

A summary of a presentation given at the joint AHPPI/BPS meeting, 14 December 2004 at the University of Newcastle
Drug development is a long, complex and expensive activity. Typical development times are between 10 and 15 years at a cost of some £500 million to £1 billion per marketed drug [1]. Surveys over the past 10 years have shown that whereas R & D expenditure is increasing almost exponentially year on year, the number of new molecular entities being registered for marketing is either static or declining [2]. Indeed the situation has become so serious that regulatory authorities, particularly the USA Federal Drugs Authority (FDA), have voiced their concerns that excessive development costs are preventing new life-saving medicines reaching the patient at an affordable price [3]. In their ‘Critical Path’ document, the FDA have asked why the tools of the last century are being used to develop drugs of the 21st. Their view is that ‘A new product development tool kit is urgently needed to improve predictability and efficiency along the critical path’.
Importance of Absorption, Distribution, Metabolism and Excretion (ADME)/Pharmacokinetics (PK) in drug development
One of the reasons for drug failures during development is suboptimal pharmacokinetics. Whilst safety, efficacy and toxicology failures dominate the reasons for drug development termination, drug metabolism could well play a role in all of these. Efficacy failures could arise through too low a concentration of drug reaching the target for an inappropriate amount of time. Safety failures could arise through the wrong concentration reaching the wrong target for too long a time period. Toxicity failures in animals may be through metabolic routes or pathways that do not occur in humans. Indeed Horobin questioned the value of animals in drug development, stating that too much focus was being placed on animal models that may not mirror what happens in humans [4].
Thus understanding the metabolism of a development drug is key to determining whether a new chemical entity (NCE) is ‘druggable’. Current methods of studying drug metabolism pathways prior to human studies rely on animal, in vitro and in silico models. When taking drugs into humans for the first time, there is always a concern that drug metabolism pathways and pharmacokinetics (PK) might differ substantially from those predicted from the model studies. Our pharmaceutical industry contacts state that allometric scaling in which animal model and in vitro PK is used to predict human PK is incorrect in approximately one in three occasions. Bioavailability is often poorly predicted, as are Phase II conjugation pathways. Whilst some metabolic differences may be of no practical consequence, others are so serious that the development programme can be taken no further. The failure rate in Phase I studies of around 30% for metabolism, safety and efficacy reasons is a figure that is both too high and too expensive to be sustainable, especially for small to medium size biotechnology companies.
The phase 0 microdosing concept to obtain early human ADME information
To address the issue of obtaining human drug metabolism PK, a new experimental approach has been developed, known as human Phase 0 or microdosing studies in which subpharmacological, trace doses of drug are administered to human subjects to obtain basic PK parameters such as clearance, volume of distribution, t1/2, etc. Microdosing is dependent on the availability of ultrasensitive analytical methods able to measure drug and metabolite concentrations in the low picogram to femotgram range. Two big nuclear physics have been applied to conduct analyses at these concentrations, viz. accelerator mass spectrometry (AMS) [5, 6] and positron emission tomography (PET) [7, 8]. Both techniques rely on the analysis of radioisotopes incorporated into the drugs under study. In the case of AMS, [14C] is the most useful isotope for drug metabolism studies whereas for PET [11C] is proving to be the most useful. It is worth noting the huge contrast in radioactive half-life of the two isotopes. [14C] has a half-life of 5740 years whilst [11C] has a half-life of 20 min. In the latter case the radiosynthesis laboratory must be in very close proximity to the volunteer or patient enrolled in the study. In contrast the stability of [14C] means that provided no radiolytic or chemical decomposition occurs, the synthesized labelled molecule is stable for many years. AMS is used for determining PK data by taking body samples over time, processing the samples in the laboratory and then analysing their drug content. PET provides primarily PD data through real-time imaging and some limited PK data. In the latter technique PK data can be obtained for only some 2 h after drug administration (i.e. six decay half-lives) whilst we have obtained PK data using AMS for up to 100 days after drug administration.
In Figure 1 a general scheme to conduct AMS microdose studies is shown. It is noteworthy that (a) a minimal toxicology package is required prior to a microdose study and hence only laboratory-scale quantities of drug substance are required, (b) the timelines to conduct a microdose study from commencement of the toxicology to obtaining human PK data are between 4 and 6 months in contrast to Phase I study timelines of 12–18 months and (c) the costs of conducting a microdose study programme are substantially less than a full Phase I study programme.
Figure 1.
