

Abstract
Aldose reductase (AR) is a ubiquitously expressed protein with pleiotrophic roles as an efficient catalyst for the reduction of toxic lipid aldehydes and mediator of hyperglycemia, cytokine and growth factor –induced redox sensitive signals that cause secondary diabetic complications. Although AR inhibition has been shown to be protective against oxidative stress signals, the role of AR in regulating nitric oxide (NO) synthesis and NO-mediated apoptosis has not been elucidated to date. We therefore investigated the role of AR in regulating lipopolysaccharide (LPS)-induced NO synthesis and apoptosis in RAW 264.7 macrophages. Inhibition or RNA interference ablation of AR suppressed LPS-stimulated production of NO and over-expression of iNOS mRNA. Inhibition or ablation of AR also prevented the LPS-induced apoptosis, cell cycle arrest, activation of caspase-3, p38-MAPK, JNK, NF-κB and AP1. In addition, AR inhibition prevented the LPS-induced down-regulation of Bcl-xl and up-regulation of Bax and Bak in macrophages. L-arginine increased and L-NAME decreased the severity of cell death caused by LPS and AR inhibitors prevented it. Furthermore, inhibition of AR prevents cell death caused by HNE and GS-HNE, but not GS-DHN. Our findings for the first time suggest that AR catalyzed lipid aldehyde-glutathione conjugates regulates the LPS-induced production of inflammatory marker NO and cytotoxicity in RAW 264.7 cells. Inhibition or ablation of AR activity may be potential therapeutic target in endotoximia and other inflammatory diseases.
Keywords: aldose reductase, sepsis, apoptosis, LPS, nitric oxide
Introduction
Bacterial lipopolysaccharide (LPS), a highly proinflammatory endotoxin, is a component of the outer envelope of all gram-negative bacteria [1, 2]. It is released from the surface of replicating bacteria into the circulation, where it is recognized by a variety of circulating cell types, triggering gene induction of pro-inflammatory cytokines, and biosynthesis of nitric oxide (NO) [3]. Increased production of NO is a key feature of inflammation, specifically in bacterial infection-induced and LPS- stimulated pathologies such as sepsis [4–6]. NO is a pleiotrophic biomolecule, a secondary signal transduction mediator, involved in a variety of physiological and pathological conditions [7, 8]. In macrophages, effective levels of NO contribute to the antimicrobial activities and help in preventing propagation of bacteria [9]. However, overwhelming levels of NO are known to be associated with oxidative stress, cell death and chronic inflammation [10–15]. On the other hand, overproduction of NO due to oxidative stress is also associated with autoimmune and a number of other diseases such as arthritis, diabetes, septic shock, hypertension and stroke [16–18]. Further, it has been shown that NO can react with various proteins and nucleic acids, and cause apoptosis of a number of cells including macrophages [12–13; 19–21]. One of the most fascinating and still unsolved question is how intracellular regulatory molecules could be involved in LPS-stimulated NO synthesis and apoptosis. The role of apoptosis in inflammatory diseases such as sepsis has not been adequately explored, but there is rapid developing evidence to suggest that increased apoptotic processes play a major role in the outcome to sepsis [22, 23]. The apoptotic death in immune cells such as T, B, NK and macrophage may contribute significantly to the risk of secondary opportunistic infections [23]. Therefore, interfering cell signaling pathways that lead to LPS-mediated apoptosis represent an important therapeutic target for sepsis.
Aldose reductase (AR), an enzyme that catalyzes first and rate limiting step of polyol pathway of glucose metabolism is implicated in the pathogenesis of secondary diabetic complications [24–26]. In addition, AR can also reduce a number of lipid peroxidation- derived aldehydes including most abundant and toxic 4-hydroxy-trans2-nonenal (HNE); [27, 28]. Recent studies have shown the presence of binding sites for redox-regulated transcription factors such as AP-1 and NF-κB in the AR gene promoter [29, 30]. This suggests that AR may be a significant component of redox signaling. AR is also induced by growth factors and cytokines which are known to generate reactive oxygen species (ROS); [24]. Expression of AR is enhanced in T-cells, macrophages, and vascular smooth muscle cells (VSMC) under oxidative stress that results in increased HNE formation, suggesting redox regulation of the AR gene in several tissues [30–33]. Further, AR inhibitors attenuate glucose-induced oxidative stress and superoxide production in retinal pericytes, bovine aortic endothelial cells and rabbit aorta [34–36]. Moreover, the strongest evidence that AR is involved in mediating growth comes from our earlier reports showing that inhibition of AR prevents proliferation of VSMC in response to fibroblast growth factor (FGF) and thrombin [37]. Our recent studies also demonstrate that AR plays a pivotal role in the proliferation of VSMC and apoptosis of vascular endothelial cells (VEC) and restenosis of rat carotid artery [38–42]. Inhibition of AR significantly decreases neointima formation in balloon-injured rat carotid arteries, and also diminishes the in situ activation of NF-κB during restenosis [43]. However, the role of AR in the regulation of inducible NO synthase (iNOS) expression, NO production and its importance in macrophage cytotoxicity are not known. Here for the first time, we report that AR regulates LPS-induced NO synthesis and apoptosis in RAW 264.7 macrophages and suggest the possible therapeutic use of AR inhibition in LPS-induced inflammatory response.
Materials and Methods
Reagents and cell culture
Dulbecco’s modified Eagle’s medium (DMEM), phosphate-buffered saline (PBS), penicillin/streptomycin solution, trypsin, and fetal bovine serum (FBS) were purchased from Invitrogen. Sorbinil and zopolrestat were gifts from Pfizer and tolrestat from American home products. Antibodies against Bcl-2 family of proteins, phospho-p38, phospho-JNK, p38, JNK and PARP were obtained from Cell Signaling Inc. Mouse anti-rabbit glyceraldehyde-3-phosphate dehydrogenase antibodies were obtained from Research Diagnostics Inc. The nitrate/nitrite assay kits were obtained from Cayman chemical company. Lipopolysaccharide (E.coli), and the reagents used in the electrophoretic mobility shift assay (EMSA) and Western blot analysis were obtained from Sigma. All other reagents used were of analytical grade. The RAW264.7 macrophage cell lines obtained from ATCC were grown in DMEM containing 10 % FBS and 1% P/S at 37°C with 5% CO2 in air atmosphere in humidified chamber.
