

Abstract
Aims
Using the fluconazole–zidovudine (AZT) interaction as a model, to determine whether inhibition of UDP–glucuronosyltransferase (UGT) catalysed drug metabolism in vivo could be predicted quantitatively from in vitro kinetic data generated in the presence and absence bovine serum albumin (BSA).
Methods
Kinetic constants for AZT glucuronidation were generated using human liver microsomes (HLM) and recombinant UGT2B7, the principal enzyme responsible for AZT glucuronidation, as the enzyme sources with and without fluconazole. Ki values were used to estimate the decrease in AZT clearance in vivo.
Results
Addition of BSA (2%) to incubations decreased the Km values for AZT glucuronidation by 85–90% for the HLM (923 ± 357 to 91 ± 9 µm) and UGT2B7 (478–70 µm) catalysed reactions, with little effect on Vmax. Fluconazole, which was shown to be a selective inhibitor of UGT2B7, competitively inhibited AZT glucuronidation by HLM and UGT2B7. Like the Km, BSA caused an 87% reduction in the Ki for fluconazole inhibition of AZT glucuronidation by HLM (1133 ± 403 to 145 ± 36 µm) and UGT2B7 (529 to 73 µm). Ki values determined for fluconazole using HLM and UGT2B7 in the presence (but not absence) of BSA predicted an interaction in vivo. The predicted magnitude of the interaction ranged from 41% to 217% of the reported AUC increase in patients, depending on the value of the in vivo fluconazole concentration employed in calculations.
Conclusions
Ki values determined under certain experimental conditions may quantitatively predict inhibition of UGT catalysed drug glucuronidation in vivo.
Keywords: drug interactions, fluconazole, glucoronidation, in vitro-in vivo correlation, UDP-glucuronosyltransferase, zidovudine
Introduction
Inhibition of drug metabolism by a coadministered drug results in decreased metabolic clearance and/or increased bioavailability. The elevated blood concentration of the parent drug may result in an enhanced and prolonged pharmacological response, with an increased likelihood of drug-induced toxicity. Indeed, drug interactions are a well-recognized cause of adverse events, within both hospitals and community-based practices [1]. Thus, the ability to predict inhibitory drug interactions is an essential consideration for the safe and efficacious use of medicines. Moreover, several drugs have been withdrawn from the market in recent years due to the occurrence of fatal inhibitory drug interactions. Apart from quality use of medicines issues, inhibitory drug interactions also represent a potential economic loss for the pharmaceutical industry [2].
The use of in vitro methodologies to predict aspects of human drug metabolism and pharmacokinetics in vivo has found increasing acceptance in recent years. At the quantitative level, an in vitro intrinsic clearance (CLint) for a metabolic pathway, generally determined from microsomal or hepatocyte kinetic data, may be extrapolated to hepatic clearance (CLH) and extraction ratio using a mathematical expression that relates these parameters [3]. In vitro approaches, based on the measurement of an inhibition constant (Ki), have also been investigated as a basis for predicting the extent of an inhibitory drug interaction in vivo[4, 5]. Predictive in vitro models potentially provide a cost-effective approach for screening inhibitory drug interactions, with reduced human drug exposure and risk [6, 7]. Although in vitro approaches have been used to predict interactions between drugs metabolized by cytochrome P450 (CYP) [2, 4–6, 8–11], in vitro–in vivo correlation for interactions involving glucuronidated drugs has not been explored in a systematic manner.
Glucuronidation involves the covalent linkage of a suitable functional group present on the substrate with glucuronic acid (derived from the cofactor UDP-glucuronic acid). The glucuronidation reaction is catalysed by the enzyme UDP-glucuronosyltransferase (UGT). Like CYP, UGT exists as an enzyme superfamily; 17 human UGT enzymes have been identified to date. The functional human UGTs exhibit distinct, but frequently overlapping, substrate and inhibitor selectivities [12]. Consistent with this heterogeneity, glucuronidation serves as an elimination mechanism for a myriad of structurally diverse endogenous compounds and xenobiotics, including many clinically used drugs. Inhibitory interactions between UGT substrates in vivo are well documented [13, 14].
Zidovudine (AZT) is cleared primarily by glucuronidation in humans [15–17] and a number of drugs are known to inhibit AZT glucuronidation in vivo. In particular, fluconazole (400 mg day−1) decreased the apparent oral clearance of AZT by glucuronidation by 47.1% (corresponding to a 1.92-fold increase in the mean area under the AZT plasma concentration–time curve (AUC) associated with clearance by glucuronidation) in patients coadministered these drugs [18]. As in vivo, the kinetics of AZT glucuronidation in vitro are well characterized and hence this compound represents a useful model for investigating in vitro–in vivo correlation. In particular, the kinetics of AZT glucuronidation by human liver microsomes (HLM) have been determined over a range of experimental conditions and a single enzyme, UGT2B7, has been shown to be responsible for AZT glucuronide (GAZT) formation [19, 20].
Previous investigations conducted in this laboratory have demonstrated that extrapolation of the CLint value determined for AZT glucuronidation by HLM under-predicted the known in vivo hepatic AZT clearance by glucuronidation, and this observation appeared consistent for other glucuronidated drugs [19]. It has been reported that the addition of bovine serum albumin (BSA) to incubations of HLM increases the CLint values for several drugs metabolized by cytochrome P4502C9 (CYP2C9) [21–23]. The higher CLint results largely from a decrease in Km. Although the mechanism of the albumin effect is unknown and the addition of albumin to microsomal incubations may appear ‘unphysiological’, the higher CLint obtained under these experimental conditions potentially improves in vitro–in vivo extrapolation and thus warrants further investigation. At present, the effect of BSA on the kinetics of drug glucuronide formation and inhibition in vitro is unknown.
This study aimed primarily to determine whether the Ki value determined for inhibition of AZT glucuronidation by fluconazole, using both HLM and UGT2B7 as the enzyme source, predicted the extent of the AZT–fluconazole interaction in vivo. Additionally, the work sought to: (i) characterize the selectivity of fluconazole as an inhibitor of human UGTs by screening for effects on individual recombinant enzymes, and (ii) determine the effects of exogenous albumin (as BSA) on the kinetics of GAZT formation and inhibition in vitro.
Materials and methods
Materials
Alamethicin (from Trichoderma viride), AZT (zidovudine; 3′-azido-3′-deoxythymidine), BSA (Fraction V, 98–99% albumin), GAZT (3′-azido-3′-deoxythymidine 5′-β-D-glucuronide), β-glucuronidase (from Escherichia coli), 4-methylumbelliferone (4 mU; sodium salt), 4-methylumbelliferone-β-D-glucuronide (4MUG), trifluoperazine (TFP; dihydrochloride salt), trifluoroacetic acid, UDP-glucuronic acid (UDPGA; trisodium salt) and cellulose dialysis membrane (molecular weight cut-off 12 000 Da) were purchased from Sigma-Aldrich Pty Ltd (Sydney, Australia). Fluconazole was a gift from Pfizer Australia (Sydney, Australia). Solvents and other reagents were of analytical reagent grade.
Methods
Human liver microsomes and expression of UGT protein Human livers (HL 10, 12, 13 and 40) were obtained from the human liver ‘bank’ of the Department of Clinical Pharmacology, Flinders Medical Centre. Approval was obtained from the Flinders Medical Centre Research Ethics Committee and from the donor next-of-kin for the procurement and use of human liver tissue in xenobiotic metabolism studies. Microsomes were prepared by differential centrifugation, as described by Bowalgaha et al.[24], and activated by the addition of the pore-forming peptide alamethicin (50 µg mg−1 of protein) with preincubation on ice for 30 min [19] prior to use in incubations.
