi, Vietnam; the Swedish Medical Product Agency, Uppsala, Sweden; and the Ethics Committee of the Medical Faculty of Uppsala University, Uppsala, Sweden. Data from 31 patients (24 male and 7 female) in the standard dose group and 28 patients (18 male and 10 female) in the escalating dose group were obtained for analysis. Owing to low saliva artemisinin concentrations, no data was available on the time-course of the compound in the remaining patients.
Table 1. Artemisinin doses in the two treatment arms.
| Dose* (mg) | |||||
|---|---|---|---|---|---|
| Dosing regimen | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 |
| Escalating | 100 | 100 | 250 | 250 | 500 |
| Standard | 500 | 500 | 500 | 500 | 500 |
On days 1 and 5 the drug was given as a single dose in two capsules. On the other days the dose was divided and given on two occasions.
Saliva sampling
A total of 12 unstimulated saliva samples were collected from each patient within 8 h after drug intake on days 1 and 5. After shipment to Sweden, samples were analysed using a validated coupled-column switching HPLC system [11]. The assay had a lower limit determination of 2 ng mL−1 saliva and an intraday coefficient of variation of 11.1% (injection volume = 1 mL) within the linear range of 2–240 ng mL−1. All samples from each patient were analysed on the same day.
Counting of parasites
Parasite number was estimated using capillary finger-tip blood followed by Giemsa staining and light microscopy. The counting was performed on samples taken immediately before and every 4 h after initiation of therapy until encountering three consecutive negative smears.
Pharmacokinetic modelling
A population PK model was fitted to the log-transformed saliva concentration-time data from all individuals simultaneously, using the first-order method (FO) without centring in the nonlinear mixed effects modelling program NONMEM, (version V) [12]. Discriminations between hierarchical models were based on the objective function value (OFV) provided by NONMEM at a significance level of 0.001, equal to a drop of 10.83 in the OFV, and graphical analysis of residuals and predictions in the model diagnostic software Xpose, version 3.1 [13]. Artemisinin concentrations at samples collected at 30 min postdose were particularly high in several patients. In the final analysis all concentration data from this time-point were considered unreliable and were not used in the model building process.
The structural model was based on the work of Gordi et al.[5], where saliva samples from healthy subjects were fitted to a semiphysiological model consisting of a PK component and an enzyme component with the former influencing the concentrations of the enzyme and the latter influencing the concentration of artemisinin.
During the early model-building phase, several variations of the original model were tested. These included one with compartments for the enzymes as well as enzyme precursor, a single enzyme compartment, linear and nonlinear effect of artemisinin liver concentration on the production rate of the enzyme precursor, and linear or nonlinear intrinsic clearance of artemisinin. The final PK model (Figure 1) consisted of one component describing the pharmacokinetics of artemisinin, and another the time-variant concentrations of inducible enzyme(s), with the two parts linked in a circular fashion. Artemisinin is ingested orally with subsequent absorption from the gut into a liver compartment. Since there is no information available on the extent of absorption of artemisinin, complete absorption of the compound without metabolism in the gut wall was assumed. Thus, structural model parameter estimates are apparent values and not the true estimate. The elimination of artemisinin was assumed to occur according to the well-stirred model in the liver compartment. The compound is distributed further into a sampling compartment (AS), which constitutes the entire body, except for the liver. Saliva concentrations are assumed to be in instantaneous equilibrium with plasma as well as all tissues but liver, to which distribution occurs [14]. The rate of change of the amount of artemisinin in the liver compartment is described by the equation:
| (1) |
where AH and CH are the amounts and concentrations of the compound in the liver compartment, ka is the absorption rate constant, and AG is the amount in the gut compartment (equal to dose at time zero). QH (liver blood flow) was set to 0.63 × body weight (in kilograms) to mimic hepatic plasma flow. The transfer rate constant kSH of artemisinin from AS to the hepatic compartment was estimated as QH/VS, VS being the volume of distribution of the sampling compartment. The hepatic volume (VH) was fixed at 1 L. The hepatic extraction ratio (EH), hepatic CL (CLH), and fraction of absorbed drug escaping hepatic first-pass extraction (FH) were defined according to the well-stirred model:
| (2) |
| (3) |
| (4) |
| (5) |
where CLint,0 and CLint,t represent the preinduced and time-variable intrinsic clearance, AENZ is the amount of enzyme(s) in the enzyme pool compartment relative to the amounts in the preinduced state, Km represents the hepatic artemisinin concentration at which CLint is half-maximal, and fu is the unbound plasma fraction of artemisinin which was set at 0.14 based on previous findings [14].