Measurement of cell viability
The cells were grown to confluency in DMEM medium, harvested and plated 5000 cells/well in a 96 well plate containing fresh medium. When the cells were 70 to 80% confluent, they were growth-arrested in 0.1% FBS. The low serum levels were maintained during growth-arrest to prevent slow apoptosis that accompanies complete serum-deprivation. After 24 h, LPS (1 to 10 μg/ml) without or with AR inhibitors (10 μM), sorbinil, tolrestat or zopolrestat, were added to the media and the cells were incubated for another 24 h. Cells incubated with the AR inhibitors alone served as control. Cell viability was determined by cell counts and MTT-assay as described earlier [38–40]. Values from 4 separate experiments for each treatment were used for statistical analysis.
Measurement of apoptosis
Apoptotic cell death was quantified using “Cell Death detection ELISA” kit (Roche Inc) according to the manufacturer’s instructions. Caspase-3 activity was measured by using the caspase-3-specific substrate Z-DEVD-AFC, (CBZ-Asp-Glu-Val-Asp-AFC) as well as by Western blot analysis of PARP protein that is in situ cleaved by activated caspase-3. The cells with fragmented and/or condensed nuclei were classified as apoptotic cells and were identified by using Hoechst 33342 dye. Values from 4 separate experiments for each treatment were used for statistical analysis.
Cell cycle analysis
The macrophages were grown in 6 well plates at a density of ~ 1.5×105 cells/well. Growth-arrested cells were pre-incubated without or with sorbinil or tolrestat 10 μM or carrier for 24 h and then stimulated with either 10 μg/ml LPS for another 24 h. For cellular DNA staining the macrophages were suspended in 250 μl of solution A, containing polyetheleneglycol (30 mg/ml), propidium iodide (0.05 mg/ml), triton-x-100 (1 μl/ml), sodium citrate 4 mM, RNAse-A 10 μg/ml and incubated at 37°C. After 20 min of incubation, the solution A was replaced with solution B containing 400 mM NaCl instead of 4 mM sodium citrate in solution A, and incubated overnight at 4°C. Cell cycle analysis was performed with a minimum of 10,000 events per analysis by using FACScan flow cytometer (Becton, Dickinson and Co., San Jose, CA, USA).
Electrophoretic mobility gel shift assay (EMSA)
The cytosolic as well as nuclear extracts were prepared as described before [38]. Consensus oligonucleotide for NF-κB and AP1 transcription factors were 5′-end labeled using T4 polynucleotide kinase. The EMSAs were performed using samples obtained from three independent experimental sets as described earlier [38]. The experiments were performed at least three times and results are reproducible.
Western blot analysis
To examine activation of caspase-3, p38 and JNK and expression of Bcl-2, Bad, Bax and Bcl-xl, Western blot analyses were carried out using samples obtained from three independent experimental sets. Briefly, equal amounts of total protein were loaded onto 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The gels were transferred to nitrocellulose membrane using an electroblotter (Bio-Rad, Richmond, CA, USA) and immunoblotted with the appropriate primary antibodies against PARP, phospho-38, phospho-JNK, non-phospho-p38, non-phospho-JNK, Bcl-2, Bad, Bax and Bcl-xl. Antigen-antibody complexes were detected by enhanced chemiluminescence (Amersham Pharmacia Biotech, NJ). GAPDH was used to monitor equal protein sample loading. The densitometry analyses of the blots were performed by using Kodak 1D image analysis software.
Determination of nitrate/nitrite levels
The nitrate/nitrite levels in macrophages were determined using commercially available microplate assay kit from Cayman Chemical according to supplier’s instructions. Briefly, to measure nitrite (NO2−), 100 μl of macrophage culture supernatant were collected, mixed with an equal volume of the Griess reagent (1% sulfanilamide/0.1% N-(1-naphthyl)-ethylenediamine dihydrochloride/2.5% H3PO4) and incubated for 10 min at room temperature. Nitrite concentration was determined by measuring the absorbance at 540 nm in an ELISA 96 well plate spectrophotometer. NaNO2 was used for external calibration. Cell-free medium alone contained 5–8 μM of nitrite; this value was determined in each experiment and subtracted from the value obtained for each cell sample.
Determination of iNOS expression in macrophages by RT-PCR
The macrophages were grown in 6 well plates at a density of approximately 3.0×105 cells/well. After 70–80% confluence macrophages were serum starved in presence or absence of sorbinil or tolrestat (10 μM) for 24 h and then stimulated with 1 μg/ml LPS. Total RNA from RAW cells was isolated by using RNeasy kit (Qiagen) as per supplier’s instructions. Aliquots of RNA (1.5 μg) isolated from each sample were reverse transcribed with Omniscript and Sensiscript reverse transcriptase one-Step RT PCR system with HotStarTaq DNA polymerase (Qiagen) at 55°C for 30 min followed by PCR amplification. The oligonucleotide primer sequences were as follows: 5′-CTGCAGGTCTTTGACGCTCG-3′ (sense) and 5′-GTGGAACACAGGGGTGATGC-3′ (antisense) for iNOS, 5′-GTG GGC CGC TCT AGG CAC CAA -3′ (sense) and 5′-CTTTAGCACGCACTGTAGTTTCTC- 3′ (antisense) for β-actin. PCR reaction was carried out in a GeneAmp 2700 thermocycler (Applied Biosystems, Foster City, CA) under the following conditions: initial denaturation at 95°C for 15 min; 35 cycles of 94°C 30 s, 62°C 30 s, 72°C 1 min, and then 72°C 5 min for final extension. PCR products were electrophoresed with 2% Agarose-1×TAE gels containing 0.5 μg/ml ethidium bromide.