UGT 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7 and 2B15 cDNAs were stably expressed in a human embryonic kidney cell line (HEK293), as described previously [25–27]. Cells were separately transfected with the individual UGT cDNAs cloned into the pEF-IRES-puro6 expression vector and incubated in Dulbecco’s modified Eagle’s medium (DMEM), which contained puromycin (1.5 mg l−1), 10% fetal calf serum and penicillin G sodium (100 U ml−1)/streptomycin sulphate (100 µg ml−1) in a humidified incubator with an atmosphere of 5% CO2, at 37°C. After growth to at least 80% confluency, cells were harvested and washed in phosphate-buffered saline. The harvested cells were lysed by sonication using a Heat Systems-Ultrasonics sonicator set at microtip limit of 4. Cells expressing UGT1A proteins were sonicated with 4 × 2-s ‘bursts’, each separated by 3 min cooling on ice. Sonication was limited to 1-s ‘bursts’ for UGT2B subfamily proteins, due to their apparently greater thermolability. The lysed samples were centrifuged at 12 000 g for 1 min at 4°C and the supernatant fraction was separated and stored at − 80°C until use.
4MU glucuronidation assay The activities of recombinant enzymes (viz. UGT 1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7 and 2B15) were confirmed with the nonselective substrate 4MU prior to use in the inhibition and kinetic studies. 4MU glucuronidation was measured according to a previously published procedure [28]. Incubations contained UDPGA (5 mm), MgCl2 (5 mm), HEK293 cell lysate, phosphate buffer (0.1 m, pH 7.4) and 4MU in a total volume of 0.6 ml. Reaction times and lysate protein concentrations for incubations with each individual isoform were as reported by Sorich et al.[25] for UGT1A1 and Uchaipichat et al.[27], for UGT 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7 and 2B15. Within-day 4MUG assay imprecision, determined by measuring product formation in five separate incubations using HLM as the enzyme source, was < 4% for 4MU concentrations in the range 20–2000 µm.
TFP glucuronidation assay TFP was used as the substrate for UGT1A4. Trifluoperazine glucuronide (TFPG) formation was measured using a modification of the method recommended by BD Gentest (http://www.bdbiosciences.com/discovery_labware/gentest/products/pdf/1A4_AAPS_S01T056R1.pdf). The incubation mixture (0.2 ml total volume) contained Tris–HCl buffer (50 mm, pH 7.4), UDPGA (5 mm), MgCl2 (5 mm), UGT1A4 HEK293 cell lysate (0.25 mg ml−1), and TFP. Reactions were initiated by the addition of UDPGA and incubations were performed at 37°C in a shaking water bath for 20 min. Incubations were terminated by the addition of 4% acetic acid/96% methanol (0.2 ml) and then centrifuged at 5000 g for 10 min. A 40-µl aliquot of the supernatant fraction was injected into the high-perfromance liquid chromatography (HPLC) column.
Measurement of TFPG formation HPLC was performed using an Agilent 1100 series instrument (Agilent Technologies, Sydney, NSW, Australia) fitted with an Ultrasphere ODS column (4.6 × 250 mm, 5 µm; Beckman Instruments, Fullerton, CA, USA). Analytes were separated using a linear gradient with flow rate of 1 ml min−1. Initial conditions were 70% 0.1% trifluoroacetic acid/water (mobile phase A) and 30% 0.1% trifluoroacetic acid in acetonitrile (mobile phase B). The proportion of mobile phase B was increased to 50% over 10 min. Column eluant was monitored by UV absorbance 256 nm. Under these conditions, retention times of TFPG and TFP were 9.1 and 9.8 min, respectively. TFPG was quantified by comparison of peak areas with those of a TFP external standard curve prepared over the concentration range 0.2–10 µm. There is evidence demonstrating that the absorption characteristics of aliphatic N+-glucuronides are similar to the aglycone [29] (http://www.bdbiosciences.com/discovery_labware/gentest/products/enzym_micro/prod_inserts/p414.shtml). Within-day overall assay reproducibility was assessed by measuring TFPG formation in eight separate incubations of the same batch of pooled HLM (from HL 10, 12, 29 and 40). Coefficients of variation were 3.8% and 5.2% for added TFP concentrations of 10 and 200 µm, respectively.
The identity of TFPG was confirmed by enzymatic and base hydrolysis. A 0.2-ml TFP glucuronidation incubation (see above) was terminated with 70% HClO4 (2 µl) and centrifuged. The aqueous sample was decanted and mixed with 20 µl of 1 m phosphate buffer (pH 7.4) (to raise the pH to 6.5) and 1800 units of β-glucuronidase. After 2 h incubation at 37°C, a 150-µl aliquot was separated and treated with 70% HClO4 (5 µl). Base hydrolysis was performed by adding an equal volume of 4 m NaOH to a 0.2-ml TFP glucuronidation incubation and heating at 75°C for 45 min, after which time a 150-µl aliquot was separated and treated with 70% HClO4 (40 µl). Following centrifugation (5000 g for 10 min), 40 µl of the supernatant fraction from each reaction was injected into the HPLC column. Both treatments resulted in loss of the TFPG peak in the chromatogram.
AZT glucuronidation assay GAZT formation was measured using a modification of the method of Boase and Miners [19]. Incubation mixtures, in a total volume 0.2 ml, contained phosphate buffer (0.1 m, pH 7.4), MgCl2 (4 mm), UDPGA (5 mm), AZT (50–4000 µm) and activated HLM (1 mg ml−1) or UGT2B7 HEK293 cell lysate (1.5 mg ml−1). Reactions were initiated by the addition of UDPGA and performed at 37°C in a shaking water bath for 60 min. Following the addition of 24% HClO4 (10 µl), samples were centrifuged (5000 g for 10 min) and 30 µl of the supernatant fraction was injected into the HPLC column.
For reactions carried out in the presence of 2% BSA, incubation mixtures contained AZT in the concentration range 10–1000 µm due to a lower Km. Incubation conditions were as described for reactions in the absence of BSA, except that the protein amount and incubation time for HLM were 0.25 mg ml−1 and 30 min, respectively. Due to the higher protein concentration, reactions were terminated by addition of 10 µl of 70% HClO4. A 0.12-ml aliquot of the supernatant fraction was transferred to a 1.5-ml Eppendorf tube containing 4 m KOH (10 µl), mixed and centrifuged at 10 000 g for 1 min. Thirty microlitres of the supernatant fraction was injected into the HPLC column.
Measurement of GAZT formation HPLC was performed using an Agilent 1100 series instrument fitted with a SecurityGuard C18 cartridge (4 × 3 mm; Phenomenex, Sydney, Australia) and a NovaPak C18 column (3.9 × 150 mm; Waters Associates, Milford, MA, USA). The mobile phase, 0.12% v/v acetic acid in 10% acetonitrile/water, was delivered at a flow rate 1.2 ml min−1. Column eluant was monitored by UV absorbance at 267 nm. Retention times of GAZT and AZT were 3 and 6.2 min, respectively. Concentrations of GAZT in incubation samples were determined by comparison of peak areas with those of GAZT standard curve with concentrations in the range 1–20 µm. Overall assay reproducibility, assessed by measuring GAZT formation in 10 separate incubations of the same batch of HLM, was 3.7%, 2.1% and 2.5% for added AZT concentrations of 50 µm, 1000 µm and 3000 µm, respectively.