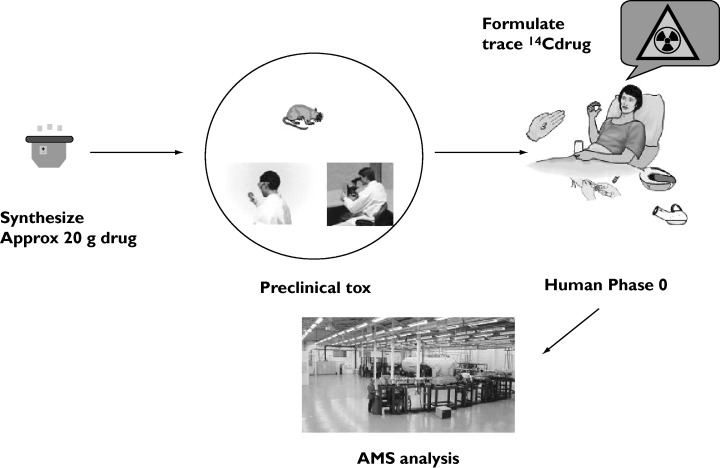
Flow diagram of microdose procedures
Uses of microdosing
The ability to conduct a truncated toxicology programme such as outlined in the EMEA Safety Working Party Position paper [9] and the recent US FDA Exploratory IND [10] ensures that microdosing studies can be used in a number of ways. Both US and European regulatory agencies have defined a microdose as the administration of 100 µg of candidate drug or 100th of the pharmacological dose determined from animal models and in vitro systems, whichever is the lesser. It is only PET and AMS that have the sensitivity to guarantee that drug and/or metabolite concentrations can be determined at these ultralow doses.
If during the drug discovery process, a number of molecules are identified which have good pharmacological activity but similar or differing animal PK, comparative human microdose studies can be conducted to establish human PK. Armed with this information, the human PK data can then be used to (a) assist in the candidate selection process, (b) determine the first dose for the subsequent phase I study on the selected candidate, (c) establish the likely pharmacological dose and (d) calculate the likely cost of goods. For a drug that is expensive to manufacture, the pharmacological dose may be so great that the drug becomes uneconomic to manufacture. This parallel way of conducting microdose studies is most appropriate when several drug candidates are available, perhaps with a common structural core where the radiolabel can be introduced into the core portion of the molecule. These parallel studies are best conducted on between two and five molecules using parallel human subject groups. Each molecule might be administered in a cross-over design such as an intravenous dose followed after a suitable washout period with an oral dose. Thus Vd and CL can be obtained as well as the other standard PK parameters. Both PET and AMS quantify the total number of labelled atoms present in a sample rather than distinguishing between parent drug and metabolite(s). In general researchers wish to know the relative proportion of both in a particular sample or study. This information can be obtained through chromatographic separation of an extract of blood or plasma followed by analysis of collected chromatography fractions. In the case of AMS, exquisite sensitivity has been achieved using this approach.
A further example of the microdosing approach is when microdosing studies are performed compound by compound in an iterative manner. Through each microdosing round, the PK and bioavailability properties of the molecule can be improved to, in the end, provide a molecule with the optimal desired PK properties. In one such study, three molecules were examined in sequence; with each microdosing round the systemic bioavailability was raised from a base value of 10% to a value of 80% in the third molecule. This iterative sequence was conducted in the space of 12 months including the radiosynthesis, toxicology, clinical study and bioanalysis.
In some cases, the drug discovery process might only yield a single molecule. Microdosing can still be useful in such circumstances as it can quickly establish if it is worth taking the molecule forward prior to committing large-scale resources to a full Phase I study. Sometimes a metabolic pathway is identified in human hepatocytes or liver microsomes, which is not seen in animals. Microdosing can be used to establish if the pathway occurs in vivo.
The pros and cons of microdosing
The database for microdosing studies is very small. This is partly due to the length of time required to get new approaches adopted, the lack of validation programmes, scientific inertia and a failure to recognize the potential benefits of microdose studies. Interestingly, (a) adoption is accelerating, (b) the regulatory climate in Europe and the USA has changed, permitting microdose studies to be conducted with a minimal toxicology package, and (c) small to medium size biotech companies are conducting microdose studies earlier than big pharma. Perhaps this reflects a greater willingness to adopt innovative approaches by these scientist-led organizations.
Radiotracer PET assay has the disadvantages of short tracer half-lives and limited specificity (assay may include metabolites). For both PET and AMS, drugs must be labelled at metabolically stable sites.