RNA interference ablation of AR
The ablation of AR mRNA was essentially carried out as described earlier [43]. Briefly, RAW264.7 cells were incubated with serum-free medium containing the AR-siRNA (AATCGGTGTCTCCAACTTCAA) or scrambled siRNA (AAAATCTCCCTAAATCATACA; control) to a final concentration of 100 nM and the RNAiFect™ transfection reagent (Qiagen). After 15 min of incubation at 25 °C, the medium was aspirated and replaced with fresh DMEM containing 10 % serum. The cells were cultured for 48 h at 37 °C, and AR expression was determined by measuring AR protein by Western blot analysis using anti-AR antibodies and by measuring AR activity in the total cell lysates [43].
Measurement of Reactive oxygen species
The serum- starved macrophages (1.5 × 104 cells/well in a 24-well plate) without or with 10 μM of sorbinil or tolrestat were treated with the ROS-sensitive dye Dihydroethidium (hydroehidine) for 15 min. Subsequently, the macrophages were exposed to LPS (1 μg/ml) for 60 min and fluorescence was measured with a Nikon Epifluorescence microscope. For quantitation, the macrophages were treated with ROS-sensitive fluorophore 2′, 7′-dichlorofluorescein diacetate for 30 min, followed by stimulation with LPS (1 μg/ml) for 60 min. The relative fluorescence was measured with a CytoFluorII fluorescence plate reader (PerSeptive Biosystems, Inc., Framingham, MA) at excitation of 485 nm and emission of 528 nm.
Preparation of GS-aldehyde esters
The cell permeable conjugate of glutathione ethyl ester with HNE (GS-HNE-ester) was prepared as described [44]. Briefly, 1μmol of [4- 3H] HNE (55000 cpm/nmol) was incubated with 5 μmol of GSH ethyl ester in 0.1 M potassium phosphate, pH 7.0 for 1 h at room temperature. The GS-HNE-ester was purified by reverse phase HPLC. The reduced form of the esterified glutathione-HNE conjugate (GS-DHN-ester) was prepared by incubating 100 nmol of GS-HNE-ester with 300 nmol of NADPH and 100 μg AR in 0.1 M potassium phosphate, pH 6.0 for 3 h at 37°C. The GS-DHN-ester was separated from GS-HNE-ester by reverse phase HPLC by using a Varian reverse phase ODS C18 column pre-equilibrated with 0.1% aqueous-trifluoroacetic acid (TFA). Chemical identities of the GS-HNE and GS-DHN-esters were established by electrospray ionization mass spectrometry (ESI/MS) as described before [44]. ESI+/MS of GS-HNE-ester and GS-DHN-ester show an m/z values of 492.2 and 494.2 (data not shown), respectively.
Statistical analysis
Data presented as mean ± SEM and p values were determined by unpaired student’s t-test using Microsoft Office Excel 2003 software.
Results
Effect of AR inhibition or ablation on LPS-induced macrophage growth
To investigate the role of AR in LPS-induced cytotoxic signals leading to RAW264.7 cell death, we determined the effect of three structurally unrelated AR inhibitors sorbinil, tolrestat, and zopolrestat on cell growth. The results shown in Fig. 1A demonstrate that the treatment of macrophages with LPS significantly diminished macrophage growth determined by MTT assay in a dose-dependent manner from 0.1 to 20 μg/ml. The decrease in growth was significantly prevented by incubation of macrophages with 10 μM of AR inhibitors sorbinil, tolrestat or zopolrestat. However, AR inhibitors alone had no effect on the growth, indicating that AR inhibition by itself does not affect macrophage growth at the concentrations used (Fig. 1A). Similar results were obtained when the cell growth was estimated by counting cell number (data not shown). Since IFN-γ is known to potentiate LPS -induced decrease in cell growth [45], we have also examined the effect of AR inhibitors on LPS+IFN-γ-induced apoptosis. The results shown in Fig. 1B suggest that AR inhibitors markedly prevented LPS+IFN-γ –induced cell death. To rule out non-specific effects of pharmacological inhibitors, we have ablated AR message by transient transfection of macrophages with AR siRNA or scrambled siRNA (control) and investigated the effect of AR ablation on LPS and LPS+IFN-γ-induced cell growth. The transfection of RAW264.7 cells with AR siRNA but not control siRNA caused >95% ablation of AR protein (Fig. 1C, insert). Consistent with the pharmacological data, transfection with AR siRNA but not control siRNA oligonucleotides prevented LPS as well as LPS+IFN-γ-induced decrease in cell growth (Fig. 1C). Together, these observations suggest that the reaction product(s) of AR catalysis may be involved in the LPS-induced cell growth.
Fig. 1. Effect of AR inhibition/ablation on LPS-induced cell viability in macrophages.

A) Growth –arrested macrophages were incubated without or with indicated AR inhibitors (10 μM) for 24 h and challenged with LPS (0–20 μg/ml) for another 24 h. B and C) cells were transfected with control or AR siRNA oligonucleotides followed by incubation with 10 μg/ml of LPS or 10 μg/ml LPS + 100 U/ml of IFN-γ for 24 h. A to C) The cell viability was determined by MTT assay and C) by cell counts as described in the Methods. The inset in Fig 1C shows the Western blot for AR protein expression. Data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to LPS-treated cells #P < 0.001 control cells. UT, untransfected; TR, Transfection Reagent; Con; Scrambled SiRNA; AR siR; AR siRNA.