Fluconazole inhibition of recombinant UGTs The selectivity of UGT inhibition by fluconazole was assessed using 4MU or TFP as the substrate. 4MU was employed as the substrate for the inhibition studies with UGT 1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7 and 2B15, whereas TFP was used as the substrate for UGT1A4. Incubations with 4MU were performed at the concentration corresponding to the apparent Km or S50 value reported for each isoform (viz. 100, 1000, 100, 15, 750, 10, 40, 350 and 300 µm for UGT 1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7 and 2B15, respectively) [25, 27]. The concentration of TFP used in incubations with UGT1A4 was 40 µm, which corresponds to the Km for this substrate [30]. Concentrations of fluconazole used in the screening experiments were 0, 10, 100, 500, 1000 and 2500 µm.
Fluconazole inhibition of AZT glucuronidation Fluconazole inhibition of AZT glucuronidation was investigated using microsomes from four human livers and with UGT2B7 HEK293 cell lysate, in the presence and absence of BSA (2%), using the incubation and assay conditions described previously. Experiments performed to determine inhibitor constants (Ki) for fluconazole included four inhibitor concentrations at each of three AZT concentrations.
Binding of AZT and fluconazole to BSA, human liver microsomes and HEK293 cell lysate Binding of AZT and fluconazole to BSA, HLM and HEK293 cell lysate and to combinations of BSA with each enzyme source was investigated using an equilibrium dialysis method [31]. One side of the dialysis apparatus contained phosphate buffer (0.1 m, pH 7.4), AZT (four to eight concentrations in the range 20–1000 µm) or fluconazole (four concentrations in the range 25–500 µm) and pooled microsomes (0.25 mg ml−1) from the four human livers used in kinetic studies or BSA (2%) or HEK293 cell lysate (1.5 mg ml−1) or the combination of BSA with each enzyme source in a total volume of 1 ml. The other compartment contained phosphate buffer (1 ml) alone. The dialysis cell assembly was immersed in a water bath maintained at 37°C and rotated at 12 r.p.m. for 3 h. Control experiments were also performed with buffer or BSA or HLM or HEK293 cell lysate or the combination of BSA with each enzyme source on both sides of the dialysis cell with high and low concentrations of both drugs to ensure that equilibrium was attained. A 0.2-ml sample was collected from each compartment, treated with 70% HClO4 (10 µl), vortex mixed, and centrifuged (5000 g for 10 min). A 0.12-ml aliquot of the supernatant fraction was transferred to a 1.5-ml Eppendorf tube containing 4 m KOH (10 µl). Mixtures were mixed and centrifuged at 10 000 g for 1 min. A 5-µl aliquot of the supernatant fraction was analysed by HPLC. The HPLC system and conditions for the AZT and fluconazole assays were essentially identical to those described previously for the measurement of GAZT, except that the content of acetonitrile in the mobile phase was increased by 3% and 15% for the AZT and fluconazole assays, respectively. Under these conditions, AZT and fluconazole eluted at 3.3 and 2.2 min, respectively. Standards in the concentration range 10–1000 µm (AZT) and 25–500 µm (fluconazole) were prepared in phosphate buffer (0.1 m, pH 7.4) and treated in the same manner as dialysis samples. The AZT or fluconazole concentrations of dialysis samples were determined by comparison of peak areas with those of the standard curve. Within-day assay imprecision was assessed by measuring AZT (10 and 500 µm) or fluconazole (50 and 300 µm) in five replicate samples containing buffer and the combination of BSA with each enzyme source. Coefficients of variation in all cases were less than 4%.
Data analysis
AZT and fluconazole kinetic parameters Kinetic constants for AZT glucuronidation by HLM or UGT2B7 HEK293 cell lysate were obtained by fitting experimental data to the Michaelis–Menten and substrate inhibition equations [27] using Enzfitter (Biosoft, Cambridge, UK).
The Michaelis–Menten equation is
| (1) |
where v is the rate of reaction, Vmax is the maximum velocity, Km is the Michaelis constant (substrate concentration at 0.5 Vmax) and [S] is the substrate concentration.
The substrate inhibition is
| (2) |
where Ksi is the constant describing the substrate inhibition interaction.
Ki values for fluconazole inhibition of AZT glucuronidation were determined by fitting experimental data to the expressions for competitive, noncompetitive and mixed inhibition using Enzfitter (Biosoft). Goodness of fit to kinetic and inhibition models was assessed from the F statistic, r2 values, parameter standard error estimates and 95% confidence intervals. Kinetic constants are reported as the value ± standard error of the parameter estimate. All data points shown in Figure 1 2 3Figures represent the mean of duplicate measurements (which invariably differed by <10%). The statistical significance of the effects of BSA on the kinetic parameters Km and Vmax were assessed using Student’s paired t-test.
Figure 1.
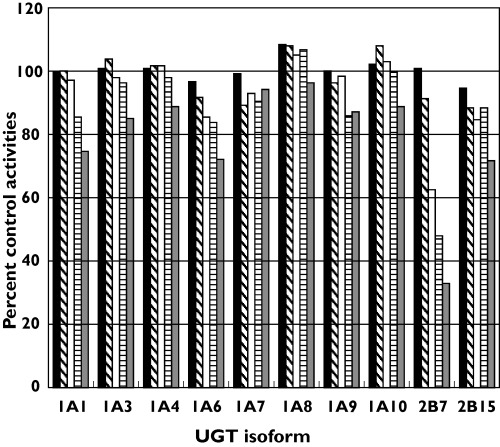
Effect of fluconazole (10–2500 µm) on the activity of human UDP-glucuronosyltransferases (UGTs). Each bar represents the mean percentage activity relative to control from duplicate measurements. 10 µM (▪), 100 µM ( ), 500 µM (□), 1000 µM (
), 500 µM (□), 1000 µM ( ), 2500 µM (
), 2500 µM ( )
)
Figure 2.
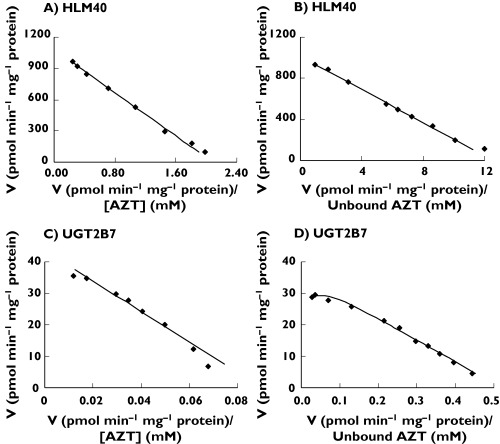
Representative Eadie–Hofstee plots for zidovudine (AZT) glucuronidation by: (A) human liver microsomes (HL40) in the absence of bovine serum albumin (BSA); (B) human liver microsomes (HL40) in the presence of BSA; (C) UGT2B7 in the absence of BSA; and (D) UGT2B7 in the presence of BSA. Points are experimentally determined values, while lines/curves are from model-fitting
Figure 3.
