

Abstract
Aims
To explore a Bayesian approach for the pharmacokinetic analysis of sirolimus concentration data arising from therapeutic drug monitoring (poorly informative concentration-time point design), and to explore possible covariate relationships for sirolimus pharmacokinetics.
Methods
Sirolimus concentration-time data were available as part of routine clinical care from 25 kidney transplant recipients. Most samples were taken at or near the trough time point at steady state. The data were analyzed using a fully conditional Bayesian approach with PKBUGS (v 1.1)/WinBUGS (v 1.3). Features of the data included noncompliance and missing concentration measurements below the limit of sensitivity of the assay. Informative priors were used.
Results
A two-compartment model with proportional residual error provided the best fit to the data (consisting of 315 sirolimus concentration-time points). The typical value for the apparent clearance (CL/F) was 12.5 l h−1 at the median age of 44 years. Apparent CL was found to be inversely related to age with a posterior probability of a clinically significant effect of 0.734.
Conclusions
A population pharmacokinetic model was developed for sirolimus using a novel approach. Bayesian modelling with informative priors allowed interpretation of a significant covariate relationship, even using poorly informative data.
Keywords: Bayesian population modelling, population pharmacokinetic analysis, prior elicitation, sirolimus, WinBUGS
Introduction
Sirolimus is an immunosuppressive drug approved for the prophylaxis of graft rejection in kidney transplant recipients. The drug is administered orally as a once daily dose [1]. Individualization of treatment with sirolimus is based on assessment of whole blood trough concentrations [1–4]. After administration, sirolimus is rapidly absorbed from the gastrointestinal tract with a mean time (± SD) to reach maximum concentration (tmax) of 1.4 h (± 1.2) [5]. Systemic whole blood concentrations then decline slowly following at least two exponential phases. A long-terminal phase half-life of about 60 h has been reported in kidney transplant recipients [5]. Although a number of studies have reported pharmacokinetic parameter values for sirolimus, the majority only provide data for apparent clearance (CL/F). Mean values of CL/F have ranged from 13 to 23 l h−1 in healthy subjects [6–9], 9–17 l h−1 in kidney transplant recipients [5, 6, 10–12] and 10 l h−1 from a single study in patients with hepatic impairment [6].
In kidney transplant recipients, body weight and body surface area were found to correlate with apparent intercompartmental clearance (Q/F), and apparent volume of distribution of a peripheral compartment (Vp/F) [12]. CL is a pharmacokinetic parameter that can be used to determine the maintenance dose required to achieve the desired steady state concentration. Marked between subject variability of sirolimus CL/F has been shown, with values of between subject variance (BSV) of up to 65%. To date possible patient characteristics that influence or explain the variability in sirolimus CL have not been reported.
Therapeutic drug monitoring of sirolimus is conducted routinely and may provide a valuable resource from which further covariate relationships can be established. However, these type of data are often clustered around a single time point. For sirolimus this is the trough concentration (at 24 h postdose), which from the perspective of model development is poorly informative about the model parameters. In this setting there are two approaches to aid data modelling. These are (i) to simplify the model (see refs [13, 14] for example) or (ii) to incorporate external data to help stabilize the modelling process. The latter approach is more appealing as more complex and realistic models can be built with better predictive performance. External information can be incorporated into the analysis by (i) fixing values for those parameters that are not likely to be estimated with accuracy [13, 14] (ii) combining the clinical data with a previous data set from intensive sampling [15], and (iii) formally incorporating prior information into the analysis using either a non-Bayesian [16] or a fully Bayesian technique [17–19]. Option (i) can lead to bias of the remaining parameter estimates if the pharmacokinetic parameter that is fixed is itself misspecified [20]. Option (ii) is generally not achievable since previous clinical data are either unavailable or not easily accessible and, in any case, they might overwhelm the sparse, poorly informative data. Bayesian techniques offer a way to incorporate prior information with current data.
To date only a few studies have implemented a Bayesian approach to analyze data obtained during routine clinical care [17–19]. Of these only the work of Gillespie et al. [18] used informative priors. We are unaware of any studies that have dealt with the use of informative priors (for pharmacokinetic and pharmacokinetic/pharmacodynamic parameters) pooled from a number of sources for population analysis. Therefore, the obtaining of prior information is an important part of the current analysis. Most reports discussing this issue have elicited opinion from expert investigators [21, 22].
The aims of the present study were (i) to explore the use of a Bayesian population modelling approach with informative priors for the analysis of poorly informative data for sirolimus (i.e. from therapeutic drug monitoring), and (ii) to explore possible covariate relationships to aid dose individualization.
Methods
Sirolimus concentration-time data were collected from 25 kidney transplant recipients who were patients at The Queen Elizabeth Hospital, Woodville, South Australia, where they received sirolimus as part of their routine care. Approval for the use of these data was obtained from the University of Queensland Medical Research Ethics Committee (Clearance no: 2002000668) and the Ethics of Human Research Committee of The Queen Elizabeth Hospital (Clearance no: 2003176). Sirolimus dosing history, drug concentration measurements, patient characteristics, concomitant drug therapy, and routine laboratory results, were extracted retrospectively from patient medical records.
All patients were administered once daily doses of sirolimus. Dose adjustment was based on whole blood trough concentration, which was measured using a high-performance liquid chromatographic assay with ultraviolet detection (HPLC-UV) [23]. The target trough concentration range was 4–12 µg l−1 when sirolimus was given concomitantly with cyclosporin-A, and was 12–20 µg l−1 without cyclosporin-A. The assay was calibrated over the concentration range of 2.5–50 µg l−1. The within-run coefficient of variation (CV) at 2.5 µg l−1 and 50 µg l−1 was 3.8% and 1.9%, respectively. The between-run variability at these same concentrations showed CVs of 9.9% and 1.1%, respectively. The lower limit of quantification (LLOQ) was 2.5 µg l−1. Concentrations below this value were reported by the laboratory as a below the LLOQ, without accompanying numerical values. The assay has also been evaluated by the external proficiency testing schemes for sirolimus (Analytical Services International Ltd [24]).
For the Bayesian population pharmacokinetic analysis, concentration-time data were described using a three-stage hierarchical model. Concentrations were modelled as a nonlinear function of individual-specific pharmacokinetic parameter values (in the first level of the hierarchy) whose distribution around the population mean is specified in the second level of hierarchy. Priors, namely pharmacokinetic data that were available prior to the current study, are given in the third level of hierarchy. The hierarchical models used are described further in Appendix A.
Elicitation of priors for sirolimus
Pharmacokinetic studies of sirolimus were identified using Medline and Embase, and the US FDA website (http://www.fda.gov). Relevant pharmacokinetic parameter values were obtained from eight studies, in which either noncompartmental [5–11] or a compartmental analyses [12] were used to describe the concentration-time data. Two-compartment model parameters were only available from the study by Ferron et al. [12]. Owing to the limited information on two-compartment model parameters, these were also derived [25] from noncompartmental variables reported in a study by Brattstrom et al. [11].