Inhibition/ablation of AR prevents LPS-induced macrophage apoptosis
To examine the nature of LPS-induced decrease in macrophage cell growth, we measured free histones released upon nucleosomal degradation, which is a hallmark of apoptotic cell death. LPS caused degradation of nucleosomal histones, suggesting that LPS causes apoptosis of macrophages (Fig. 2). Pre-incubation of the cells with sorbinil, tolrestat or zopolrestat (Fig. 2A) and ablation of AR by siRNA (Fig. 2B) prevented these changes. Further in the absence of LPS, neither AR inhibition nor ablation caused apoptosis of macrophages. For additional confirmation of apoptosis, we stained macrophages with Hoechst 33342 which can detect apoptotic cells with morphological changes leading to nuclear fragmentation and counter stained with propidium iodide, which can differentiate apoptotic and necrotic cells, respectively. As shown in Fig. 3A, cells treated with LPS displayed nuclear fragmentation and condensation, whereas, pre-incubation with sorbinil or tolrestat prevented the cells from undergoing apoptosis induced by LPS. Under identical conditions, no significant necrotic cells were found either with LPS or LPS+AR inhibitors (Fig. 3B).
Fig. 2. Effect of AR inhibition on LPS-induced apoptosis in macrophages.

Growth –arrested macrophages were A) incubated without or with indicated AR inhibitors (10 μM), B) transfected with control or AR siRNA oligonucleotides and both A and B were challenged with LPS (10 μg/ml) for 24 h. The apoptosis was determined by measuring nucleosomal degradation using ELISA kit. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to LPS-treated cells #P < 0.001 control cells.
Fig. 3. Effect of AR inhibition on LPS-induced morphological changes in macrophages.
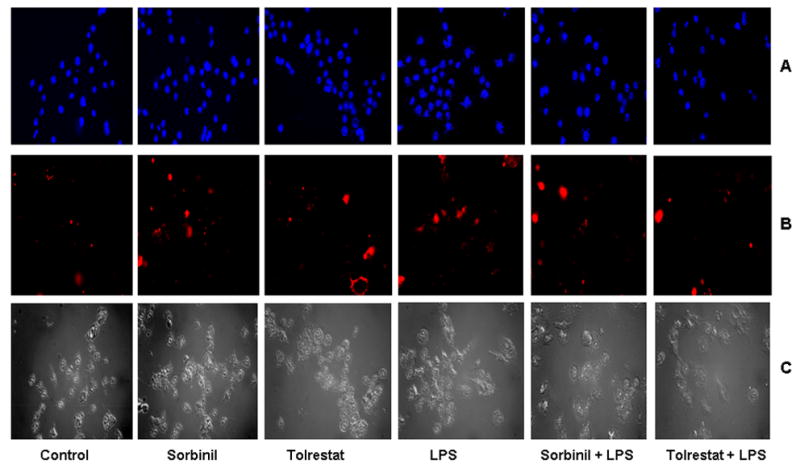
Growth –arrested macrophages were incubated without or with indicated AR inhibitors (10 μM), and challenged with LPS (10 μg/ml) for 24 h. Subsequently, the cells were stained with A) 5 μg/ml of Hoechst 33342 and B) 1 μg/ml of propidium iodide for 30 min at 4°C to morphologically identify apoptotic cells (blue) and Necrotic cells (red), respectively. C) The cells shown in bright filed. All the pictures were taken at 40X magnifications using a Nikon epifluorescence microscope.
Inhibition of AR prevents LPS-induced activation of caspase-3, p38-MAPK and JNK
Incubation of macrophages with LPS caused ~2 fold increase in caspase-3 activity as measured by using caspase-3 specific synthetic peptide fluorimetrically and inhibition or ablation of AR significantly (>85%) prevented it (Fig. 4A and B). In situ activation of caspase-3 was further confirmed by the cleavage of 116-kDa PARP substrate to an 85-kDa (Fig. 4C). These results suggest that the inhibition or ablation of AR prevents LPS-induced apoptosis by preventing caspase-3 activation. In addition, incubation of macrophages with LPS caused phosphorylation of p38 and JNK within 10 and 20 min, respectively (Fig. 5A and C; left). Inhibition of AR prevented the LPS-induced phosphorylation of p38 and JNK (Fig. 5A and C; right). However, LPS alone, AR inhibitor alone or LPS + AR inhibitor did not alter the expression of total p38-MAPK and JNK (Fig. 5B and D).
Fig. 4. Effect of AR inhibition/ablation on LPS-induced activation of caspase-3 in macrophages.

Growth –arrested macrophages were A & C) incubated without or with indicated AR inhibitors (10 μM), B) transfected with control or AR siRNA oligonucleotides and challenged with LPS (10 μg/ml) for 24 h. The caspase-3 activity was determined by A and B) in vitro ELISA kit and C) in situ PARP cleavage by Western blot using antibodies against PARP as described in the Methods. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to LPS-treated cells #P < 0.001 control cells.
Fig. 5. AR inhibition prevents LPS-induced activation of p38 MAPK and JNK in macrophages.

Growth –arrested macrophages were incubated without or with AR inhibitor, sorbinil (10 μM), and challenged with LPS (1 μg/ml) for indicated times. A-D) Western blots were developed using antibodies against A) phospho-p38 MAPK, B) total p38 MAPK, C) phospho-JNK and D) total JNK. Antigen-antibody complex was detected by enhanced chemiluminescence.
AR inhibition prevents LPS-induced expression of Bcl-2 family of proteins in macrophages
Next, we determined the effect of AR inhibition on the expression of Bcl-2 family proteins, whose expression has been shown to alter significantly during apoptosis caused by various oxidant stimuli [46, 47]. Stimulation of macrophages with LPS down -regulated the expression of Bcl-xl but not Bcl-2 (Fig. 6A and B) and up-regulated the expression of Bax and Bak (Fig. 6C and D). However, stimulation with LPS did not affect Bad levels (Fig. 6E). AR inhibitors significantly prevented the down-regulation of Bcl-xl and up-regulation of Bax and Bak by LPS but had no effect on basal levels of these proteins in the absence of LPS.