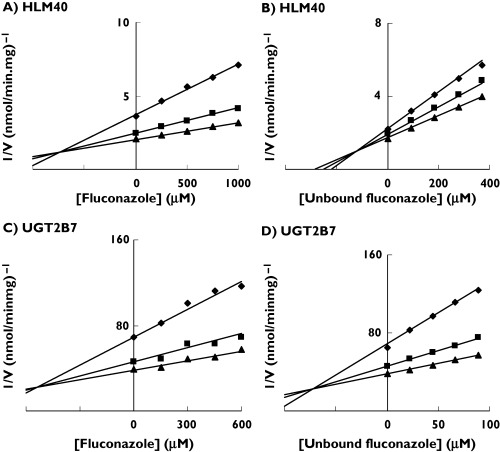
Representative Dixon plots for fluconazole inhibition of zidovudine (AZT) glucuronidation by: (A)human liver microsomes (HL40) in the absence of bovine serum albumin (BSA) 250 µM AZT (♦), 500 µM AZT (▪), 750 µM AZT (▴); (B) human liver microsomes (HL40) in the presence of BSA, 60 µM AZT (♦), 80 µM AZT (▪), 100 µM AZT (▴); (C) UGT2B7 in the absence of BSA, 200 µM AZT (♦), 400 µM AZT (▪), 600 µM AZT (▴); and (D) UGT2B7 in the presence of BSA, 25 µM AZT (♦), 50 µM AZT (▪), 75 µM AZT (▴). Points are experimentally determined values, while lines are from model-fitting
Prediction of AZT glucuronidation clearance Microsomal AZT glucuronidation intrinsic clearance, CLint, was calculated as Vmax/Km (units of µl min−1 mg−1 microsomal protein) and subsequently scaled to the whole liver CLint assuming a liver weight of 1500 g and a microsome yield of 45 mg microsomal protein g−1 of liver [3]. In vivo CLH was then predicted using expressions for the well-stirred, parallel-tube and dispersion models.
Well stirred model:
| (3) |
where fu is fraction unbound in blood and QH is liver blood flow, assumed to be 90 l h−1.
Parallel-tube model:
| (4) |
Dispersion model:
| (5) |
and
CLH = QH (1 − FH)
DN, the dispersion number, may be taken as 0.17 [32] and a = (1 + 4.RN.DN)1/2. RN, the efficiency, is given by
The fraction of drug unbound in blood was evaluated as fu = fu,p /RB, where RB is the blood to plasma concentration ratio and fu,p is the fraction unbound in plasma. For AZT, fu,p was taken as 0.77 and RB as 0.86 [33]. In vivo CLH for AZT glucuronidation was taken from the literature. Mean AZT systemic clearance ranges from 77 to 114 l h−1 per 70 kg [15–17], giving an average value of 94 l h−1 per 70 kg. Since 80% of the recovered dose is accounted for as AZT glucuronide in urine, with the majority of the remainder being unchanged drug [17], the plasma AZT clearance by glucuronidation in vivo may be taken as 75 l h−1 per 70 kg. Taking into account the blood to plasma concentration ratio (RB), the blood AZT clearance by glucuronidation calculated from the plasma clearance and RB was 87 l h−1.
Quantitative prediction of the AZT–fluconazole interaction The extent of inhibition of AZT hepatic clearance by fluconazole (determined as the ratio of the areas under the plasma AZT concentration–time curves with and without fluconazole coadministration, R = AUC(+fluconazole)/AUC(control)), was predicted using the equation for oral administration of a high hepatic clearance drug [5]:
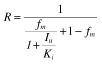 |
(6) |
where Iu is the unbound fluconazole concentration at the enzyme active site; fm is the fraction of AZT metabolism via glucuronidation in the liver, and Ki is the inhibition constant for fluconazole generated in vitro. The extent of inhibition was calculated based on the maximum unbound (Imax,u) and the average unbound (Iav,u) concentrations of fluconazole in serum. The average Imax and Iav values measured in patients who participated in the in vivo fluconazole–AZT interaction study were 77.7 µm and 60.1 µm, respectively [18]. (Patients were studied after 7 days treatment with fluconazole, 400 mg once daily.) The unbound fraction of fluconazole in plasma has been reported as 0.89 [34]. In addition to systemic fluconazole concentrations, the predicted in vivo AUC ratio was estimated based on the unbound fluconazole hepatic inlet concentration (Iinlet,u), calculated as the sum of the maximal unbound plasma concentration in circulating blood (Imax × fu) and the contribution from gastrointestinal absorption after oral administration ( fu × [ka.Fa.Dose/QH]), assuming that the unbound liver concentration equates to that in plasma [5]. The Iinlet,u can be calculated from the equation:
| (7) |
where fu, ka and Fa are the unbound fraction in plasma, the absorption rate constant and the fraction absorbed from the gastrointestinal tract into the portal vein, respectively. The bioavailability of fluconazole is close to 100%[35]. Values for the absorption rate constant of fluconazole have been reported by Demuria et al.[35].
Results
Binding of AZT and fluconazole to human liver microsomes, HEK293 cell lysate and BSA
The binding of AZT and fluconazole was determined as the drug concentration in the buffer compartment divided by the drug concentration in the protein compartment, and expressed as the fraction unbound to incubation constituents ( fuinc). With both AZT and fluconazole, fuinc was independent of concentration. Mean data are shown in Table 1. The binding of AZT to HLM and HEK293 cell lysate was negligible, both in the absence and presence of BSA, despite measurable binding (approximately 5%) to BSA alone. Similarly, fluconazole did not bind nonspecifically to HLM or HEK293 cell lysate. However, binding to BSA (2%), alone and in the presence of HLM and HEK293 cell lysate, ranged from approximately 10% to 15%. Thus, concentrations of fluconazole added to incubation mixtures containing BSA were corrected for binding in experiments that determined a Ki value.
Table 1.
Fraction unbound (fuinc) aof zidovudine (AZT) and fluconazole in presence of human liver microsomes, HEK293 cell lysate, bovine serum albumin (BSA) (2%) and the combination of BSA with each enzyme source
| AZT | Fluconazole | |
|---|---|---|
| HLM (0.25 mg ml−1)b | 1.042 ± 0.017 | 1.043 ± 0.020 |
| Lysate (1.5 mg ml−1)c | 1.010 ± 0.005 | 1.016 ± 0.009 |
| BSA (2%) | 0.942 ± 0.009 | 0.848 ± 0.013 |
| HLM + BSA | 0.980 ± 0.009 | 0.919 ± 0.027 |
| Lysate + BSA | 0.995 ± 0.016 | 0.892 ± 0.020 |
Data presented as mean ± SD.
HLM, human liver microsomes.
Lysate, HEK293 cell lysate.
Inhibition of human UGTs by fluconazole
Fluconazole was screened for inhibition of UGT 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7 and 2B15 using TFP (UGT1A4) or 4MU (all other enzymes) as the ‘probe’ substrates. Inhibition was assessed at the reported Km or S50 value for these substrates with the respective enzymes. Fluconazole inhibited UGT2B7 in a concentration-dependent manner (Figure 1). Inhibition of the other enzymes by fluconazole concentrations in the range 10–1000 µm was negligible or minor (0–15%). At the highest concentration of fluconazole assessed (viz. 2500 µm), inhibition of UGT 1A3, 1A4, 1A7, 1A8, 1A9 and 1A10 was ≤ 14%, although this concentration of fluconazole inhibited UGT 1A1, 1A6 and 2B15 by approximately 25%.