Figure 1.

Schematic presentation of the proposed PK/PD model 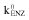 , kenz, zero-order production rate and first-order elimination rate of induced enzymes; ka, absorption rate constant; QH, hepatic plasma flow; FH, hepatic bioavailability; VH, hepatic volume of distribution; kSH, transfer rate of artemisinin from the sampling to the hepatic compartment; CLH, hepatic clearance; kRING, rate constant for transfer of schizonts to early forms (rings); kPAR, rate constant for transfer of mature parasites (trophozoits) to invisible parasites (schizonts); kPINJ, the second-order transfer rate constant from trophozoit and schizont forms to injured parasites caused by artemisinin; kEINJ, parasite removal rate constant from blood by spleen
, kenz, zero-order production rate and first-order elimination rate of induced enzymes; ka, absorption rate constant; QH, hepatic plasma flow; FH, hepatic bioavailability; VH, hepatic volume of distribution; kSH, transfer rate of artemisinin from the sampling to the hepatic compartment; CLH, hepatic clearance; kRING, rate constant for transfer of schizonts to early forms (rings); kPAR, rate constant for transfer of mature parasites (trophozoits) to invisible parasites (schizonts); kPINJ, the second-order transfer rate constant from trophozoit and schizont forms to injured parasites caused by artemisinin; kEINJ, parasite removal rate constant from blood by spleen
The rate of change of the amounts of drug in the sampling compartment was defined as:
| (6) |
The expressions for total volume of distribution at steady-state (VSS) and clearance (CL) of artemisinin are given by Equations 7 and 8. Although this is a two-compartment model, the distribution half-life with the corresponding slope constant λ1, will be short and the distribution phase of insignificant quantitative importance.
| (7) |
| (8) |
The enzyme component was modelled according to an indirect response model [15], which consisted of one enzyme pool compartment with zero-order production 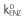 and first-order elimination (kENZ) rates. The amount of enzyme prior to treatment (AENZ,0) was set to unity. The change in the pool with time was modelled by allowing the amount of drug in the liver to increase the enzyme formation rate:
and first-order elimination (kENZ) rates. The amount of enzyme prior to treatment (AENZ,0) was set to unity. The change in the pool with time was modelled by allowing the amount of drug in the liver to increase the enzyme formation rate:
| (9) |
| (10) |
where IND is the inductive effect of artemisinin present in the liver, INDMAX is the maximum induction rate, and IND50 is the amount of artemisinin in the liver that produces 50% of maximum induction. The model was parameterized using the elimination half-life of the enzymes, t1/2,ENZ, rather than kENZ.
The absorption lag-time (tlag) and enzyme half-life (t1/2,ENZ) were given values obtained from a previous study involving more frequent sampling from healthy subjects [5].
An exponential variance model was used to describe the interindividual variability (IIV) of VS and Km, and the interoccasional variability (IOV) of tlag and ka. A residual error model with a proportional component was applied in the final model with the log-transformed data.
The effects of the covariates’ body weight and sex on different parameters were analysed using general additive models (GAM) implemented in Xpose. The final model incorporated the effect of sex on CLint.
Pharmacodynamic modelling
A semimechanistic model was fitted to the log-transformed parasite count vs. time data from all individuals simultaneously. The subroutine AVDAN6 TRANS1 with first-order method (FO) without centring in NONMEM (version V) was used. Discriminations between hierarchical models were made as described for the PK model. Post-hoc individual PK parameter estimates from the PK model were fixed, functioning as the driving force for the observed antiparasitic effect of artemisinin.