Fig. 6. AR inhibition prevents LPS-induced regulation of BCl-2 family of proteins.

Growth –arrested macrophages were incubated without or with AR inhibitors (10 μM), and challenged with LPS (10 μg/ml) for 24 h. A–F) Western blots were developed using antibodies against A) Bcl-2, B) Bcl-xl, C) Bax, D) Bak, E) Bad and F) GAPDH. Antigen-antibody complex was detected by enhanced chemiluminescence.
Effect of AR inhibition on LPS-induced cell cycle arrest in macrophages
Treatment of macrophages with LPS caused a significant decrease in the cells entering the synthetic (S)- phase of cell cycle (Fig. 7) and cells accumulated in the G0-G1 phase suggesting cell cycle arrest at S-phase. Inhibition of AR in the presence of LPS caused accumulation of cells in S-and G1 phases, suggesting that AR inhibition prevents LPS-induced arrest of synthetic phase of cell cycle in RAW264.7 macrophages.
Fig. 7. Inhibition of AR prevents LPS-induced arrest of synthesis phase of cell cycle in macrophages.

Growth-arrested macrophages were pre-incubated with sorbinil or tolrestat or carrier for 24 h followed by stimulation with of LPS for 24 h and cell cycle analysis was performed by FACS.
Inhibition of AR prevents LPS-induced production of nitric oxide and expression of iNOS in macrophages
It is known that nitric oxide is the major cause of macrophage cell death induced by LPS [9–13], hence we have investigated the effect of AR inhibition/ablation on LPS –induced NO levels and iNOS expression in macrophages. As shown in Fig. 8A and B, treatment of macrophages with 1 μg/ml of LPS for 24 h caused ~9- fold increase in the levels of nitrite/nitrate and inhibition or ablation of AR significantly (>80%) prevented these changes. A ~6.0 fold increase in the LPS-induced iNOS protein levels in macrophages was also significantly (>80%) prevented by AR inhibitors (Fig. 9A and B). LPS caused a significant (~2.5 fold) induction of iNOS mRNA within 8 h and sorbinil or tolrestat significantly (>85%) inhibited the same (Fig. 9D and E) suggesting that AR inhibition prevents transcriptional activation of iNOS induced by LPS in macrophages.
Fig. 8. AR inhibition prevents LPS-induced nitric oxide production in macrophages.

Growth-arrested macrophages were A) pre-incubated with AR inhibitors or carrier for 24 h, B) transfected with control or AR siRNA oligonucleotides and both A and B were challenged with LPS (1 μg/ml) for 24 h. In the culture media Nitrate/nitrite levels were measured by using specific ELISA kits as described in the methods. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to LPS-treated cells #P < 0.001 control cells.
Fig. 9. AR inhibition prevents LPS-induced iNOS expression in macrophages.

Growth-arrested macrophages were pre-incubated with AR inhibitors or carrier for 24 h followed by the incubation with LPS (1 μg/ml) for additional 24 h. In the cell extracts iNOS expression was measured by B) Western blot analysis and E) RT-PCR as described in the methods. A & D) Densitometric analysis of B and E. C & F) Loading controls. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to LPS-treated cells #P < 0.001 control cells.
AR inhibition prevents LPS –induced activation of NF-κB and AP1
Since redox sensitive transcription factors such as NF-κB and AP1 transcribe various inflammatory markers including iNOS [48, 49], we next examined the effect of AR inhibition or ablation on LPS-induced activation of NF-κB and AP1 in macrophages. As shown in Fig. 10A–D, LPS caused profound activation of NF-κB and AP1 and sorbinil, tolrestat and zopolrestat as well as AR siRNA significantly prevented it. However, AR inhibitors alone did not affect the basal NF-κB and AP1 levels in the macrophages. These results suggest that by modulating the LPS-induced activation of redox-sensitive transcription factors, AR inhibition could prevent LPS –induced production of nitric oxide and associated cytotoxicity leading to apoptosis.
Fig. 10. AR inhibition prevents LPS-induced activation of NF-κB and AP1 in macrophages.

Growth-arrested macrophages were A & C) pre-incubated with AR inhibitors or carrier for 24 h and B & D) transfected with control or AR siRNA oligonucleotides. Subsequently all the four (A–D) were incubated with LPS (1 μg/ml) for 2 h. Equal amounts of nuclear extracts were subjected to EMSA for A & B) NF-κB and C & D) AP1 as described in the Methods.
Effect of L-arginine and L-NAME on the LPS-induced apoptosis of macrophages
Since, it is well known that nitric oxide causes apoptosis in LPS-induced macrophages [8–14]; we next examined the effect of iNOS substrate, L-arginine and iNOS inhibitor, L-NAME on the LPS –induced apoptosis of macrophages in the absence and presence of sorbinil. As shown in Fig. 11, L-arginine increased and L-NAME decreased LPS –induced apoptosis of macrophages, and inhibition of AR significantly prevented the effects of L-arginine + LPS but not L-NAME +LPS. These results suggest that AR could mediate the LPS-induced production of NO which is responsible for apoptotic death of macrophages.
Fig. 11. Effect of NO in LPS-induced macrophage viability.

Growth-arrested macrophages were pre-incubated with sorbinil for 24 h followed by incubation with L-arginine or L-NAME for 2 h. Subsequently the macrophages were incubated with LPS (10 □g/ml) for 24h. The cell viability was determined by MTT assay as described in the methods. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to LPS-treated cells, #P < 0.001 control cells and ##P < 0.01 as compared to LPS-treated cells.