Formation and inhibition kinetics of AZT glucuronidation by human liver microsomes and UGT2B7
AZT glucuronidation by HLM and UGT2B7 in the absence of BSA followed Michaelis–Menten kinetics (Figure 2A,C). Mean (± SD) derived Km and Vmax values for the four livers investigated were 923 ± 357 µm and 1066 ± 325 pmol min−1 mg−1, respectively (Table 2). The Km for AZT glucuronidation by UGT2B7 was 478 µm. Kinetic constants for AZT glucuronidation determined in the absence of BSA are generally similar to those reported previously for this reaction by this laboratory [19] and by Court et al.[20]. The addition of 0.2, 2 and 4% BSA increased the rate of AZT glucuronidation (at a substrate concentration of 500 µm) by 2.7-, 3.5-and 3.6-fold, respectively. Thus, subsequent experiments that investigated the effects of BSA on AZT glucuronidation kinetics and inhibition by fluconazole included 2% BSA. Addition of 2% BSA to incubations caused a 90% reduction in the mean Km (to 91 ± 9 µm) for AZT glucuronidation by HLM, without significantly affecting Vmax (1166 ± 484 pmol min−1 mg−1) (Table 2). Similarly, BSA (2%) caused an approximately 85% reduction in the Km (to 70 µm) for AZT glucuronidation by UGT2B7, with only a minor effect on Vmax (19% reduction) (Table 2). Interestingly, AZT glucuronidation by UGT2B7 in the presence of BSA exhibited weak substrate inhibition (Ksi 5168 µm), whereas the kinetic model for this reaction by HLM (i.e. Michaelis–Menten) was not changed by BSA (Figure 2B,D). When considered in terms of intrinsic clearance, BSA increased this parameter 10-fold and 5.7-fold for the human liver microsomal and UGT2B7 catalysed reactions, respectively (Table 2).
Table 2.
Kinetic constants for zidovudine (AZT) glucuronidation by alamethicin-treated human liver microsomes and UGT2B7 in the absence and presence of bovine serum albumin (BSA)
| Without BSAa,b | With 2% BSAa,c | |||||
|---|---|---|---|---|---|---|
| Km (µm) | Vmax (pmol min−1 mg−1 protein) | cCLintd (µl min−1 mg−1) | Kme (µm) | Vmax (pmol min−1 mg−1 protein) | CLintd (µl min−1 mg−1) | |
| HL10 | 743 ± 2.9 | 731 ± 0.9 | 0.98 | 94 ± 0.01 | 782 ± 0.03 | 8.3 |
| HL12 | 1133 ± 24 | 1503 ± 12 | 1.33 | 90 ± 1.1 | 1876 ± 8.9 | 20.8 |
| HL13 | 1299 ± 17 | 949 ± 5.5 | 0.73 | 103 ± 0.5 | 999 ± 1.8 | 9.7 |
| HL40 | 518 ± 1.4 | 1082 ± 0.8 | 2.09 | 80 ± 0.5 | 1009 ± 2.2 | 12.6 |
| UGT2B7 | 478 ± 14 | 43 ± 0.5 | 0.09 | 70 ± 2.3 | 36 ± 0.6 | 0.51 |
Data presented as mean ± SE of parameter fit.
Best fit to Michaelis–Menten model for all data.
Best fit to Michaelis–Menten model for all data except for UGT2B7, which was fitted to the substrate inhibition expression (Ksi 5168 ± 596 µm).
CLint calculated as Vmax/Km.
Km values expressed as unbound AZT.
Fluconazole was a competitive inhibitor of AZT glucuronidation by HLM and UGT2B7 (Figure 3). The respective Ki values determined in the absence of BSA were 1133 ± 403 µm (mean ± SD) and 529 µm (Table 3). The addition of BSA (2%) caused an 86% reduction in the Ki values for fluconazole inhibition of AZT glucuronidation by HLM (to 145 ± 36 µm) and by UGT2B7 (to 73 µm) (Table 3).
Table 3.
Inhibitor constants for fluconazole inhibition of zidovudine (AZT) glucuronidation by alamethicin-treated human liver microsomes and UGT2B7 in the absence and presence of bovine serum albumin (BSA)
| Without BSA Ki (µm)a,b | With 2% BSA Ki (µm)a,b,c | |
|---|---|---|
| HL10 | 893 ± 12 | 125 ± 7.6 |
| HL12 | 1309 ± 61 | 134 ± 1.5 |
| HL13 | 1609 ± 32 | 199 ± 6.7 |
| HL40 | 719 ± 22 | 122 ± 5.2 |
| UGT2B7 | 529 ± 46 | 73 ± 2.3 |
aCompetitive inhibition.
Data presented as mean ± SE of parameter fit.
Ki values expressed as unbound fluconazole.
In vitro–in vivo correlation
The microsomal CLint values for AZT glucuronidation shown in Table 2 were extrapolated to blood AZT hepatic clearances as described in Data analysis. For kinetic constants determined in the absence of BSA, mean (± SD) predicted hepatic AZT clearances by glucuronidation calculated using the well-stirred (equation 3), parallel-tube (equation 4) and dispersion (equation 5) models were 3.29 ± 1.45 l h−1, 3.35 ± 1.51 l h−1 and 3.33 ± 1.49 l h−1, respectively. For kinetic constants determined in the presence of BSA, respective predicted hepatic clearances calculated using the three models were 24.2 ± 7.34 l h−1, 28.0 ± 9.7 l h−1 and 26.8 ± 8.9 l h−1. As noted in Data analysis, the reported mean hepatic AZT clearance by glucuronidation is approximately 87 l h−1 per 70 kg.
Similarly, the predicted in vivo ratios of the AZT AUC values in the presence and absence of fluconazole were estimated from the Ki values shown in Table 3 using equation 6 (Data analysis). Since glucuronidation is responsible for all but a very minor proportion of hepatic clearance, fm (the fraction of the metabolic process subject to inhibition) was taken as 1. The in vivo AUC ratio was calculated using several estimates of the unbound concentration of inhibitor (i.e. fluconazole); Imax,u, Iav,u and Iinlet,u (Data analysis). The expression for the latter includes the absorption rate constant, ka. Three different values of ka were used to calculate Iinlet,u: the maximum and average values reported in vivo (viz. 0.0318 and 0.0164 min−1) [35] and 0.1 min−1, which is the theoretical maximum assuming absorption is rapid and gastric emptying rate is rate-limiting [5, 11]. Predicted AUC ratios are shown in Table 4. The known mean AUC ratio (viz. 1.92; see Introduction) associated with the AZT–fluconazole interaction was underpredicted (by 85–95%) using Ki values determined with both HLM and UGT2B7 as the enzyme source in the absence of BSA. However, the use of Ki values determined in the presence of BSA predicted mean AUC ratios ranging from 1.38 to 2.05 and 1.73 to 3.0 with HLM and UGT2B7 as the enzyme sources, respectively (Table 4).
Table 4.