If more than one study design was used (e.g. comparisons of two treatment groups or different patient groups), information from each group was treated naïvely as if it came from a separate study. The study arm in which diltiazem and sirolimus were co-administered in the work of Böttiger et al. [9] was excluded because the CL/F of sirolimus decreased by approximately 38% during co-administration with diltiazem.
A total of 14 sets of pharmacokinetic parameter data for sirolimus (mean and variance) were available for computation of the priors. Appendix B contains the relevant equations and details of the method used for determining the informative prior pharmacokinetic parameters. Briefly, a meta-analytic technique with a reciprocal variance weighting approach was used to calculate the weighted means of structural pharmacokinetic parameters and their between subject variance (BSV). A standard equation for weighted standard deviation [26] was applied, which was then used to compute the precision of the structural pharmacokinetic model parameter values. Simulation was used to estimate the precision of BSV (see appendix B).
Bayesian analysis
A fully conditional hierarchical Bayesian analysis was undertaken using PKBUGS (v 1.1), an interface of the software program WinBUGS 1.3 (MRC Biostatistics Unit, Cambridge, UK). In WinBUGS, Markov chain Monte Carlo (MCMC) techniques are used to make inferences about posterior distributions of the parameters of interest. MCMC is an iterative simulation based approach, Duffull et al. [27] and Lunn et al. [28] review Bayesian modelling of pharmacokinetic data that involves these techniques.
Using the default approach in PKBUGS, models were parameterized in terms of the natural log of the parameters values (e.g. ln(CL), ln(V) [29]. In the present study, two MCMC chains were run simultaneously. The initial estimates of chain-1 were the same as the prior means, and the initial estimates of chain-2 were perturbed by at least 50%. Convergence of the MCMC chains were assessed using the Gelman & Rubin convergence diagnostic (GR value). Both MCMC chains were assumed to have reached the stationary distribution if GR values were less than 1.2 for all parameters [30]. Furthermore, the trace history of MCMC samples for both chains were examined visually for all parameters, in which a ‘fuzzy caterpillar’ [29] suggests that MCMC chains had reached a stationary distribution.
The two MCMC chains were run for 20 000 samples (excluding the 4000 samples that are discarded during the burn in phase). The two MCMC chains were pooled to provide the population pharmacokinetic parameter values, and residual error standard deviation. An extra 1000 updates were subsequently performed, in which individual pharmacokinetic parameters were monitored, which reduced run times. The posterior median of individual parameter estimates was obtained and used for screening the possible relationships with covariates. Additionally, 1000 values of the individual parameter estimates were exported into NCSS software (NCSS Statistical Software., Kaysville, Utah, USA) for graphical representation.
Model selection
Bayesian model selection was generally based on the deviance value (equivalent to −2 log likelihood). Empirically, the model providing the smaller deviance was preferred.
To decide whether a one- or a two-compartment model provided the better fit to the data, a mixture parameter called Mix, was used to discriminate between competing models when both were fitted simultaneously to the data [31–33]. Thus,
| (1) |
where  and
and 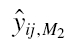 are the predicted concentration for the jth observation of the ith individual based for model 1 and model 2, respectively.
are the predicted concentration for the jth observation of the ith individual based for model 1 and model 2, respectively.
Mix provides the probability that the data supports a two-compartment model. The prior for Mix was assumed to be noninformative, and to have a uniform distribution between 0 and 1 [Mix ∼ U(0, 1)].
Model building
A population pharmacokinetic analysis for sirolimus was conducted using following steps: (i) identification of the structural and residual error models; (ii) estimation of the compliance factor (CFi) for patients who were suspected to be non-compliant as noted by the clinical team; and (iii) covariate modelling. For all analyses, missing concentration-time pairs (where values were below the LLOQ) were included (see below). One- and two-compartment models were initially explored. Three different types of residual error variance models (additive, proportional, and mixed additive and proportional error) were tested.
Sirolimus concentrations below the LLOQ were treated as missing observations. Their influence was assessed by computing their expected contribution to the likelihood using the MCMC integration process. This is a relatively simple procedure using PKBUGS/WinBUGS by limiting the possible range of values for the missing data to between zero and the LLOQ. This provides the boundaries for computing the expected value. The MCMC process then automatically evaluates the expected value from within the boundary range [29].
Noncompliance
Noncompliant patients were identified from information documented by the clinician in the medical notes. All patients were considered to have been initially fully compliant, but some were noted to have become noncompliant during their continuing treatment. During the MCMC sampling, a compliance factor (CFi) was estimated and used as a correction factor (equation 2). This is a relatively simplistic approach where the percent dose ingested is estimated. A more sophisticated Bayesian approach to noncompliance is provided by Mu & Ludden [34]. Thus, if concentration data were obtained before noncompliance was noted, CFi was given the value ‘1’, otherwise CFi was assumed to arise from a lognormal distribution centred on a geometric mean of 1. Once noncompliance was suspected, an average CFi was estimated for the remainder of their course of treatment.
| (2) |
where  is the jth predicted concentration for the ith individual and f(θi, xij) is the structural model predicted concentration.
is the jth predicted concentration for the ith individual and f(θi, xij) is the structural model predicted concentration.
Covariate modelling
Scatter plots showing the relationship between the posterior median value of individual pharmacokinetic parameters and a range of variables were visually investigated to screen for potential covariates. Variables investigated were age, total bilirubin, albumin, haematocrit, body weight, postoperative days, and gender. Covariates for which a trend with pharmacokinetic parameters was demonstrated were considered for inclusion into the final model using the log-linear relationship,
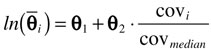 |
(3) |
where θ1 and θ2 are the population regression coefficients when a covariate was included in the model, 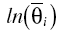 represents the natural log of the typical value for an individual with those characteristics, covi is the median covariate value of the ith individual and covmedian is the median covariate over the population.
represents the natural log of the typical value for an individual with those characteristics, covi is the median covariate value of the ith individual and covmedian is the median covariate over the population.
Model selection for inclusion of a covariate in the final base model was based on (i) the decrease in the deviance values, (ii) the decrease in between subject variance and (iii) the probability of a clinically significant influence on the parameter values, defined as a change in the population parameter of at least 20% between subjects with the lowest and the highest covariate value in the data set.
A different model was used for haematocrit (HCT). As sirolimus is highly bound to red blood cells, the higher the haematocrit, the higher the expected total sirolimus concentration should be. However, since sirolimus toxicity may result in depressed haematocrit concentrations, such a relationship is naturally confounded. The influence of haematocrit on the residual error variance was explored, in which the residual variance of the observed concentrations is expected to be partly explained by variability in haematocrit values.