Several structural models were tested during the model-building process. These included a bio-phase model and a parasite model with two, three, or four compartments describing the parasite life-cycle, with artemisinin concentrations in the sampling compartment increasing the elimination rate from different parasite compartments. Other models tested included an indirect response model where artemisinin increased the natural elimination rate of parasites, a model without any spleen compartment, and one model with artemisinin exerting a saturable increase in the injury rate of the parasites (kPINJ).
The final PD model consisted of four compartments, reflecting the life-cycle of falciparum parasites during artemisinin therapy. Compartment 1 (AP1) represents the mature trophozoits, visible to the microscopist and susceptible to artemisinin action, whereas compartment 2 (AP2) represents the sequestered schizonts, also susceptible to the drug but not visible under the microscope. Compartment 3 (AP3) represents the ring stage of parasites, when the parasites are assumed to be insensitive to artemisinin. Compartment 4 (AP4) represents injured parasites, which are cleared from the circulation by the spleen. The total number of visible parasites is the sum of compartments 1, 3, and 4. The rate of change of parasite number in each compartment is given by the expressions:
| (11) |
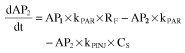 |
(12) |
| (13) |
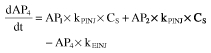 |
(14) |
where CS is the salivary artemisinin concentration (set equal to AS/ VS) and kPAR is the rate constant for transfer of mature parasites (trophozoits) to invisible parasites (schizonts), and from schizonts to early forms (rings). The model was parameterized in order that MTTPAR (parasite mean transit time) was estimated, and kPAR was set to 2/MTTPAR. RF is the replication factor for each parasite from visible to invisible stages (set to 15), kPINJ is the second-order transfer rate constant from trophozoit and schizont forms to drug damaged parasites, and kEINJ is the parasite removal rate constant from blood by spleen. The parasite life-cycle was restricted to 48 h. The rate constant for transfer of parasites from the ring form (AP3) to trophozoits (AP1) kRING was calculated from the expression 1/(48-MTTPAR). The first parasite count was assumed to be equal to the initial value of AP1, whereas the initial number of parasites in the invisible stage (AP2) was estimated. The initial number of parasites in the ring and injured stages were assumed to be negligible.
An exponential variance model was used to describe the interindividual variability (IIV) in TPAR and the initial number of invisible parasites. A residual error model with proportional components was applied in the final model.
Results
The observed saliva concentration-time profiles after the first and last administration of artemisinin in all subjects are presented in Figure 2. The data were adequately described by the proposed final model (Figures 3 and 4). The parameters from the latter are similar to those from the original model [5] and had relatively low CV% values (Table 2), indicating an overall good performance of the model. The intrinsic clearance of artemisinin at low concentrations (CH < < Km) was estimated to be 1550 L h−1 before the onset of induction, giving a hepatic extraction ratio of 0.87. The volume of distribution, based on saliva sampling was estimated to be 27 L. The contribution of drug from the liver compartment to VSS is negligible (less than 4%) and therefore the latter was approximately equal to VS. The model-predicted saliva elimination half-life for artemisinin was 0.7 h in the uninduced state. Female patients were found to have higher CLint values than males.
Figure 2.

Artemisinin saliva concentration vs. time in patients in the standard dosing group (STD) or the escalating dosing group (ESC)
Figure 3.

Population predictions, individual predictions, and individual weighted residuals vs. observed saliva artemisinin concentrations
Figure 4.

Observed artemisinin concentrations and model fits for two typical subjects: subject #208, receiving the standard dosing regimen, and subject #331, receiving the escalating dosage regimen
Table 2. Artemisinin pharmacokinetic parameters and associated interoccasional (IOV) and interindividual (IIV) variability in Vietnamese malaria patients.