AR catalyzed GS-aldehydes mediates cytotoxic signals in macrophages
We examined the effect of AR inhibition on LPS-induced generation of ROS. As shown in Fig. 12A, incubation of macrophages with LPS caused a significant increase in ROS levels as determined by fluorescence microscopy and inhibition of AR prevented it. Further, we have also measured ROS levels quantitatively by using 2′, 7′-dichlorofluorescein diacetate. The relative fluorescent units of 15.5 ± 1.3 were observed in control cells which upon stimulation with LPS increased to 28 ± 2.5. However in the cells treated with sorbinil and tolrestat followed by stimulation with LPS the fluorescent units corresponding to ROS levels were significantly lower, 19 ±1.7 and 18.4 ± 1.4, respectively. Since AR catalyzes the reduction of lipid peroxidation derived aldehydes (such as HNE) and their conjugates with glutathione (such as GS-HNE), we next examined the effect of inhibition of AR on HNE-, GS-HNE- and GS-DHN- induced apoptosis as well as generation of NO in macrophages. The results shown in Fig. 12B demonstrate that HNE, GS-HNE and GS-DHN caused decrease in the macrophage growth and inhibition of AR prevented HNE- and GS-HNE –induced decrease in cell growth but not GS-DHN. Similarly inhibition of AR prevented the increase in the HNE- and GS-HNE –induced NO levels but had no effect on the GS-DHN (Fig. 12C). This indicates that GS-DHN could be mediator cytotoxic signals in macrophages.
Fig. 12. Effect of AR inhibition on GS-aldehydes –induced cytotoxicity in macrophages.

A) The growth arrested macrophages with or without sorbinil or tolrestat were treated with Dihydroethidium (hydroehidine) for 15 min followed by LPS for 60 min. The fluorescence intensity was evaluated under Nikon Epifluorescence microscope (40X magnification). I) Control, II) Sorbinil, III) Tolrestat, IV) LPS, V) LPS+Sorbinil and VI) LPS+Tolrestat. B and C) Growth-arrested macrophages were pre-incubated with AR inhibitors or carrier for 24 h followed by the incubation with HNE, GS-HNE-ester or GS-DHN-ester (1 μM) for additional 24 h. B) The cell viability was determined by MTT assay and C) the nitrate/nitrite levels were measured by using specific ELISA kits as described in the methods. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to aldehyde-treated cells #P < 0.01 control cells.
Discussion
Gram–negative bacterial- induced septic shock is the most common cause of death among the patients in medical and surgical intensive care units and severely burnt [1, 2]. The most common cause of septic shock in infections is due to the release of bacterial cell wall endotoxin, LPS [2]. LPS induces the production of many inflammatory cytokines and chemokines which by autocrine and paracrine manner increase the severity of the disease [3]. Further, NO is one of the most important inflammatory marker involved in the pathophysiology of sepsis [4, 5]. LPS is known to induce iNOS gene expression which increases the levels of NO in various cells, including macrophages. Moreover, NO interacts with proteins and nucleic acids, and causes apoptosis of macrophages [10–12]. NO-mediated apoptosis is considered to be involved in the multi-organ failure observed in sepsis [13–15]. Therefore, it is necessary to identify the intracellular molecules involved in the regulation of NO synthesis and apoptosis. Here we demonstrate that AR mediates LPS-induced NO synthesis and apoptosis in RAW 264.7 macrophages.
We present several lines of experimental evidence to suggest that LPS predominantly induces apoptosis in murine macrophages which can be inhibited by either pharmacological inhibition of AR or by using RNA interference ablation of AR message. Inhibition of AR also prevented the LPS –induced increase in caspase-3, abundance of Bax and Bak and the down-regulation of Bcl-xl, indicating that inhibition of AR attenuates the apoptotic signaling of LPS. In various types of cells, activation of caspases has been shown to be involved in apoptosis [50–52]. An increase in the activity of caspase-3 along with nitric oxide has been reported in apoptosis of T-cells induced by Fas [53], macrophages induced by LPS [14, 15] and endothelial cells induced by cytokines [54]. Furthermore, caspase-3 inhibition has been shown to suppress NO-mediated apoptotic cell death in macrophages [55]. Earlier studies have shown that the activation of caspase-3 by TNF-α in endothelial cells [41] and LPS in rat hippocampus [56] and macrophages [57] is in part due to increased generation of ROS. The increased ROS could be the mediator of LPS signaling leading to the up-regulation of Bax and Bak and down-regulation of the Bcl-xl in macrophages. The critical role of ROS in the mediation of LPS signaling is further confirmed by the observations that treatment with antioxidants simultaneously blocks LPS-induced activation of NF-κB and stress kinases [58, 59]. Thus, inhibition or ablation of AR could also be preventing multiple pathways of LPS signaling by minimizing changes in the redox state. The demonstration that inhibition of AR prevents the activation of stress kinases and apoptotic signaling provides new avenues for understanding and managing inflammation induced by Gram-negative bacterial infections.
Our studies suggest that inhibition of AR may be beneficial since it could attenuate inflammatory signals mediated by transcription factors (NF-κB and AP-1) and NO. LPS-induced stimulation of NF-κB activity via phosphorylation and degradation of IκB-α, is the major mechanism of increased synthesis of NO in a variety of immune cells [59, 60]. Along with NF-κB activation, AP-1 activation is important for the expression of the pro-inflammatory genes. Both NF-κB and AP-1 are sensitive to ROS [61] and antioxidants, over-expression of catalase, superoxide dismutase and γ-glutamyl transpeptidase prevent their activation [62–64]. We have recently shown that inhibition of AR prevents high glucose or cytokines-induced activation of AP-1 and NF-κB in various cell types, including VSMC, HLEC and HUVEC [38–41]. Our demonstration that prevention of LPS-induced increase in the DNA binding activity of NF-κB and AP-1 by AR ablation or inhibition indicates that AR mediates the inflammatory signals induced by Gram-negative bacteria. Numerous reports indicate a functional connection between NO synthesis and apoptosis in a number of cell types [10–15]. Our results showing a marked decrease in LPS-induced macrophage death by L-NAME, an inhibitor of NO synthase, and a significant increase of macrophage death by L-arginine, a substrate of NO synthase, suggest a definitive involvement of NO in LPS-induced death of macrophages. Under similar conditions, inhibition of AR prevented LPS alone or LPS+L-arginine –induced cell death, suggesting that AR regulates the LPS-induced production of NO in macrophages probably by modulating the transcriptional activation of iNOS via NF-κB. Similar to our findings, recently, it has been shown that NO mediates galactosamine-induced apoptosis in primary rat hepatocytes [65] and LPS-induced apoptosis in murine macrophages [55]. Indeed, Nakagawa and Yamagucgi have shown that LPS-induced cell death was significantly blocked by L-NAME, in in vitro cultured rat kidney proximal epithelial cells [66] and in post traumatic spinal cord injury [67].