The model for residual error variance without HCT (shown only for the proportional error variance as in the final base model (number 6, Table 3)) is represented by equation 4 and that with HCT by equation 5:
Table 3.
Posterior medians and 95% credible intervals of deviance
| Number | Model | Deviance median | 95% credible interval |
|---|---|---|---|
| 1 | 1-cpt with additive residual error | 1866 | 1849–1889 |
| 2 | 1-cpt with proportional residual error | 1730 | 1712–1751 |
| 3 | 1-cpt with mixed residual error | 1731 | 1714–1752 |
| 4 | 2-cpt with proportional residual error | 1717 | 1701–1737 |
| 5 | 2-cpt with proportional residual error +CFi | 1681 | 1664–1702 |
| 6 | 2-cpt with proportional residual error +CFi + age-CL | 1681 | 1663–1702 |
1-cpt = one-compartment model; 2-cpt = two-compartment model; CFi = compliance factor.
| (4) |
| (5) |
where τij is the precision of prediction for the jth observations of the ith individual, σ is the residual standard deviation, based on a uniform distribution (U) [σ1 ∼ U(0, 0.5), σ2 ∼ U(0, 10)] and HCTij is the haematocrit value obtained on the jth observation of the ith individual.
Sensitivity analysis
The sensitivity of the posterior distribution of the parameter estimates to specification of the priors was evaluated in the following settings:
Scenario 1:
The sensitivity of the posterior distribution to the choice of a multivariate normal distribution for the individual parameters around the population value.
The distributions of individual parameter estimates were allowed to have arisen from a multivariate t-distribution, rather than a multivariate normal distribution. The degrees of freedom (ν) of the p-dimensional multivariate t-distribution (MVtp) were set empirically to 10. Low degrees of freedom were chosen to provide heavy tails in order to accommodate possible outliers. The values of the prior mean and variance were the same as used in the final model.
Scenario 2:
The sensitivity of the posterior distribution to the informativeness of the priors.
The analysis was repeated using less informative priors. The latter were obtained by decreasing the precision of the pharmacokinetic parameters (Σθ−1) and of the BSV (ρ). Σθ−1 was calculated using a value of twice the original standard deviation of the weighted mean for the structural pharmacokinetic model parameters. For the Wishart distribution, ρ was re-estimated using a value 50% higher than the original observed 75th percentile of between subject variance (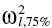 ). The 25th percentile was not included in the calculation since in this setting the informativeness of the Wishart is related to the upper interval of the simulation matrices (see Appendix B).
). The 25th percentile was not included in the calculation since in this setting the informativeness of the Wishart is related to the upper interval of the simulation matrices (see Appendix B).
Results
Patients
The demographics of the 25 kidney transplant recipients studied (14M/11F) are summarized in Table 1. Twenty-one of these were ‘rescued’ patients (i.e. sirolimus was prescribed after an episode of graft rejection, delayed graft function or after the occurrence of toxicity while receiving other immunosuppressive drug(s)). The other four patients were prescribed sirolimus as part of a primary immunosuppressive regimen following transplantation. Over the time frame that data were reviewed, three patients were suspected to be noncompliant with therapy by the clinical team.
Table 1.
Patient demographics (25 patients)
| Mean ± SDa | Range | |
|---|---|---|
| Age (years) | 44 ± 13 | 22–73 |
| Days after transplantation (days) | 618 ± 847 | 2–3918 |
| Weight (kg) | 73 ± 15 | 50–115 |
| Body mass index (kg m−2) | 27 ± 5.5 | 19–44 |
| Liver function | ||
| Total bilirubin (µmol l−1) | 10 ± 4.7 | 2–50 |
| ALP (U l−1) | 83 ± 30 | 28–332 |
| AST (U l−1) | 25 ± 13 | 5–336 |
| GGT (U l−1) | 56 ± 45 | 7–355 |
| ALT (U l−1) | 39 ± 44 | 4–751 |
| Renal function | ||
| Serum creatinine (mmol l−1) | 0.24 ± 0.16 | 0.07–1.01 |
| CLcrb (ml min−1) | 45.3 ± 21.6 | 7.1–141 |
| Others | ||
| Albumin (g l−1) | 37 ± 4.6 | 17–49 |
| Globulins (g l−1) | 28 ± 4.8 | 5–51 |
| Haematocrit | 0.33 ± 0.05 | 0.2–0.51 |
| WBC (x 109 l−1) | 7.4 ± 2.5 | 1–21 |
| PLT (x 109 l−1) | 233 ± 78.6 | 37–766 |
| Glucose (mmol l−1) | 7 ± 3 | 2–43.8 |
Computed from the average value for each individual.
CLcrwas calculated using Cockcroft & Gault formula [38].
The mean (± SD) sirolimus dose over the data collection period was 6 (± 3) mg day−1 (range 2–20 mg day−1). Patients studied were receiving combination immunosuppressive therapy including sirolimus with mycophenolate mofetil and corticosteroids (n = 15), cyclosporin-A and corticosteroids (n = 3), tacrolimus and corticosteroids (n = 4), corticosteroids (n = 2) and mycophenolate mofetil (n = 1).
Concentration-time data
Three hundred and fifteen concentration-time points were available from the 25 patients. These data were obtained at a median of 278 days after transplantation (from 2 days to 10.7 years), and ranged from 3 to 20 observations per patient. The median blood sampling time was 23.7 h postdose with a range of 1.6–77 h. A plot of sirolimus concentration vs. time postdose is shown in Figure 1.
Figure 1.

Scatter plot of sirolimus concentrations (µg l−1) vs. time postdose in 25 kidney transplant recipients (315 observations). A single postdose concentration was obtained at about 77 h after the previous dose in one patient
Fourteen of the concentrations were from three patients suspected of noncompliance. In addition, 13 observations were below the LLOQ of 2.5 µg l−1. Ten of these were from patients whose compliance had been questioned.
Priors
Priors elicited from 14 sets of pharmacokinetic data are presented in Table 2. CL and volume of distribution (V) terms are apparent terms because their values are scaled by bioavailability (F), e.g. CL/F or V/F for one-compartment model. For simplicity, F is omitted from further descriptions.
Table 2.
Priors for the Bayesian population analysis of sirolimus concentration data

The noninformative residual error standard deviation for the additive and proportional error models were given as σadd ∼ U(0, 6), and σprop ∼ U(0, 0.5), respectively. The upper boundaries were empirically chosen, in which the maximum residual standard deviation was about half of the mean concentration for the additive model, and in which the variation was about 50% for the proportional error model. There are no residual error variance data for sirolimus. A residual error of 14.4% has been reported for tacrolimus [35], another immunosuppressant sharing a similar structure to sirolimus. Therefore, using a value of 50% for sirolimus is erring on the side of caution.