| Parameter (unit) | Estimate (RSE%) | IIV (RSE%) | IOV (RSE%) |
|---|---|---|---|
| t1/2,ENZ (h) | 37.9 (Fixed) | – | – |
| INDMAX (L ng−1) | 13.4 (25) | – | – |
| IND50 (ng mL−1) | 140 (62) | – | – |
| CLint,0 (L h−1) | 1550 (13) | – | – |
| Female effect on CLint (L h−1) | 438 (61) | – | – |
| VS (L) | 27.0 (14) | 1.1 (40) | – |
| Lag-time (h) | 0.50 (Fixed) | – | 1.5 (31) |
| ka (h−1) | 0.3 (23) | – | 0.53 (43) |
| Km (ng mL−1) | 1960 (53) | 0.67 (54) | – |
| Residual error | 0.65 (12) | – | – |
t1/2,ENZ, enzyme elimination half-life; INDMAX, maximum induction rate of artemisinin hepatic concentration on the production rate of enzymes; IND50, liver artemisinin concentrations that produce 50% of the maximum induction; CLint, intrinsic clearance; VS, volume of sampling compartment; Lag-time, absorption lag-time; ka, absorption constant rate; Km, hepatic artemisinin concentration resulting in 50% of maximum intrinsic clearance; RSE%, Relative standard error in per cent.
The time-course of the number of parasites (Figure 5) was appropriately described by the final pharmacodynamic model (Figures 6 and 7). The model was able to predict an initial increase, observed in several but not all patients, as well as the decline of the number of parasites over several orders of magnitude during the course of treatment. The typical elimination rate constant for injured parasites was estimated to be 0.26 h−1, resulting in a half-life of 2.7 h. The model parameters are listed in Table 3.
Figure 5.

Time course of parasite elimination in the two study arms (STD: standard dosing group, ESC: escalating dosage group)
Figure 6.

Population predictions, individual predictions, and individual weighed residuals vs. observed number of parasites
Figure 7.

Observed parasite numbers and model fits for two typical subjects: subject #331, receiving the standard dosing regimen (STD), and subject # 208, receiving the escalating dosage regimen (ESC)
Table 3. Artemisinin pharmacodynamic parameters and associated interindividual (IIV) variability in Vietnamese malaria patients.
| Parameter (unit) | Estimate (RSE%) | IIV (RSE%) |
|---|---|---|
| TPAR (h) | 34.4 (5.5) | 0.26 (70) |
| TRING (h) | 13.6 | – |
| kPINJ (ng mL−1 h−1) | 1.58 (59) | – |
| RF | 15 (Fixed) | – |
| kEINJ (h−1) | 0.26 (9.1) | – |
| INV | 26200 (28) | 2.1 (20) |
| Proportional residual error | 0.88 (6.3) | – |
TPAR, transfer time of parasites from trophozoits to schizonts and further to rings; TRING, transfer time of parasites from rings to schizonts, equal to 48-TPAR; kPINJ, injury rate of trophozoits and schizonts; RF, parasite replication factor per life-cycle; kEINJ, elimination rate of injured parasites; INV, initial value of invisible parasites; RSE%, Relative standard error in per cent.
Discussion
Artemisinin and its derivatives have been used for treatment of falciparum malaria infections for more than two decades. Their main advantage is the lack of resistance amongst malaria parasites, which makes them the first-line treatment in many areas of the world, where strains resistant to other antimalarials have developed [7]. Moreover, artemisinin and its derivatives offer the most rapid rate of parasite killing, decreasing the number of parasites to below the detection limit within 1–2 days in a majority of patients [16]. Furthermore, artemisinin compounds have proved to be very safe with few and mild side-effects [17].
The pharmacokinetics of artemisinin have been studied in both healthy subjects [2, 18, 19] and malaria patients [4, 6, 20]. Artemisinin induces its own metabolism even after a single dose, resulting in decreased concentrations after repeated administration. Only a single report characterizing the time-course of the autoinduction of artemisinin metabolism has been published [5], where data from healthy subjects receiving two different dosing regimens were fitted using a semimechanistic model. The present work used this model with minor modifications. Gordi et al. were able to collect saliva samples on 4 out of the 6 days that the drug was administered, which allowed for a comprehensive investigation of the multiple dose pharmacokinetics of artemisinin. Increasing amounts of artemisinin in the liver compartment increased the rate of production of an enzyme precursor in a linear fashion, resulting in greater amounts of enzyme. In our present study in patients, we were able to collect samples on the first and last days of administration only, although in a larger group of subjects. Such sampling did not allow for the estimation of all model parameters and thus the elimination half-life of the enzymes as well as artemisinin's absorption lag-time were fixed at values based on the original model. The elimination half-life of the induced enzyme(s) is a systemic parameter independent of artemisinin concentration and can thus be assumed to be constant. The rationale for fixing the absorption lag-time was the lack of sufficient data to estimate this parameter, especially since all samples taken at 30 min postabsorption were excluded from the data analysis. In the present model, inclusion of an enzyme precursor compartment did not result in a significant improvement of the fit of the model. Moreover, we were able to characterize the saturation of induction, whereas the original model proposed a linear increase in the rate of precursor production. In setting a value for enzyme elimination half-life based on previous data, we have assumed that the pharmacokinetics of artemisinin and the induction process are similar in healthy subjects and malaria patients [2, 3, 5, 6].