Prediction of in vivo interaction between zidovudine (AZT) and fluconazole (cf. observed in vivo AUC ratio of 1.92)
| BSA (2%) | Enzyme source | Ki (µm) | Predicted AUC ratio based on | ||||
|---|---|---|---|---|---|---|---|
| Imax,u | Iav,u | Iinlet,ua | Iinlet,ub | Iinlet,uc | |||
| Absent | HLM (mean ± SD) | 1132 ± 402 | 1.07 ± 0.02 | 1.05 ± 0.02 | 1.08 ± 0.03 | 1.09 ± 0.03 | 1.14 ± 0.05 |
| UGT2B7 | 529 | 1.13 | 1.10 | 1.15 | 1.18 | 1.28 | |
| Present | HLM (mean ± SD) | 145 ± 36 | 1.49 ± 0.10 | 1.38 ± 0.08 | 1.59 ± 0.12 | 1.67 ± 0.14 | 2.05 ± 0.21 |
| UGT2B7 | 73 | 1.94 | 1.73 | 2.11 | 2.28 | 3.0 | |
Discussion
The magnitude of an inhibitory drug interaction in vivo may theoretically be predicted from the ratio of the inhibitor concentration and the inhibition constant (Ki). Although values of Ki determined in vitro, typically using HLM as the enzyme source, have been used to predict interactions arising from inhibition of CYP-mediated drug metabolism [2, 4–6, 8–11], the application of this approach to glucuronidated drugs has received less attention. Thus, we investigated whether interactions arising from inhibition of UGT-catalysed drug metabolism could be determined quantitatively from in vitro kinetic data using the fluconazole–AZT interaction as the model. It was demonstrated that the in vivo interaction may be predicted from in vitro inhibition data, generated using both HLM and recombinant UGT2B7, but only for Ki values determined in the presence of BSA.
Initial studies investigated the selectivity of human UGT inhibition by fluconazole. Fluconazole was a relatively selective inhibitor of UGT2B7, and this provides a mechanistic basis for the AZT–fluconazole interaction since AZT is known to be glucuronidated almost exclusively by UGT2B7 [20]. This observation also indicates that fluconazole may be of use for the reaction phenotyping of human liver microsomal xenobiotic glucuronidation by this UGT2B7. Subsequent experiments showed that fluconazole inhibition of AZT glucuronidation by both HLM and UGT2B7 was competitive. This is consistent with the observation that a small proportion (viz. 6.5%) of orally administered fluconazole is recovered as fluconazole glucuronide in humans [36].
The addition of BSA, at least at low concentrations, to microsomal incubations has been shown to increase the CLint values, via a reduction in Km, of several drugs metabolized by CYP2C9 [21–23]. Furthermore, BSA (1.8–2.25%) has been reported to enhance the rates of human liver microsomal AZT and fenoldopam glucuronidation, up to 15-fold [37, 38]. Here, BSA (2%) increased the mean CLint for human liver microsomal-and UGT2B7-catalysed AZT glucuronidation by 10-and 5.7-fold, respectively, due primarily to a reduction in Km.
Apart from predicting the extent of inhibitory drug interactions, quantitative in vitro–in vivo extrapolation has been employed extensively to calculate in vivo CLH from CLint determined in vitro. Typically, the CLint obtained from microsomal or hepatocyte kinetic data is scaled to a whole liver value and then substituted in the mathematical expressions for models of hepatic clearance. Although the validity of this strategy has been demonstrated for some drugs eliminated by CYP, the approach underpredicts the CLH of AZT and other glucuronidated drugs by one to two orders of magnitude [12, 19, 39, 40]. Despite the increase in microsomal CLint for GAZT formation in the presence of BSA, values of AZT glucuronidation hepatic clearances determined using the well-stirred, parallel-tube and dispersion models still underpredicted the reported mean in vivo CLH for AZT glucuronidation by approximately 3.5-fold. While the difference between the predicted and known in vivo CLH was decreased using in vitro kinetic data generated in the presence of BSA, AZT was not predicted to be a ‘high’ hepatic clearance drug. (It should be noted that, while different scaling factors have been reported for the extrapolation of microsomal CLint, the use of lower or higher estimates of microsome yield would not substantially alter predictivity.) The use of human hepatocytes has been promoted for the generation of in vitro CLint (for example [40]) given the underprediction of in vivo CLH using HLM as the enzyme source. Human kidney microsomes exhibit high activity towards several UGT2B7 substrates [40] and a contribution of the kidney to metabolic clearance in vivo would also affect in vitro–in vivo clearance extrapolation based on human liver microsomal kinetic data. There is no evidence at present for a significant component of renal metabolic clearance to AZT elimination in vivo.
Like Km, the Ki for fluconazole inhibition of human liver microsomal and UGT2B7-catalysed AZT glucuronidation was reduced by almost 90% in the presence of BSA. When substituted in equation 6, the Ki values determined in the absence of BSA did not predict an interaction between fluconazole and AZT. (Unlike CLint, the extrapolation of Ki is not dependent on a microsomal scaling factor.) However, an interaction was predicted by the Ki generated in the presence of BSA, using both HLM and UGT2B7 as the enzyme source. The in vivo AUC ratio was predicted for various concentrations of fluconazole (see below), and ranged from 1.38 to 2.05 and 1.73 to 3.0 using the Ki determined with HLM and UGT2B7, respectively. The mean AZT AUC ratio (in the presence and absence of fluconazole), calculated from apparent oral clearances via glucuronidation, in patients coadministered AZT (200 mg 8-hourly) and fluconazole (400 mg once daily) is 1.92 [18].
In addition to Ki, the predicted in vivo AUC ratio depends on the inhibitor concentration, I, at the enzyme active site. Several estimates of I have been used for in vitro–in vivo correlation, including total and unbound drug plasma concentrations, hepatic input concentration and the concentration in liver tissue [2]. In this work, unbound concentrations in plasma were used to calculate the AZT AUC ratio (equation 6), since it is generally assumed that only unbound drug is available to the enzyme active site. (It should be noted, however, that since the plasma protein binding of fluconazole is low, the use of total plasma concentrations increases the AZT AUC ratios shown in Table 4 by < 10%.) Based on plasma fluconazole concentrations reported in the fluconazole–AZT interaction study [18] and Ki values determined with HLM (in the presence of BSA) as the enzyme source, inclusion of maximum and average unbound fluconazole concentrations in equation 6 under-estimated the known mean increase in AZT AUC by 47% and 59%, respectively. The use of the hepatic input concentration has been recommended as a measure of I[5, 11, 41] but, as shown in equation 7, calculation of this parameter requires additional data, particularly the absorption rate constant (ka). Three values of ka were used to estimate Iinlet,u: the mean and maximum reported absorption rate constants for fluconazole [35] and the theoretical maximum (0.1 min−1) [5]. The predicted increases in the AZT AUCs calculated using inhibitor concentrations based on the experimental rate constants were lower than the mean in vivo value (by 28–36%), whereas use of the theoretical maximum absorption rate constant marginally overestimated the magnitude of the in vivo interaction (by 14%).
Interestingly, the Ki value determined with UGT2B7 as the enzyme source (in the presence of BSA) also predicted inhibition of AZT glucuronidation by fluconazole, although the magnitude of the interaction was generally over-estimated. Nevertheless, this observation suggests that recombinant UGTs may be useful for screening inhibitory interactions between glucuronidated drugs, at least where the selectivity of a reaction is known.