Structural and residual error model
As shown in Table 3, for a one-compartment model, the proportional residual error model (number 2) and the mixed residual error model (number 3) provided a better fit (lower deviance) compared with the additive residual error model (number 1). A two-compartment model with proportional residual error variance was then fitted to the data. The posterior median of the deviance was found to be lower than that of the one-compartment model (1717 vs. 1730). When both one- and two-compartment models were fitted simultaneously, the posterior probability in favour of the two-compartment model was greater than 0.975, and the posterior median of the mixture parameter (Mix) was 0.891 with a 95% credible interval of 0.6638–0.9915. The latter refers to the 95th percentile range of the parameter distribution. These results strongly supported the use of the more complex model with an odds ratio of 8.2. Hence, a two-compartment model was chosen for further analysis.
Modelling in the presence of non-compliance
The addition of a compliance factor (CFi) resulted in a drop in the posterior median of the deviance by 36 points (from 1717 to 1681, number 4 vs. number 5 in Table 3). Thus model 5 was used for the investigation of covariate relationships.
The median (95% credible interval) compliance factors for individual patients 3, 4, and 22 were 0.5 (0.3–0.7), 0.2 (0.1–0.4), and 0.3 (0.2–0.6), respectively. These values imply that, on average, about 50%, 20% and 30% of the nominal doses were taken by these patients, respectively.
Covariate modelling
Covariate relationships were investigated for CL only, due to the sparseness of the information about the other parameters.
Scatter plots of the posterior median of individual CL and individual median covariate values are shown in Figure 2. Among the screened covariates, a trend to a decrease in CL with advancing age was seen. The posterior distribution of individual ln(CL) vs. age is presented in Figure 3A and for comparison the median of the posterior is shown in Figure 3B. The influence of age on ln(CL) was explored by the use of a log-linear model (equation 3). The median age in this data set was 44 years. The informative priors (as used in the final base model) were applied for the intercept value (θ1) of CL-age model, and also for Q, Vc, Vp, and ka, whereas a noninformative prior was used for the regression coefficient (slope, θ2). Prior mean and precision of θ2 were set to 0 and 0.01, respectively.
Figure 2.

Plots of posterior median of individual ln(CL) vs. covariates [gender 0 = male, 1 = female; Tbil = total bilirubin; POD = post-transplantation days; ALB = albumin; WT = body weight]
Figure 3.

Plots of ln(CL) vs. age. Panel A: the whole posterior distribution of individual ln(CL), in which the open circle (○) is the median; the vertical line represents the interquartile range; and the shaded area represents the posterior distribution, reflected around the vertical axis. Panel B: posterior median of individual ln(CL). Note that the scale on the x-axis is the same for both panels
When age was included in the model, the posterior median of deviance (95% credible interval) remained at 1681 (1663–1702). The posterior median of BSV(CL) (95% credible interval) decreased from 0.1925 (0.1595–0.236) in the final base model to 0.1903 (0.1571–0.2337) with the inclusion of age. The probability that age had a clinically significant effect on CL was 0.734 and this covariate was included in the final model. For a person of 44 years of age, the typical CL/F was 12.5 l h−1.
The influence of total bilirubin (Tbil), as an index of liver function, and time after transplantation (post-transplantation days (POD)) seemed to be driven by one extreme value in each case (Tbil = 28 µmol l−1, POD = 3909 days). The relationship between CL and albumin, weight and gender appeared minimal. Hence they were not included in the model.
The inclusion of haematocrit concentration did not further decrease the posterior median of the deviance (1682, 95% credible interval = 1664–1704) compared with the previous best model (number 6, Table 3). The residual error variance from the final base model decreased by 14% after the inclusion of HCT, which was considered to be of minimal significance. Thus, HCT was not included in the final model.
In summary, the model providing the best fit for this sirolimus data set was a two-compartment model with a proportional residual error variance and with the inclusion of age as a covariate for predicting CL. The scatter plots of the individual predicted vs. observed concentrations obtained from this model, and the weighted residual vs. predicted concentrations are shown in Figure 4. No outliers were observed based on the weighted residual plots. The pharmacokinetic parameters from the final model are shown in Table 4.
Figure 4.

A: Plots of the posterior median of individual predicted concentration vs. observed concentrations obtained from the final model (two-compartment with proportional residual error variance, with the inclusion of age for the parameter CL. The solid line is the line of identity. B: The weighted residual vs. the posterior median of individual predicted concentrations
Table 4.
Sirolimus population pharmacokinetic parameters
| Posterior median of population PK parameters (95% credible interval) | |
|---|---|
| CL/F (l h−1) | 12.94a (9.46, 16.43) |
| Q/F (l h−1) | 20.4 (13.1, 32.4) |
| Vc/F (l) | 117 (99.4, 138) |
| Vp/F (l) | 583 (459, 736) |
| ka*b (h−1) | 2.195 (2.123, 2.269) |
| Posterior median of regression parameters for covariate model (95% credible interval) | |
|---|---|
 |
θ1 = 2.827 (2.408, 3.249)c |
| θ2 = −0.2987 (−0.726, 0.119)c | |
| agemedian = 44 years | |
| Between subject variance-covariance matrix (95% credible interval) | |||||
|---|---|---|---|---|---|
| CL/F | Q/F | Vc/F | Vp/F | ka* | |
| CL/F | 0.1903 (0.1571, 0.2337) | ||||
| Q/F | 1.91E-4 (−0.0202, 0.0209) | 0.1035 (0.08448, 0.1282) | |||
| Vc/F | −1.35E-5 (−0.0356, 0.0356) | 4.21E-4 (−0.0264, 0.0270) | 0.305 (0.250, 0.378) | ||
| Vp/F | 3.52E-4 (−0.0160, 0.0170) | −4.46E-4 (−0.0131, 0.0119) | −2.73E-4 (−0.0217, 0.0206) | 0.0654 (0.0533, 0.0814) | |
| ka* | 9.98E-5 (−0.0242, 0.0242) | 6.22E-5 (−0.0185, 0.0185) | 4.03E-5 (−0.0317, 0.0321) | 2.93E-5 (−0.0147, 0.0148) | 0.1449 (0.1186, 0.1792) |
| Residual error standard deviation (95% credible interval) |
|---|
| 0.3768 (0.3451, 0.4146) |
results are the median of exponential of the typical individual In(CL) for each pair of θ1and θ2when age was included in the model.
ka = ka*+ CL/Vc, ka≈2.305 h–1.
natural log values: exp(θ1) = 16.9 (11.1, 25.8) and exp(θ2) = 0.742 (0.484, 1.13).
Sensitivity analysis
Results for CL from the sensitivity analysis are presented in Figure 5.
Figure 5.