We as well as other investigators have shown that AR mediates cell growth or death induced by a variety of stimuli such as TNF-α, FGF, PDGF, angiotensin-II and high glucose. We have further shown that AR inhibition prevents high glucose and cytokine-induced PKC and NF-κB activation [38–41]. In agreement with our findings, others have shown that AR inhibition prevents high glucose -induced MAP kinase [68], JAK2 [69], and TGF-beta –induced p38, JNK and AP-1 activation [70]. These studies indicate that AR could be an essential mediator of stress signals that activate NF-κB and AP-1. It is known that LPS activates TAK1-dependent pathway which in turn activates NF-κB via assembly of IKKα/β/γ complex and activation of TAK1 also known to activate AP-1 via JNK and p38 MAPK [71]. The demonstration that activation of NF-κB and AP-1, and phosphorylation of both the JNK and p38 kinases in the macrophages can be prevented by AR inhibition further suggests that the signals upstream of TAK1 activation could be prevented by AR inhibitors. LPS-triggered signaling events further upstream to TAK1 activation are mediated by the activation of PKC because macrophage PKC activity is increased by LPS-stimulation and PKC inhibition prevents LPS-induced NF-κB activation and NO production [72]. In agreement with a central role of PKC, we have shown earlier that AR inhibition or ablation prevents cytokine- and high glucose -induced activation of PKC [38, 73]. Although the mechanisms by which AR inhibition prevents PKC activation remains unclear, we propose that inhibition of AR prevents the events that could lead to the activation of PLC isozymes which are activated by LPS similar to high glucose- treated VSMCs [73]. Alternatively, AR inhibition could affect signaling due to lipid peroxidation- derived aldehydes and their glutathione (GSH) conjugates. AR reduces lipid peroxidation- derived aldehydes such as HNE and their GSH conjugates (such as GS-HNE) with a Km 10–30 uM, which is significantly (>1000 fold) lower than that for aldo-sugars (Km 50–100 mM), suggesting that AR is an excellent catalyst for the reduction of lipid aldehydes and GS- lipid aldehydes (24). Indeed, we have recently crystallized an AR ternary complex with NADPH and a GSH analog (S-1,2-dicarboxyethyl-glutathione) and have solved the crystal structure (74). The structural analysis suggests that AR active site has both GSH and carbonyl binding sites and confirms our earlier observations that AR is catalytically more efficient towards GS-aldehydes than parent aldehydes (24). We now investigated the effect of HNE, GS-HNE and GS-DHN on macrophage apoptosis. Results shown in Fig 12B and C suggest that AR catalyzed reaction product GS-DHN mediates cytotoxic signaling in macrophages. Our earlier observations also suggest that inhibition of AR prevents GS-HNE but not GS-DHN –induced proliferation and activation of NF-κB in VSMC (44). However, it is not clear how GS-DHN could mediates cytotoxic signaling, further investigations are required to understand the significance of GS-aldehyde conjugates in oxidative stress signaling that modulates cell function. In summary, our results provide evidence for an unanticipated role of an aldehyde-metabolizing enzyme, aldose reductase in mediating acute inflammatory responses and provide a novel concept that inhibition of AR could be therapeutically useful in preventing NO release and its dependent cell death as observed in inflammatory processes induced by Gram-negative bacterial infections.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants GM71036 (to KVR) and DK36118 (to SKS).
The abbreviations used are
- AR
aldose reductase
- HNE
4-hydroxy-trans-2-nonenal
- GSH
glutathione
- GS-HNE
glutathionyl-4-hydroxynonanal
- GS-DHN
glutathionyl-1,4-dihydroxynonane
- LPS
lipopolysaccharide
- AP1
activator protein-1
- iNOS
inducible nitric oxide synthase
- NO
nitric oxide
- NF-kB
nuclear factor kappa binding protein
- PARP
Poly(ADP-Ribose) Polymerase
- PKC
protein Kinase C
- PLC
phospholipase C
- siRNA
small interference RNA
- TR
transfection reagent
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Raetz CR, Ulevitch RJ, Wright SD, Sibley CH, Ding A, Nathan CF. Gram-negative endotoxin: an extraordinary lipid with profound effects on eukaryotic signal transduction. FASEB J. 1991;5:2652–2660. doi: 10.1096/fasebj.5.12.1916089. [DOI] [PubMed] [Google Scholar]
- 2.Heine H, Rietschel ET, Ulmer AJ. The biology of endotoxin. Mol Biotechnol. 2001;19:279–296. doi: 10.1385/MB:19:3:279. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Bojorquez LN, Dehesa AZ, Reyes-Teran G. Molecular mechanisms involved in the pathogenesis of septic shock. Arch Med Res. 2004;35:465–479. doi: 10.1016/j.arcmed.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Wolkow PP. Involvement and dual effects of nitric oxide in septic shock. Inflamm Res. 1998;47:52–166. doi: 10.1007/s000110050309. [DOI] [PubMed] [Google Scholar]