The addition of BSA to incubations of HLM has been reported to reduce the Km of a number of drugs metabolized by CYP2C9 [21–23]. The mechanism of this effect is unknown, but altered protein conformation and the ‘mopping up’ of endogenous inhibitors present in microsomal incubations have been proposed [23]. In the present study, BSA decreased the Km for AZT glucuronidation by both HLM and UGT2B7 (expressed in the mammalian HEK293 cell line). It is possible that components of the commercial BSA preparation used here, such as globulins and fatty acids, may contribute to the effect on AZT glucuronidation, as has been demonstrated recently for CYP2C9 [42]. However, there is recent evidence demonstrating that UGT1A1 binds directly to albumin in vitro[43], and it might be speculated that an interaction between albumin and UGTs enhances substrate binding in vitro and in vivo. Although there are similarities between the effects of BSA on CYP2C9 and UGT2B7 activities, differences are also apparent. The decrease in the Km of CYP2C9 substrates occurs only at low BSA concentrations, with reversal of the effect at 4% BSA [23]. In contrast, the effect of BSA on the activity of UGTB7 was observed to plateau in the range 2–4%. Studies are underway to elucidate the mechanism and universality of the effect of BSA and HSA on xenobiotic glucuronidation.
In summary, fluconazole was shown to be a relatively selective competitive inhibitor of UGT2B7, consistent with the inhibitory interaction between AZT and fluconazole reported in vivo. The addition of BSA to incubations decreased the Km for AZT glucuronidation by both HLM and recombinant UGT2B7, independent of any effect on protein binding. Similarly, BSA (2%) also reduced the Ki for inhibition of HLM-and UGT2B7-catalysed AZT glucuronidation by fluconazole by almost 90%. The Ki values generated in the presence, but not absence, of BSA predicted an interaction between the two drugs to an extent dependent on estimates of the fluconazole concentration in vivo. These data provide preliminary evidence to suggest that a Ki value determined in vitro may predict the magnitude of an inhibitory interaction involving glucuronidated drugs.
This work was supported by grants from Pfizer Global Research and Development and the National Health & Medical Research Council of Australia. V.U. is the recipient of a Flinders University International Postgraduate Research Scholarship.
References
- 1.Halkin H, Katzir I, Kurman I, Jan J, Malkin BB. Preventing drug interactions by online prescription screening in community pharmacies and medical practices. Clin Pharmacol Ther. 2001;69:260–5. doi: 10.1067/mcp.2001.114228. [DOI] [PubMed] [Google Scholar]
- 2.Lin JH. Sense and nonsense in the prediction of drug–drug interactions. Curr Drug Metab. 2000;1:305–31. doi: 10.2174/1389200003338947. [DOI] [PubMed] [Google Scholar]
- 3.Houston JB. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;47:1469–79. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 4.Ito K, Iwatsubo T, Kanamitsu S, Nakajima Y, Sugiyama Y. Quantitative prediction of in vivo drug clearance and drug interactions from in vitro data on metabolism, together with binding and transport. Annu Rev Pharmacol Toxicol. 1998;38:461–99. doi: 10.1146/annurev.pharmtox.38.1.461. [DOI] [PubMed] [Google Scholar]
- 5.Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. Prediction of pharmacokinetic alterations caused by drug–drug interactions: metabolic interaction in the liver. Pharmacol Rev. 1998;50:387–412. [PubMed] [Google Scholar]
- 6.von Moltke LL, Greenblatt DJ, Schmider J, Wright CE, Harmatz JS, Shader RI. In vitro approaches to predicting drug interactions in vivo. Biochem Pharmacol. 1998;55:113–22. doi: 10.1016/s0006-2952(97)00239-6. [DOI] [PubMed] [Google Scholar]
- 7.Yuan R, Parmelee T, Balian JD, Uppoor RS, Ajayi F, Burnett A, Lesko LJ, Marroum P. In vitro metabolic interaction studies: experience of the Food and Drug Administration. Clin Pharmacol Ther. 1999;66:9–15. doi: 10.1016/S0009-9236(99)70048-2. [DOI] [PubMed] [Google Scholar]
- 8.Schmider J, von Moltke LL, Shader RI, Harmatz JS, Greenblatt DJ. Extrapolating in vitro data on drug metabolism to in vivo pharmacokinetics: evaluation of the pharmacokinetic interaction between amitriptyline and fluoxetine. Drug Metab Rev. 1999;31:545–60. doi: 10.1081/dmr-100101935. [DOI] [PubMed] [Google Scholar]
- 9.Komatsu K, Ito K, Nakajima Y, Kanamitsu S, Imaoka S, Funae Y, Green CE, Tyson CA, Shimada N, Sugiyama Y. Prediction of in vivo drug–drug interactions between tolbutamide and various sulfonamides in humans based on in vitro experiments. Drug Metab Dispos. 2000;28:475–81. [PubMed] [Google Scholar]
- 10.Yao C, Levy RH. Inhibition-based metabolic drug–drug interactions: predictions from in vitro data. J Pharm Sci. 2002;91:1923–35. doi: 10.1002/jps.10179. [DOI] [PubMed] [Google Scholar]
- 11.Ito K, Brown HS, Houston JB. Database analyses for the prediction of in vivo drug–drug interactions from in vitro data. Br J Clin Pharmacol. 2004;57:473–86. doi: 10.1111/j.1365-2125.2003.02041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miners JO, Smith PA, Sorich MJ, McKinnon RA, Mackenzie PI. Predicting human drug glucuronidation parameters: application of in vitro and in silico modelling approaches. Annu Rev Pharmacol Toxicol. 2004;44:1–25. doi: 10.1146/annurev.pharmtox.44.101802.121546. [DOI] [PubMed] [Google Scholar]
- 13.Miners JO, Mackenzie PI. Drug glucuronidation in humans. Pharmacol Ther. 1991;51:347–69. doi: 10.1016/0163-7258(91)90065-t. [DOI] [PubMed] [Google Scholar]
- 14.Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, Ball SE. Drug–drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32:1201–8. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- 15.Klecker RW, Jr, Collins JM, Yarchoan R, Thomas R, Jenkins JF, Broder S, Myers CE. Plasma and cerebrospinal fluid pharmacokinetics of 3′-azido-3′-deoxythymidine: a novel pyrimidine analog with potential application for the treatment of patients with AIDS and related diseases. Clin Pharmacol Ther. 1987;41:407–12. doi: 10.1038/clpt.1987.49. [DOI] [PubMed] [Google Scholar]
- 16.Blum MR, Liao SH, Good SS, de Miranda P. Pharmacokinetics and bioavailability of zidovudine in humans. Am J Med. 1988;85:189–94. [PubMed] [Google Scholar]
- 17.Stagg MP, Cretton EM, Kidd L, Diasio RB, Sommadossi JP. Clinical pharmacokinetics of 3′-azido-3′-deoxythymidine (zidovudine) and catabolites with formation of a toxic catabolite, 3′-amino-3′-deoxythymidine. Clin Pharmacol Ther. 1992;51:668–76. doi: 10.1038/clpt.1992.79. [DOI] [PubMed] [Google Scholar]
- 18.Sahai J, Gallicano K, Pakuts A, Cameron DW. Effect of fluconazole on zidovudine pharmacokinetics in patients infected with human immunodeficiency virus. J Infect Dis. 1994;169:1103–7. doi: 10.1093/infdis/169.5.1103. [DOI] [PubMed] [Google Scholar]