Priors and 95% credible interval of posterior of the intercept (θ1, top panel) and slope parameter (θ2, bottom panel) of covariate model for CL. In scenario 1 (left panel), the distributional assumption about the parameter values for all parameters was evaluated (normal vs. t-distribution). In scenario 2 (right panel), the sensitivity to informativeness of the prior was evaluated. θi = individual PK parameter estimates; ∼MVN = random sample from a multivariate normal distribution; ∼MVt = random sample from a multivariate t distribution
In scenario 1, when individual parameter estimates were assumed to arise from a multivariate t-distribution, the results showed that the posterior distribution of θ1 was similar to that from the final model, in which a multivariate normal distribution was assumed (top left panel, Figure 5). This was also shown for the regression coefficient, θ2 (bottom left panel, Figure 5). These findings suggested that inference about the pharmacokinetic parameter estimates from the final model were robust to the assumption of normal distribution.
In scenario 2, less informative priors were used for all structural model parameters (including θ1 in the log-linear CL-age model, Q, Vc, Vp and ka), while a noninformative prior was retained for θ2 as in previous analyses. Although less informative priors were used for all other parameters, the inverse relationship between CL and age remained significant (bottom right panel, Figure 5). The posterior median for the regression coefficient of ln(CL), θ2 was −0.4664, with a 95% credible interval of −0.9974 to 0.0765 with less informative priors, compared with −0.2987 (95% credible interval of −0.726 to 0.119) from the final best model (with informative priors). The posterior median of θ1 was 3.003, with a 95% credible interval of 2.43 to 3.551 compared with 2.827 (95% credible interval of 2.408 to 3.249) from the final best model. Changing the prior to be less informative did not appear to affect the ability to quantify the relationship between age and CL.
Discussion
We have explored the use of a fully conditional Bayesian modelling approach to analyse sirolimus concentration-time data gathered from routine clinical monitoring. Observations were clustered around the 24 h after dosing time point. This data set contained observations below the lower limit of quantification (LLOQ) and from noncompliant patients. In contrast to well-controlled studies in the early phase of drug development, data that are obtained from routine clinical monitoring are often sparse and poorly informative (i.e. mostly trough concentrations).
We have shown with the use of informative priors that model pharmacokinetic parameters for sirolimus can be estimated for a relatively complex model. An unexpected finding was that a two-compartment model provided a better fit to such a poorly informative design with high process noise. When both one- and two-compartment model were fitted simultaneously, the odds of a two-compartment model being preferred was 8.2, which suggested a strong preference for the more complex model. We were also able to define a relationship between sirolimus CL and age.
Several issues relating to the analysis of poorly informative data have been addressed in this study. First, data below the LLOQ were modelled by averaging over the predicted missing values during MCMC sampling. This process happens naturally as a part of the Bayesian framework which allows computation of the expected missing concentrations with minimal extra coding (by setting the EVID item to 2 in the data file as well as providing upper and lower boundaries for the missing concentration data items). Second, data from noncompliant patients were incorporated into the analysis, and a mean compliance factor was estimated based on data obtained after noncompliance was first suspected.
Implementation of a fully conditional Bayesian approach for the analysis allows the posterior distribution of parameters to be estimated, conditional on informative priors. The choice for the use of either informative or noninformative priors depends on the nature of data and the desired experimental outcomes. It has been argued that the use of priors incorporates a subjective element to an analysis. Many researchers aim to overcome this criticism by using so-called noninformative priors, although it is apparent that no proper prior is truly noninformative (although many can be very poorly informative). The corollary to this argument is that informative priors that are elicited in an objective manner can provide an important contribution to our understanding of (pharmacokinetic) processes in circumstances where the data may otherwise have yielded little valuable information. A test of any prior is how well the model performs with respect to an important point of inference, when the priors are subsequently set to be less informative.
In our study when less informative priors were applied to all parameters, the posterior distribution of CL and the relationship with age remained largely unaffected. The relationship of CL and age was observed to be weaker when informative priors were used, compared with the use of less informative priors (θ2 = −0.2987 vs.−0.4664, respectively). This is possibly because the informative priors were collated from the information in which age had not been identified and included in the model. Hence, there may be a lower likelihood of successfully identifying possible covariate relationships. Nonetheless a significant relationship was observed even when using these priors.
The other concern regarding the use of informative priors relates to how precisely parameters have been estimated in previous studies. For sirolimus, no standard errors of estimation have been reported. Data for elicitation of priors for the two-compartment model parameters were derived from only two studies [11, 12]. Sirolimus blood concentrations were collected before, and at 0.5, 1, 2, 3, 6, 12, 24, 48, 72, 96, 120, and 144 h after a single dose of sirolimus from 36 and12 kidney transplant patients in the studies by Ferron et al. [12] and Brattstrom et al. [11], respectively. Hence, we considered that these data were relatively similar and would provide reasonably precise pharmacokinetic parameter estimates.
A finding of this analysis, and most likely for similar analyses where essentially only trough concentration-time data were available, was the small change in the posterior distributions (relative to the prior distributions) of the parameters relating to absorption and central volume of distribution. The posterior median of Vc (95% distribution) was 117 l (99.4, 138), and the prior was 117 l (99.1, 138). Similarly, the posterior median of ka (95% distribution) was 2.195 h−1 (2.123, 2.269), and the prior was 2.195 h−1 (2.124, 2.268). This could mean either that the prior distribution described the data exactly and hence the prior and posterior were exchangeable, or that the data contained almost no information on these two parameters. The latter is the most likely explanation. In contrast, a change posterior median and/or a tightening of posterior distribution was observed for CL, Q, and Vp. The posterior median of CL was lower than its prior value by 0.7 l h−1, and a tighter 95% credible distribution was clearly shown [the posterior (95% distribution) value was 12.94 l h−1 (9.46, 16.43) compared with 13.6 l h−1 (7.9, 23.5) for priors]. The posterior median of Q and Vp was different from their priors, and the 95% range was tighter for Q, but not for Vp. The posterior median of Q (95% distribution) was 20.4 l h−1 (13.1, 32.4) whereas the prior was 28.3 l h−1 (16.8, 47.8). The posterior median of Vp (95% distribution) was 583 l (459, 736) compared with a prior value of 480 l (363, 635). This suggested that data were informative for CL, Q, and Vp, although from a covariate modelling perspective only CL was of interest. For the population mean of between subject variance, a slight shift from the priors for CL was observed for its distributions. The posterior median for the BSV of CL (95% range) was 0.19 (0.1571, 0.2337), whereas its prior distribution was 0.21 (0.17, 0.26). The data were less informative for the BSV of the other parameters.
Our results indicated an inverse relationship between patient age and CL. In our study, clinical significance was defined as a change in CL equal to or greater than 20% between subjects with the lowest and the highest covariate value in the data set. The value of 20% was chosen as representing a difference that might be important enough clinically to warrant a change in dosage. Patient age can be a good predictor of sirolimus CL in kidney transplant recipients. However, in the present analysis, only 25% of patients were older than 50 years. The influence of this covariate should be assessed further to determine whether this relationship continues at ages that are outside of the range of the present study and whether age is surrogate for a more biologically meaningful explanatory variable in future studies. It is unclear why age has a particularly strong influence on sirolimus CL, although a number of physiological processes are known to decline with age, such as organ blood flow, kidney function and perhaps to a lesser extent hepatic function.