The time course of drug action against malaria parasites has been investigated previously. In general, two approaches have been utilized in relating drug concentrations or exposure to the decline in the number of parasites after drug therapy. In the first approach, direct relationships between drug concentration/exposure and various PD markers have been sought, which has lead to discrepant results. Whereas some authors found no relationship between drug concentrations and/or AUC values and the main clinical endpoints [4], others have reported nonlinear relationships between various PK and PD parameters [21]. The second approach involves compartmental modelling, where mathematical models have been developed to describe the time-course of parasite numbers in drug-free individuals [22] or after treatment with chloroquine [23], mefloquine [24], artemisinin derivatives [9], or combination therapy [10, 25–27].
Several complicating factors arise when data from falciparum malaria infections are modelled. The life-cycle of the parasites includes a period of sequestration, during which they cannot be detected by microscopy. Thus, the number of visible parasites does not necessarily represent the total load. A decrease in the number of parasites could be due to two mechanisms, namely clearance from the blood by spleen or sequestration. Unlike other forms of malaria, synchronized falciparum infections are uncommon and the different forms of the parasite may be present in a patient at any time. Owing to the high risk of mortality, patients with falciparum malaria are generally treated immediately the parasites are detected, and thus there are no data on the natural course of the parasites, which would allow information on the progression of the disease to be added to any mechanistic model.
It is common to observe an initial increase in the number of parasites after the first dose of an antimalarial compound [28, 29] and several patients in the present study were found to have higher numbers of parasites in samples collected at 4 and 8 h after the first dose of artemisinin. Such an increase could be due to factors including a delay in drug action due to its distribution to the site of action, production of active metabolite, rapid influx of parasites from the site of sequestration to the circulation, or to a nonsensitive and visible period of the parasite life-cycle. The first two mechanisms seem unlikely for artemisinin. Malaria parasites are present in the blood and thus the drug can readily reach its site of action through a rapid distribution into red blood cells [30]. Moreover, it is generally believed that artemisinin is rapidly converted to free radical after distribution into the red blood cells [31], although recent data suggest that such activation is not essential for the antimalarial activity of the compound [32].
Our proposed model features compartments representing different stages of the parasite life-cycle. The total number of parasites was assumed to consist of a drug sensitive and an insensitive pool as well as injured parasites. The last group are eliminated at a different rate than the process causing injury. The final model also included a nondetectable pool of parasites, within which replication occurs and from which the parasites are transferred to the circulation. Our model successfully described this process. A similar approach with visible and invisible parasite pools has been proposed by Gravenor et al.[26].
There are contradicting opinions on whether artemisinin is active against all or parts of the blood stages of the falciparum life-cycle. Our proposed model includes an insensitive compartment representing early ring-forms of the parasites. It was assumed in the current model that the mean transit time of parasites through one life-cycle was 48 h. The mean transit time of parasites from the sensitive and invisible stage to the early ring-form was estimated to be the longest during the life-cycle. A fixed replication factor of 15 was used in the final model, which results in the shortest mean transit time from visible to invisible parasites during the life-cycle. It is generally believed that falciparum parasites replicate at a rate of 10–25 times during each cycle. During the model building process, two variations of the final model were tested where the replication factor was fixed to 10 or 20. However, the different replication factors, did not result in any significant changes in other parameter estimates and a value of 15 was chosen based on model selection criteria described in the Methods section.