A ‘microdose’ may not predict the behaviour of clinical doses, although we have a body of evidence that for many drugs linearity or near-linearity is approached. Figure 2 demonstrates in a comparative study the linear clearance kinetics of a microdose and pharmacological dose for an intravenously administered drug. A 100-fold difference in dose administered results in drug plasma concentrations that are also 100-fold different. Nonlinearities may be induced when binding, metabolizing or eliminating systems become saturated. To address this issue, a collaborative industry-sponsored trial has been undertaken in which several drugs, whose human PK is difficult to predict due to, for example, high first pass effects, were dosed at microdose and pharmacological dose concentrations. Comparative PK data were obtained in this study known as the CREAM trial. Of the five drugs studied, microdose PK data reflected pharmacological dose PK for three and gave important metabolism data for one (one compound was a no-test). Whilst this study was not exhaustive, it demonstrated an approximately 70% correspondence between microdose and pharmacological dose PK. Other unpublished studies we are aware of support this percentage predictivity.
Figure 2.
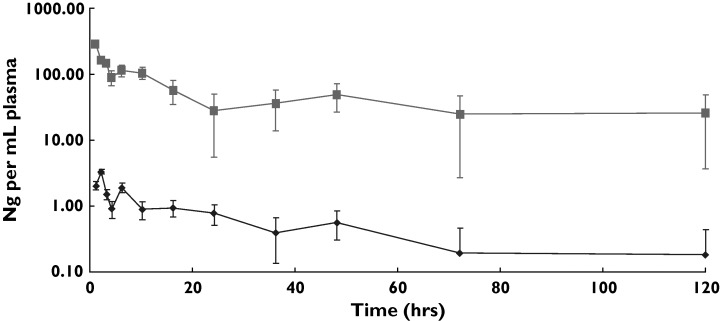
Plasma clearance of parent drug in microdose vs. pharmacological dose study. Results are the geometric mean data from six male volunteers. Blood samples were taken at fixed time intervals, extracted, HPLC profiled and the parent drug fraction collected and analysed by AMS. Therapeutic dose = 10 mg IV, microdose = 100 microgram IV. Micro iv (ng/mL) ( ), ther iv (ng/mL) (░)
), ther iv (ng/mL) (░)
In conclusion, it is our view that microdosing will become an accepted approach in drug development and that eventually all first in human studies will commence with a Phase 0 study. Is it ethical to expose human subjects unnecessarily to a pharmacological dose of potential drug that has poor PK properties, whose development is terminated as a result, when the same information could have been obtained in a microdose study? Has there not been an unnecessary use of animals, including dogs and primates, on the terminated compound? Microdosing will make a contribution to smarter drug development by enabling early human data to be obtained. Drug selection as a result will become more human based and therefore more predictive.
References
- 1.DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Hlth Econ. 2003;22:151–85. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 2.Parexel’s Pharmaceutical R & D. Statistical Sourcebook. Waltham Massachusetts, USA: Parexel; 2003. 2004. [Google Scholar]
- 3.Food and Drug Administration. Innovation or Stagnation. Challenge and Opportunity on the Critical Path to New Medical Products. Washington DC, USA: Food and Drug Administration; 2004. [Google Scholar]
- 4.Horrobin DF. Modern biomedical research: an internally self-consistent universe with little contact with medical reality? Nature Rev Drug Discovery. 2003;2:151–4. doi: 10.1038/nrd1012. [DOI] [PubMed] [Google Scholar]
- 5.Lappin G, Garner RC. Big physics, small doses: the use of AMS and PET in human microdosing of development drugs. Nature Rev Drug Discovery. 2003;2:233–40. doi: 10.1038/nrd1037. [DOI] [PubMed] [Google Scholar]
- 6.Lappin G, Garner RC. Current perspectives of 14C-isotope measurement in biomedical accelerator mass spectrometry. Anal Bioanal Chem. 2004;378:356–64. doi: 10.1007/s00216-003-2348-5. [DOI] [PubMed] [Google Scholar]
- 7.Aboagye EO, Price PM, Jones T. In vivo pharmacokinetics and pharmacodynamics in drug development using positron-emission tomography. Drug Discovery Today. 2001;6:293–302. doi: 10.1016/s1359-6446(01)01684-1. [DOI] [PubMed] [Google Scholar]
- 8.Bergström M, Grahnén A, Langström B. Positron emission tomography microdosing: a new concept with application in tracer and early clinical drug development. Eur J Clin Pharmacol. 2003;59:357–66. doi: 10.1007/s00228-003-0643-x. [DOI] [PubMed] [Google Scholar]
- 9.Committee for Medicinal Products for Human Use. 2004. Position Paper on non-clinical safety studies to support clinical trials with a single microdose. EMEA CPMP/SWP/2599/02/Rev 1,
- 10.Guidance For Industry. Exploratory IND Studies. Guidance. Washington DC, USA: US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research; 2006. [Google Scholar]