Others have not shown that age is a determinant of sirolimus clearance [12, 36]. Recently, Boni et al. [36] characterized the pharmacokinetic profiles of temsirolimus (CCI-779) and its major metabolite, sirolimus, in patients with advance renal cancer. It was found that CCI-779 dose was a significant covariate of sirolimus clearance, whereas haematocrit concentration was a significant covariate of the volume of distribution of the peripheral compartment. Age, sex, and race were not found to be significant. A lack of ability to identify an age-clearance relationship even if concentration-time data were considered to be more informative, may be, in part, due to the differences in the population studied by Boni et al. [36] compared with our population which had a greater age range. It is also possible that age-clearance relationship is weaker in patients with advance renal cell carcinoma. Nonetheless, studies aimed to confirm our finding are needed.
In summary, the use of a Bayesian analysis framework allows incorporation of prior information about pharmacokinetic parameters, which helps stabilize the modelling process in a population analysis of poorly informative data available from routine sirolimus monitoring. In addition, covariates were identified from these sparse data that otherwise may have been missed without the use of Bayesian analysis with informative priors. A population pharmacokinetic model, with covariates, was successfully developed for sirolimus, using data readily available from routine clinical monitoring.
Acknowledgments
This work was financially supported by a grant funded by the NHMRC (#210173).
Appendix A: Bayesian hierarchical models
Stage 1: model for the residual error variance
The jth observed concentration for the ith individual (yij) was assumed to be normally distributed around the individual predicted concentration (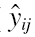 ), with an unknown precision of prediction (τ). Note that in Bayesian analysis, variance is modelled in terms of precision, e.g. τ is the inverse of residual error variance, σ2.
), with an unknown precision of prediction (τ). Note that in Bayesian analysis, variance is modelled in terms of precision, e.g. τ is the inverse of residual error variance, σ2.
| (a1) |
In this notation, and elsewhere, N is used to denote a normal distribution; ‘∼’ is used to signify ‘a random sample from’.
Stage 2: model for the between subject variability of PK parameters
The distribution of an individuals’ PK parameter values (θi) around the population mean (θ) was described using a multivariate normal distribution as shown below.
| (a2) |
Bold notation is used to indicate a vector or matrix. θi is a p× 1 vector of individual PK model parameters; MVNp denotes a p-dimensional multivariate normal distribution; θ is a p× 1 vector of mean population PK model parameters; and Ω−1 is the inverse of the p×p variance-covariance matrix of between subject variability.
Stage 3: priors
Priors for θ, Ω−1, and τ were supplied at this stage. The population PK parameter (θ) was assumed to arise from a multivariate normal distribution with a prior mean (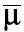 ) and precision (Σθ−1) (a3).
) and precision (Σθ−1) (a3).
| (a3) |
where 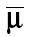 and Σθ−1 is a p × 1 vector of prior means and precision for structural PK model parameters, respectively.
and Σθ−1 is a p × 1 vector of prior means and precision for structural PK model parameters, respectively.
The inverse of the between subject variance (Ω), denoted Ω−1, was assumed to arise from a Wishart distribution (a4);
| (a4) |
where Ω−1 is the inverse of the variance-covariance matrix of between subject variability; W denotes a Wishart distribution; ρ is the degree of freedom of the Wishart distribution, which indicates the informativeness of between subject variance; and  is a matrix of the expectations of the p × p elements of the between subject variances and covariances.
is a matrix of the expectations of the p × p elements of the between subject variances and covariances.
The precision of the residual error was generated from the inverse of the square of the residual standard deviation (σ). The prior for the residual standard deviation was described using a uniform distribution (U) with noninformative boundaries, such that
| (a5) |
where a is chosen to be small but greater than zero and b to be sufficiently large so as not to truncate the value of σ during sampling.
Priors that were elicited for the current analysis included (i) the structural form of the distribution, e.g. normal distribution and (ii) the parameter(s) describing the location or central tendencies (i.e. mean) and the dispersion of each distribution (i.e. variance, degrees of freedom).
Appendix B: Computation of priors
Priors that were elicited for the current analysis included the parameter(s) describing the location or central tendencies (i.e. mean) and the dispersion of each distribution (i.e. variance, degrees of freedom). They were (i) the mean and precision of the population PK parameters ( and Σθ−1, respectively), and (ii) the between subject variance-covariance matrix (
and Σθ−1, respectively), and (ii) the between subject variance-covariance matrix (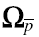 ) and degrees of freedom of the Wishart distribution (ρ).
) and degrees of freedom of the Wishart distribution (ρ).
Two steps were undertaken in order to elicit the priors. These included (1) compiling the existing information; and (2) computation of priors for each of one- and two- compartment model. Detail of computation of priors is given here.
1. Computation of the prior means ( ,
, )
)
Computation of the prior means was based on a meta-analytic technique with a reciprocal variance weighting approach (b1) [37].
| (mb1) |
where ‘k’ represents the kth data set (k = 1, 2, 3, …, K); wk is the weight applied to the kth PK parameter values (θk); Var(θk) and SE(θk) represent the variance, and standard error of θk, respectively.
Prior means of structural PK parameters and each between subject variance of the parameter were calculated as the ‘weighted mean’ [26] with the number of subjects (Nk) used as a weight (b2–b3).
 |
(mb2) |
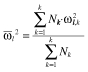 |
(mb3) |
where l is the index of PK parameter; µl,k and 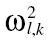 denote the mean structural PK parameter and between subject variance for the lth parameter obtained from the kth data set, respectively;
denote the mean structural PK parameter and between subject variance for the lth parameter obtained from the kth data set, respectively; 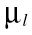 and
and 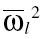 were weighted mean of the structural PK parameter and between subject variance for the lth parameter, respectively. Note that
were weighted mean of the structural PK parameter and between subject variance for the lth parameter, respectively. Note that  form the diagonal components of the variance-covariance matrix of the between subject variability.
form the diagonal components of the variance-covariance matrix of the between subject variability.
 is used to represent a variance-covariance matrix of prior between subject variability (p × p dimension), in which the diagonal part of the matrix is set to the weighted mean of between subject variance for each parameter (
is used to represent a variance-covariance matrix of prior between subject variability (p × p dimension), in which the diagonal part of the matrix is set to the weighted mean of between subject variance for each parameter ( ) and off-diagonal part is set to zero.
) and off-diagonal part is set to zero.
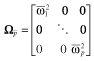 |
(mb4) |
The number of subjects (Nk) was used as the weight because standard error values were not reported.