- 19.Boase S, Miners JO. In vitro–in vivo correlations for drugs eliminated by glucuronidation: investigations with the model substrate zidovudine. Br J Clin Pharmacol. 2002;54:493–503. doi: 10.1046/j.1365-2125.2002.01669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Court MH, Krishnaswamy S, Hao Q, Duan SX, Patten CJ, Von Moltke LL, Greenblatt DJ. Evaluation of 3′-azido-3′-deoxythymidine, morphine, and codeine as probe substrates for UDP-glucuronosyltransferase 2B7 (UGT2B7) in human liver microsomes: specificity and influence of the UGT2B7*2 polymorphism. Drug Metab Dispos. 2003;31:1125–33. doi: 10.1124/dmd.31.9.1125. [DOI] [PubMed] [Google Scholar]
- 21.Ludden LK, Ludden TM, Collins JM, Pentikis HS, Strong JM. Effect of albumin on the estimation, in vitro, of phenytoin Vmax and Km values: implications for clinical correlation. J Pharmacol Exp Ther. 1997;282:391–6. [PubMed] [Google Scholar]
- 22.Carlile DJ, Hakooz N, Bayliss MK, Houston JB. Microsomal prediction of in vivo clearance of CYP2C9 substrates in humans. Br J Clin Pharmacol. 1999;47:625–35. doi: 10.1046/j.1365-2125.1999.00935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang C, Lin Y, Rodrigues AD, Lin JH. Effect of albumin on phenytoin and tolbutamide metabolism in human liver microsomes: an impact more than protein binding. Drug Metab Dispos. 2002;30:648–54. doi: 10.1124/dmd.30.6.648. [DOI] [PubMed] [Google Scholar]
- 24.Bowalgaha K, Elliot DJ, Mackenzie PI, Knights KM, Swedmark S, Miners JO. Naproxen and desmethylnaproxen glucuronidation by human liver microsomes and recombinant human UDP-glucuronosyltransferases (UGT): role of UGT2B7 in the elimination of naproxen. Br J Clin Pharmacol. 2005;60:423–33. doi: 10.1111/j.1365-2125.2005.02446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sorich MJ, Smith PA, McKinnon RA, Miners JO. Pharmacophore and quantitative structure activity relationship modelling of UDP-glucuronosyltransferase 1A1 (UGT1A1) substrates. Pharmacogenetics. 2002;12:635–45. doi: 10.1097/00008571-200211000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Stone AN, Mackenzie PI, Galetin A, Houston JB, Miners JO. Isoform selectivity and kinetics of morphine 3-and 6-glucuronidation by human udp-glucuronosyltransferases: evidence for atypical glucuronidation kinetics by UGT2B7. Drug Metab Dispos. 2003;31:1086–9. doi: 10.1124/dmd.31.9.1086. [erratum appears in Drug Metab Dispos. 2003; 31: 1541]. [DOI] [PubMed] [Google Scholar]
- 27.Uchaipichat V, Mackenzie PI, Guo XH, Gardener-Stephen D, Galetin A, Houston JB, Miners JO. Human UDP-glucuronosyltransferases. Isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab Dispos. 2004;32:413–23. doi: 10.1124/dmd.32.4.413. [DOI] [PubMed] [Google Scholar]
- 28.Miners JO, Lillywhite KJ, Matthews AP, Jones ME, Birkett DJ. Kinetic and inhibitor studies of 4-methylumbelliferone and 1-naphthol glucuronidation in human liver microsomes. Biochem Pharmacol. 1988;37:665–71. doi: 10.1016/0006-2952(88)90140-2. [DOI] [PubMed] [Google Scholar]
- 29.Breyer-Pfaff U, Becher B, Nusser E, Nill K, Baier-Weber B, Zaunbrecher D, Wachsmuth H, Prox A. Quaternary N-glucuronides of 10-hydroxylated amitriptyline metabolites in human urine. Xenobiotica. 1990;20:727–38. doi: 10.3109/00498259009046888. [DOI] [PubMed] [Google Scholar]
- 30.Uchaipichat V, Mackenzie PI, Elliot DJ, Miners JO. Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine and sulfinpyrazone) ‘probes’ for human UDP-glucuronosyltransferases. Drug Metab Dispos. doi: 10.1124/dmd.105.007369. in press. [DOI] [PubMed] [Google Scholar]
- 31.McLure JA, Miners JO, Birkett DJ. Nonspecific binding of drugs to human liver microsomes. Br J Clin Pharmacol. 2000;49:453–61. doi: 10.1046/j.1365-2125.2000.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roberts MS, Rowland M. Correlation between in-vitro microsomal enzyme activity and whole organ hepatic elimination kinetics: analysis with a dispersion model. J Pharm Pharmacol. 1986;38:177–81. doi: 10.1111/j.2042-7158.1986.tb04540.x. [DOI] [PubMed] [Google Scholar]
- 33.Luzier A, Morse GD. Intravascular distribution of zidovudine: role of plasma proteins and whole blood components. Antiviral Res. 1993;21:267–80. doi: 10.1016/0166-3542(93)90032-e. [DOI] [PubMed] [Google Scholar]
- 34.Debruyne D, Ryckelynck JP. Clinical pharmacokinetics of fluconazole. Clin Pharmacokinet. 1993;24:10–27. doi: 10.2165/00003088-199324010-00002. [DOI] [PubMed] [Google Scholar]
- 35.DeMuria D, Forrest A, Rich J, Scavone JM, Cohen LG, Kazanjian PH. Pharmacokinetics and bioavailability of fluconazole in patients with AIDS. Antimicrob Agents Chemother. 1993;37:2187–92. doi: 10.1128/aac.37.10.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brammer KW, Coakley AJ, Jezequel SG, Tarbit MH. The disposition and metabolism of [14C]fluconazole in humans. Drug Metab Dispos. 1991;19:764–7. [PubMed] [Google Scholar]
- 37.Klecker RW, Collins JM. Stereoselective metabolism of fenoldopam and its metabolites in human liver microsomes, cytosol, and slices. J Cardiovasc Pharmacol. 1997;30:69–74. doi: 10.1097/00005344-199707000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Trapnell CB, Klecker RW, Jamis-Dow C, Collins JM. Glucuronidation of 3′-azido-3′-deoxythymidine (zidovudine) by human liver microsomes: relevance to clinical pharmacokinetic interactions with atovaquone, fluconazole, methadone, and valproic acid. Antimicrob Agents Chemother. 1998;42:1592–6. doi: 10.1128/aac.42.7.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mistry M, Houston JB. Glucuronidation in vitro and in vivo. Comparison of intestinal and hepatic conjugation of morphine, naloxone, and buprenorphine. Drug Metab Dispos. 1987;15:710–7. [PubMed] [Google Scholar]
- 40.Soars MG, Burchell B, Riley RJ. In vitro analysis of human drug glucuronidation and prediction of in vivo metabolic clearance. J Pharmacol Exp Ther. 2002;301:382–90. doi: 10.1124/jpet.301.1.382. [DOI] [PubMed] [Google Scholar]
- 41.Kanamitsu S, Ito K, Sugiyama Y. Quantitative prediction of in vivo drug–drug interactions from in vitro data based on physiological pharmacokinetics: use of maximum unbound concentration of inhibitor at the inlet to the liver. Pharm Res. 2000;17:336–43. doi: 10.1023/a:1007509324428. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Q, Matsumoto S, Ding LR, Fischer NE, Inaba T. The comparative interaction of human and bovine serum albumins with CYP2C9 in human liver microsomes. Life Sci. 2004;75:2145–55. doi: 10.1016/j.lfs.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 43.Ohta Y, Fukushima S, Yamashita N, Niimi T, Kubota T, Akizawa E, Koiwai O. UDP-glucuronosyltransferase1A1 directly binds to albumin. Hepatol Res. 2005;31:241–5. doi: 10.1016/j.hepres.2004.12.007. [DOI] [PubMed] [Google Scholar]