When applying the model to data, the pretreatment observations were presented to the system as the initial number of parasites in the compartment of visible and sensitive parasites (AP1). Thus, it was assumed that the visible pretreatment parasites were all in the mature trophozoit form, excluding the presence of rings or naturally injured parasites. Although a differentiation of rings and trophozoit by microscopy is possible, it is a laborious task and was not undertaken during the course of the original study. Parasites spend a relatively short period of their life as rings and thus the proportion at this stage of the lifecycle was probably negligible in comparison with the other forms. Furthermore, it is virtually impossible to separate the healthy and injured parasites by microscopy. Although patients in endemic areas have some degree of natural immunity to malaria, any decline in the number of parasites due to such immunity is negligible in comparison with their large replication rate and/or the antiparasitic action of artemisinin.
Large variability was observed in the graph of parasite numbers vs. time profiles as observed, which partly contributed to the substantial variation in the estimates of parameters of the PD model. Both pharmacokinetic and pharmacodynamic factors may explain these observations. Salivary artemisinin concentrations were measured in samples collected up to 8 h postadministration on days 1 and 5 of the treatment period only. Thus, for most patients, there are no concentration data available during the period of parasite decline. Moreover, artemisinin pharmacokinetics exhibits large interoccasional variability, resulting in uncertainty in the estimation of drug concentrations on days 2–4 when most of the pharmacodynamic activity takes place. There is also inherent variability associated with the life-cycle of the parasite. Since falciparum malaria is treated upon detection, most patients do not experience synchronized infections. This results in the presence of all forms of P. falciparum, at any time in a given patient. Moreover, an increase in the number of parasites can be due to replication as well as transfer from the sequestered site to the circulation, whereas a decrease in their number may be due to sequestration or drug action.
Low artemisinin concentrations toward the end of treatment might influence the outcome of the therapy. However, the major factor determining the effectiveness of antimalarial treatment is the relationship between parasite life-cycle and drug concentration, as discussed by White [28]. For therapy to result in high cure rates, i.e. total parasite clearance and no recrudescence, the antimalarial compound must be present in the circulation for at least four parasite life-cycles, i.e. 8 days. Artemisinin has a short half-life, which necessitates treatment over 8 days, making monotherapy practically impossible due to an increased risk of poor compliance. This has lead to the advancement of combination therapies of artemisinin compounds with other antimalarials with longer half-lives, where artemisinin or its derivatives significantly decrease the parasite load after one or two doses and the longer half-life of the second drug ensures the presence of antimalarial activity for more than 8 days.
In conclusion, we have presented a semimechanistic PK/PD model that describes the time course of both saliva drug concentrations and parasite numbers after treatment with artemisinin. Although based on data from monotherapy with artemisinin, the structural parasite life-cycle model is independent of the antimalarial compound and can be applied to concentration-effect data obtained from other drugs or combination therapies.
Acknowledgments
The authors would like to thank Dr M.B. Hoshen for valuable discussions during the model-building process. This work was partially supported by NIH Grant No.GM 56980.
References