An empirical additional weighting factor was applied to information gathered from different populations. Information arising from kidney transplant recipients were weighted according to Nk, whereas for data from nonkidney transplant recipients (i.e. data from healthy volunteers or patients with hepatic impairment) a further weighting factor of 0.5 was used (i.e. multiplying the number of subjects by 0.5). The number of 0.5 was empirically chosen. The sensitivity of this weighting factor was tested, using weighting factors of 0.25 and 0.75 in place of 0.5. Results showed a slight change in the estimated prior mean and precision only for parameter CL and V, but Vc, Vp, Q, and ka. An application of weighting factor of 0.25 resulted in a decrease in the mean CL (3%), and V (7%). The precision of parameter CL and V was however, increased by 9%, and 17%, respectively. The utilization of weighting factor of 0.75 increased the mean CL (2%) and V (5%), while decrease in the precision of parameter CL (4%) and V (2%) was shown.
2. Computation of precision for the structural PK model parameters (Σθ−1)
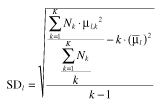
Σθ−1 was computed as the inverse of weighted variance [26]. In this study, PK parameter values from each study were assumed to be log-normally distributed around the weighted mean, μl. Relative weighted variance [b5] was then applied, in which the value was approximated as the square of the quotient of the weighted standard deviation (SDl, b6) and weighted mean (μl) for the lth PK parameter.
| (mb5) |
where
 |
(mb6) |
3. Computation of degrees of freedom of the Wishart distribution (ρ)
The inverse of the between subject variance-covariance matrix (Ω−1) was assumed to arise from a Wishart distribution, in which the Wishart distribution was parameterized in terms of mean (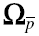 ) and the precision is given by the degrees of freedom (ρ). We used simulation to explore the properties of the Wishart distribution in a 3-step process.
) and the precision is given by the degrees of freedom (ρ). We used simulation to explore the properties of the Wishart distribution in a 3-step process.
Step 1: The between subject variance of each parameter for the kth data set (ω2l,k) was gathered from literature. The 25th and 75th percentiles of these observed BSV values were computed for each of the l elements of the p × p variance-covariance matrix. Their values were denoted using 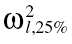 and
and 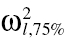 .
.
Step 2: Inverse variance-covariance matrices (Ω−1) were simulated from the Wishart distribution using MATLAB version 6.5 (The MathWorks, Natick, MA, USA) from a specified value of the degrees of freedom (ρ). For simulation, the diagonal part of variance-covariance matrix was set to the prior mean values of the BSV ( ) and the off-diagonal part was set to be zero, i.e. covariances were zero (as shown in b4). The value of ρ was changed to obtain the desired characteristics of the dispersion of simulated matrices of Ω−1. For each value of ρ chosen, 10 000 values of Ω−1 were simulated from the Wishart distribution, and the 25th and 75th percentiles of the diagonal elements of its inverse were computed. Their values are denoted by
) and the off-diagonal part was set to be zero, i.e. covariances were zero (as shown in b4). The value of ρ was changed to obtain the desired characteristics of the dispersion of simulated matrices of Ω−1. For each value of ρ chosen, 10 000 values of Ω−1 were simulated from the Wishart distribution, and the 25th and 75th percentiles of the diagonal elements of its inverse were computed. Their values are denoted by  and
and 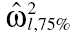 , respectively.
, respectively.
Step 3: The objective function value (OF) for estimation of ρ was defined as the sum of squared differences between the observed and simulated 25th and 75th percentiles for the p-diagonal elements of variance-covariance matrix.
| (mb7) |
Steps 2 and 3 were repeatedly performed using different values of ρ to find the value that minimized expression b7. Its value was then used as a prior for Bayesian analysis.
Using methods detailed above, computation of priors for a one- and two-compartment model with first order absorption was performed and used for Bayesian analysis.
References
- 1.MacDonald A, Scarola J, Burke JT, Zimmerman JJ. Clinical pharmacokinetics and therapeutic drug monitoring of sirolimus. Clin Ther. 2000;22(Suppl B):B101–21. doi: 10.1016/s0149-2918(00)89027-x. [DOI] [PubMed] [Google Scholar]
- 2.Kahan BD, Napoli KL, Podbielski J, Hussein I, Katz SH, Van Buren CT. Therapeutic drug monitoring of sirolimus for optimal renal transplant outcomes. Transplant Proc. 2001;33:1278. doi: 10.1016/s0041-1345(00)02476-3. [DOI] [PubMed] [Google Scholar]
- 3.Kahan BD, Keown P, Levy GA, Johnston A. Therapeutic drug monitoring of immunosuppressant drugs in clinical practice. Clin Ther. 2002;24:330–50. doi: 10.1016/s0149-2918(02)85038-x. discussion 329. [DOI] [PubMed] [Google Scholar]
- 4.Yatscoff RW, Boeckx R, Holt DW, Kahan BD, LeGatt DF, Sehgal S, Soldin SJ, Napoli K, Stiller C. Consensus guidelines for therapeutic drug monitoring of rapamycin: report of the consensus panel. Ther Drug Monit. 1995;17:676–80. doi: 10.1097/00007691-199512000-00022. [DOI] [PubMed] [Google Scholar]
- 5.Zimmerman JJ, Kahan BD. Pharmacokinetics of sirolimus in stable renal transplant patients after multiple oral dose administration. J Clin Pharmacol. 1997;37:405–15. doi: 10.1002/j.1552-4604.1997.tb04318.x. [DOI] [PubMed] [Google Scholar]
- 6.Kumi KA. Approval package for 021083: Clinical pharmacology and biopharmaceutics review(s). http://www.fda.gov/cder/foi/nda/99/21083A_Rapamune_clinphrmr.pdf.
- 7.Zimmerman JJ, Ferron GM, Lim HK, Parker V. The effect of a high-fat meal on the oral bioavailability of the immunosuppressant sirolimus (rapamycin) J Clin Pharmacol. 1999;39:1155–61. [PubMed] [Google Scholar]
- 8.Brattstrom C, Sawe J, Jansson B, Lonnebo A, Nordin J, Zimmerman JJ, Burke JT, Groth CG. Pharmacokinetics and safety of single oral doses of sirolimus (rapamycin) in healthy male volunteers. Ther Drug Monit. 2000;22:537–44. doi: 10.1097/00007691-200010000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Bottiger Y, Sawe J, Brattstrom C, Tollemar J, Burke JT, Hass G, Zimmerman JJ. Pharmacokinetic interaction between single oral doses of diltiazem and sirolimus in healthy volunteers. Clin Pharmacol Ther. 2001;69:32–40. doi: 10.1067/mcp.2001.112513. [DOI] [PubMed] [Google Scholar]
- 10.Yatscoff RW. Pharmacokinetics of rapamycin. Transplant Proc. 1996;28:970–3. [PubMed] [Google Scholar]
- 11.Brattstrom C, Sawe J, Tyden G, Herlenius G, Claesson K, Zimmerman J, Groth CG. Kinetics and dynamics of single oral doses of sirolimus in sixteen renal transplant recipients. Ther Drug Monit. 1997;19:397–406. doi: 10.1097/00007691-199708000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Ferron GM, Mishina EV, Zimmerman JJ, Jusko WJ. Population pharmacokinetics of sirolimus in kidney transplant patients. Clin Pharmacol Ther. 1997;61:416–28. doi: 10.1016/S0009-9236(97)90192-2. [DOI] [PubMed] [Google Scholar]
- 13.Staatz CE, Willis C, Taylor PJ, Lynch SV, Tett SE. Toward better outcomes with tacrolimus therapy: Population pharmacokinetics and individualized dosage prediction in adult liver transplantation. Liver Transpl. 2003;9:130–7. doi: 10.1053/jlts.2003.50023. [DOI] [PubMed] [Google Scholar]
- 14.Staatz CE, Willis C, Taylor PJ, Tett SE. Population pharmacokinetics of tacrolimus in adult kidney transplant recipients. Clin Pharmacol Ther. 2002;72:660–9. doi: 10.1067/mcp.2002.129304. [DOI] [PubMed] [Google Scholar]
- 15.Bruno R, Vivler N, Vergniol JC, De Phillips SL, Montay G, Sheiner LB. A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J Pharmacokinet Biopharm. 1996;24:153–72. doi: 10.1007/BF02353487. [DOI] [PubMed] [Google Scholar]
- 16.Gisleskog PO, Karlsson MO, Beal SL. Use of prior information to stabilize a population data analysis. J Pharmacokinet Pharmacodyn. 2002;29:473–505. doi: 10.1023/a:1022972420004. [DOI] [PubMed] [Google Scholar]
- 17.Wakefield J, Bennett J. The Bayesian modeling of covariates for population pharmacokinetic models. J Am Stat Assoc. 1996;91:917–27. [Google Scholar]
- 18.Gillespie WR, Khoo KC. Population pharmacokinetics in pediatric patients using Bayesian approaches with informative prior distributions based on adults. Clin Pharmacol Ther. 1999;65:140. [Abstract PI-93] [Google Scholar]
- 19.Stickland MD, Kirkpatrick CM, Begg EJ, Duffull SB, Oddie SJ, Darlow BA. An extended interval dosing method for gentamicin in neonates. J Antimicrob Chemother. 2001;48:887–93. doi: 10.1093/jac/48.6.887. [DOI] [PubMed] [Google Scholar]
- 20.Wade JR, Kelman AW, Howie CA, Whiting B. Effect of misspecification of the absorption process on subsequent parameter estimation in population analysis. J Pharmacokinet Biopharm. 1993;21:209–22. doi: 10.1007/BF01059771. [DOI] [PubMed] [Google Scholar]
- 21.Chaloner K, Church T, Louis TA, Matts JP. Graphical elicitation of a prior distribution for a clinical trial. Statistician. 1993;42:341–53. [Google Scholar]
- 22.Tan SB, Chung YF, Tai BC, Cheung YB, Machin D. Elicitation of prior distributions for a phase III randomized controlled trial of adjuvant therapy with surgery for hepatocellular carcinoma. Control Clin Trials. 2003;24:110–21. doi: 10.1016/s0197-2456(02)00318-5. [DOI] [PubMed] [Google Scholar]
- 23.French DC, Saltzgueber M, Hicks DR, Cowper AL, Holt DW. HPLC assay with ultraviolet detection for therapeutic drug monitoring of sirolimus. Clin Chem. 2001;47:1316–9. [PubMed] [Google Scholar]
- 24.Sirolimus international proficiency testing scheme. Analytical Services International Ltd. http://www.bioanalytics.co.uk.
- 25.Dansirikul C, Choi M, Duffull SB. Estimation of pharmacokinetic parameters from non-compartmental variables using Microsoft Excel (R) Comput Biol Med. 2005;35:389–403. doi: 10.1016/j.compbiomed.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Bland JM, Kerry SM. Statistics notes. Weighted comparison of means. BMJ. 1998;316:129. doi: 10.1136/bmj.316.7125.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duffull SB, Kirkpatrick CMJ, Green B, Holford NHG. Analysis of population pharmacokinetic data using NONMEM and WinBUGS. J Biopharm Stat. 2005;15:53–73. doi: 10.1081/bip-200040824. [DOI] [PubMed] [Google Scholar]
- 28.Lunn DJ, Best N, Thomas A, Wakefield J, Spiegelhalter D. Bayesian analysis of population PK/PD models: General concepts and software. J Pharmacokinet Pharmacodyn. 2002;29:271–307. doi: 10.1023/a:1020206907668. [DOI] [PubMed] [Google Scholar]
- 29.Lunn DJ, Wakefield J, Thomas A, Best NG, Spiegelhalter DJ. Pkbugs User Guide. London: Imperial College; 1999. Version 1.1. [Google Scholar]
- 30.Kass RE, Carlin BP, Gelman A, Neal RM. Markov chain Monte Carlo in practice: a roundtable discussion. Am Stat. 1998;52:93–100. [Google Scholar]
- 31.Friberg LE, Dansirikul C, Duffull SB. Uppsala, Sweden: Population Approach Group in Europe (PAGE); 2004. Simultaneous fit of competing models as a model discrimination tool in Bayesian analysis [abstract] [Google Scholar]
- 32.Gelman A. Bayesian Data Analysis. 2. Boca Raton, Fla: Chapman & Hall/CRC; 2004. [Google Scholar]
- 33.Friberg LE, Isbister GK, Hackett LP, Duffull SB. The population pharmacokinetics of citalopram after deliberate self-poisoning: a Bayesian approach. J Pharmacokinet Pharmacodyn. 2005 doi: 10.1007/s10928-005-0022-6. in press. [DOI] [PubMed] [Google Scholar]
- 34.Mu S, Ludden TM. Estimation of population pharmacokinetic parameters in the presence of non-compliance. J Pharmacokinet Pharmacodyn. 2003;30:53–81. doi: 10.1023/a:1023297426153. [DOI] [PubMed] [Google Scholar]
- 35.Dansirikul C, Staatz CE, Duffull SB, Taylor PJ, Lynch SV, Tett SE. Sampling times for monitoring tacrolimus in stable adult liver transplant recipients. Ther Drug Monit. 2004;26:593–9. doi: 10.1097/00007691-200412000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Boni JP, Leister C, Bender G, Fitzpatrick V, Twine N, Stover J, Dorner A, Immermann F, Burczynski ME. Population pharmacokinetics of CCI-779: correlations to safety and pharmacogenomic responses in patients with advanced renal cancer. Clin Pharmacol Ther. 2005;77:76–89. doi: 10.1016/j.clpt.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 37.Altman DG, Smith GD, Egger M. Systematic Reviews in Health Care: Meta-Analysis in Context. 2. London: British Medical Journal; 2001. [Google Scholar]
- 38.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]