- 1.Price RN. Artemisinin drugs: novel antimalarial agents. Expert Opin Invest Drugs. 2000;9:1815–27. doi: 10.1517/13543784.9.8.1815. [DOI] [PubMed] [Google Scholar]
- 2.Ashton M, Hai TN, Sy ND, Huong DX, Huong NV, Nieu Nt Cong LD. Artemisinin pharmacokinetics is time-dependent during repeated oral administration in healthy male adults. Drug Metab Dispos. 1998;26:25–7. [PubMed] [Google Scholar]
- 3.Alin MH, Ashton M, Kihamia CM, Mtey GJ, Björkman A. Clinical efficacy and pharmacokinetics of artemisinin monotherapy and in combination with mefloquine in patients with falciparum malaria. Br J Clin Pharmacol. 1996;41:587–92. doi: 10.1046/j.1365-2125.1996.35116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashton M, Sy ND, Huong NV, Gordi T, Hai TN, Huong DX, Niêu NT, Công LD. Artemisinin kinetics and dynamics during oral and rectal treatment of uncomplicated malaria. Clin Pharmacol Ther. 1998;63:482–93. doi: 10.1016/S0009-9236(98)90044-3. [DOI] [PubMed] [Google Scholar]
- 5.Gordi T, Xie R, Huong NV, Huong DX, Karlsson M, Ashton M. A semi-physiological pharmacokinetic model for artemisinin in healthy subjects incorporating autoinduction of metabolism and saturable first-pass hepatic extraction. Br J Clin Pharmacol. 2005;59:189–98. doi: 10.1111/j.1365-2125.2004.02321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gordi T, Huong DX, Hai TN, Nieu NT, Ashton M. Artemisinin pharmacokinetics and efficacy in uncomplicated-malaria patients treated with two different dosage regimens. Antimicrob Agent Chemother. 2002;46:1026–31. doi: 10.1128/AAC.46.4.1026-1031.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Agtmael MA, Eggelte TA, van Boxtel CJ. Artemisinin drugs in the treatment of malaria: from medicinal herb to registered medication. Trends Pharmacol Sci. 1999;20:199–205. doi: 10.1016/s0165-6147(99)01302-4. [DOI] [PubMed] [Google Scholar]
- 8.Dhingra V, Vishweshwar RK, Lakshmi NM. Current status of artemisinin and its derivatives as antimalarial drugs. Life Sci. 2000;66:279–300. doi: 10.1016/s0024-3205(99)00356-2. [DOI] [PubMed] [Google Scholar]
- 9.Hoshen MB, Na-Bangchang K, Stein WD, Ginsburg H. Mathematical modelling of the chemotherapy of Plasmodium falciparum malaria with artesunate: postulation of ′dormancy', a partial cytostatic effect of the drug, and its implication for treatment regimens. Parasitol. 2000;12:237–46. doi: 10.1017/s0031182099006332. [DOI] [PubMed] [Google Scholar]
- 10.Svensson US, Alin H, Karlsson MO, Bergqvist Y, Ashton M. Population pharmacokinetic and pharmacodynamic modelling of artemisinin and mefloquine enantiomers in patients with falciparum malaria. Eur J Clin Pharmacol. 2002;58:339–51. doi: 10.1007/s00228-002-0485-y. [DOI] [PubMed] [Google Scholar]
- 11.Gordi T, Nielsen E, Yu Z, Westerlund D, Ashton M. Direct analysis of artemisinin in plasma and saliva using coupled-column high-performance liquid chromatography with a restricted-access material pre-column. J Chromatogr B Biomed Sci Appl. 2000;742:155–62. doi: 10.1016/s0378-4347(00)00156-0. [DOI] [PubMed] [Google Scholar]
- 12.Beal SL, Sheiner LB. NONMEM Users GuideIntroductory Guide. San Francisco: NONMEM Project Group; 1998. [Google Scholar]
- 13.Jonsson EN, Karlsson MO. Xpose- an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comp Meth Prog Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 14.Gordi T, Hai TN, Hoai NM, Thyberg M, Ashton M. Use of saliva and capillary blood samples as substitutes for venous blood sampling in pharmacokinetic investigations of artemisinin. Eur J Clin Pharmacol. 2000;56:561–6. doi: 10.1007/s002280000179. [DOI] [PubMed] [Google Scholar]
- 15.Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21:457–78. doi: 10.1007/BF01061691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White NJ. Clinical pharmacokinetics and pharmacodynamics of artemisinin and derivatives. Trans R Soc Trop Med Hyg. 1994;88(Suppl 1):S41–3. doi: 10.1016/0035-9203(94)90471-5. [DOI] [PubMed] [Google Scholar]
- 17.Gordi T, Lepist EI. Artemisinin derivatives: toxic for laboratory animals, safe for humans? Toxicol Lett. 2004;147:99–107. doi: 10.1016/j.toxlet.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Ashton M, Gordi T, Trinh NH, Nguyen VH, Nguyen DS, Nguyen TN, Dinh XH, Johansson M, Le DC. Artemisinin pharmacokinetics in healthy adults after 250, 500 and 1000 mg single oral doses. Biopharm Drug Disp. 1998;19:245–250. doi: 10.1002/(sici)1099-081x(199805)19:4<245::aid-bdd99>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 19.Svensson US, Ashton M, Trinh NH, Bertilsson L, Dinh XH, Nguyen VH, Nguyen TN, Nguyen DS, Lykkesfeldt J, Le DC. Artemisinin induces omeprazole metabolism in human beings. Clin Pharmacol Ther. 1998;64:160–7. doi: 10.1016/S0009-9236(98)90149-7. [DOI] [PubMed] [Google Scholar]
- 20.Hassan Alin M, Ashton M, Kihamia CM, Mtey GJ, Bjorkman A. Multiple dose pharmacokinetics of oral artemisinin and comparison of its efficacy with that of oral artesunate in falciparum malaria patients. Trans R Soc Trop Med Hyg. 1996;90:61–5. doi: 10.1016/s0035-9203(96)90480-0. [DOI] [PubMed] [Google Scholar]
- 21.Angus BJ, Thaiaporn I, Chanthapadith K, Suputtamongkol Y, White NJ. Oral artesunate dose–response relationship in acute falciparum malaria. Antimicrob Agents Chemother. 2002;46:778–82. doi: 10.1128/AAC.46.3.778-782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoshen MB, Heinrich R, Stein WD, Ginsburg H. Mathematical modelling of the within-host dynamics of Plasmodium falciparum. Parasitology. 2000;121:227–35. doi: 10.1017/s0031182099006368. [DOI] [PubMed] [Google Scholar]
- 23.Hoshen MB, Stein WD, Ginsburg H. Modelling the chloroquine chemotherapy of falciparum malaria: the value of spacing a split dose. Parasitology. 1998;116:407–16. doi: 10.1017/s0031182098002480. [DOI] [PubMed] [Google Scholar]
- 24.Hoshen MB, Stein WD, Ginsburg HD. Pharmacokinetic-pharmacodynamic modelling of the antimalarial activity of mefloquine. Parasitology. 2001;123:337–46. doi: 10.1017/s003118200100854x. [DOI] [PubMed] [Google Scholar]
- 25.Hoshen MB, Stein WD, Ginsburg H. Mathematical modelling of malaria chemotherapy: combining artesunate and mefloquine. Parasitology. 2002;124:9–15. doi: 10.1017/s0031182001008952. [DOI] [PubMed] [Google Scholar]
- 26.Gravenor MB, van Hensbroek MB, Kwiatkowski D. Estimating sequestered parasite population dynamics in cerebral malaria. Proc Natl Acad Sci USA. 1998;95:7620–4. doi: 10.1073/pnas.95.13.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Vries PJ, Bich NN, Van Thien H, Hung LN, Anh TK, Kager PA, Heisterkamp SH. Combinations of artemisinin and quinine for uncomplicated falciparum malaria: efficacy and pharmacodynamics. Antimicrob Agents Chemother. 2000;44:1302–8. doi: 10.1128/aac.44.5.1302-1308.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White NJ. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob Agents Chemother. 1997;41:1413–22. doi: 10.1128/aac.41.7.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silachamroon U, Phumratanaprapin W, Krudsood S, Treeprasertsuk S, Budsaratid V, Pornpininworakij K, Wilairatan P, Looareesuwan S. Frequency of early rising parasitemia in falciparum malaria treated with artemisinin derivatives. Southeast Asian J Trop Med Public Health. 2001;32:50–6. [PubMed] [Google Scholar]
- 30.Bakhshi HB, Gordi T, Ashton M. In vitro interaction of artemisinin with intact human erythrocytes, erythrocyte ghosts, haemoglobin and carbonic anhydrase. J Pharm Pharmacol. 1997;49:223–6. doi: 10.1111/j.2042-7158.1997.tb06784.x. [DOI] [PubMed] [Google Scholar]
- 31.Meshnick SR. Artemisinin mechanisms of action, resistance and toxicity. Int J Parasitol. 2002;32:1655–60. doi: 10.1016/s0020-7519(02)00194-7. [DOI] [PubMed] [Google Scholar]
- 32.Parapini S, Basilico N, Mondani M, Olliaro P, Taramelli D, Monti D. Evidence that haem iron in the malaria parasite is not needed for the antimalarial effects of artemisinin. FEBS Lett. 2004;575:91–4. doi: 10.1016/j.febslet.2004.08.039. [DOI] [PubMed] [Google Scholar]