- 5.Baumgarten G, Knuefermann P, Schuhmacher G, Vervolgyi V, von Rappard J, Dreiner U, Fink K, Djoufack C, Hoeft A, Grohe C, Knowlton AA, Meyer R. Toll-like receptor 4, nitric oxide, and myocardial depression in endotoxemia. Shock. 2006;25:43–49. doi: 10.1097/01.shk.0000196498.57306.a6. [DOI] [PubMed] [Google Scholar]
- 6.Yamada M, Ichikawa T, Ii M, Sunamoto M, Itoh K, Tamura N, Kitazaki T. Discovery of novel and potent small-molecule inhibitors of NO and cytokine production as antisepsis agents: synthesis and biological activity of alkyl 6-(N-substituted sulfamoyl)cyclohex-1-ene-1-carboxylate. J Med Chem. 2005;48:7457–7467. doi: 10.1021/jm050623t. [DOI] [PubMed] [Google Scholar]
- 7.Cary SP, Winger JA, Derbyshire ER, Marletta MA. Nitric oxide signaling: no longer simply on or off. Trends Biochem Sci. 2006;31:231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Saraiva RM, Hare JM. Nitric oxide signaling in the cardiovascular system: implications for heart failure. Curr Opin Cardiol. 2006;21:221–228. doi: 10.1097/01.hco.0000221584.56372.dc. [DOI] [PubMed] [Google Scholar]
- 9.Alam MS, Akaike T, Okamoto S, Kubota T, Yoshitake J, Sawa T, Miyamoto Y, Tamura F, Maeda H. Role of nitric oxide in host defense in murine salmonellosis as a function of its antibacterial and antiapoptotic activities. Infect Immun. 2002;70:3130–3142. doi: 10.1128/IAI.70.6.3130-3142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tinker AC, Wallace AV. Selective inhibitors of inducible nitric oxide synthase: potential agents for the treatment of inflammatory diseases? Curr Top Med Chem. 2006;6:77–92. doi: 10.2174/156802606775270297. [DOI] [PubMed] [Google Scholar]
- 11.Ulett GC, Adderson EE. Nitric oxide is a key determinant of group B streptococcus-induced murine macrophage apoptosis. J Infect Dis. 2005;191:1761–1770. doi: 10.1086/429693. [DOI] [PubMed] [Google Scholar]
- 12.Luo G, Peng D, Zheng J, Chen X, Wu J, Elster E, Tadaki D. The role of NO in macrophage dysfunction at early stage after burn injury. Burns. 2005;31:138–144. doi: 10.1016/j.burns.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Storling J, Binzer J, Andersson AK, Zullig RA, Tonnesen M, Lehmann R, Spinas GA, Sandler S, Billestrup N, Mandrup-Poulsen T. Nitric oxide contributes to cytokine-induced apoptosis in pancreatic beta cells via potentiation of JNK activity and inhibition of Akt. Diabetologia. 2005;48:2039–2050. doi: 10.1007/s00125-005-1912-2. [DOI] [PubMed] [Google Scholar]
- 14.Marriott HM, Ali F, Read RC, Mitchell TJ, Whyte MK, Dockrell DH. Nitric oxide levels regulate macrophage commitment to apoptosis or necrosis during pneumococcal infection. FASEB J. 2004;18:1126–1128. doi: 10.1096/fj.03-1450fje. [DOI] [PubMed] [Google Scholar]
- 15.Zamora R, Bult H, Herman AG. The role of prostaglandin E2 and nitric oxide in cell death in J774 murine macrophages. Eur J Pharmacol. 1998;349:307–315. doi: 10.1016/s0014-2999(98)00211-8. [DOI] [PubMed] [Google Scholar]
- 16.Konukoglu D, Serin O, Turhan MS. Plasma Leptin and its Relationship with Lipid Peroxidation and Nitric Oxide in Obese Female Patients with or without Hypertension. Arch Med Res. 2006;37:602–606. doi: 10.1016/j.arcmed.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Nagareddy PR, Xia Z, MacLeod KM, McNeill JH. N-acetylcysteine prevents nitrosative stress-associated depression of blood pressure and heart rate in streptozotocin diabetic rats. J Cardiovasc Pharmacol. 2006;47:513–520. doi: 10.1097/01.fjc.0000211744.93701.25. [DOI] [PubMed] [Google Scholar]
- 18.Nagai H, Kumamoto H, Fukuda M, Takahashi T. Inducible nitric oxide synthase and apoptosis-related factors in the synovial tissues of temporomandibular joints with internal derangement and osteoarthritis. J Oral Maxillofac Surg. 2003;61:801–807. doi: 10.1016/s0278-2391(03)00155-1. [DOI] [PubMed] [Google Scholar]
- 19.Chen Q, Casali B, Pattacini L, Boiardi L, Salvarani C. Tumor necrosis factor-alpha protects synovial cells from nitric oxide induced apoptosis through phosphoinositide 3-kinase Akt signal transduction. J Rheumatol. 2006;33:1061–1068. [PubMed] [Google Scholar]
- 20.Vicente S, Perez-Rodriguez R, Olivan AM, Martinez Palacian A, Gonzalez MP, Oset-Gasque MJ. Nitric oxide and peroxynitrite induce cellular death in bovine chromaffin cells: Evidence for a mixed necrotic and apoptotic mechanism with caspases activation. J Neurosci Res. 2006;84:78–96. doi: 10.1002/jnr.20853. [DOI] [PubMed] [Google Scholar]
- 21.Tiwari MM, Messer KJ, Mayeux PR. Inducible nitric oxide synthase and apoptosis in murine proximal tubule epithelial cells. Toxicol Sci. 2006;91:493–500. doi: 10.1093/toxsci/kfj168. [DOI] [PubMed] [Google Scholar]
- 22.Hotchkiss RS, McConnell KW, Bullok K, Davis CG, Chang KC, Schwulst SJ, Dunne JC, Dietz GP, Bahr M, McDunn JE, Karl IE, Wagner TH, Cobb JP, Coopersmith CM, Piwnica-Worms D. TAT-BH4 and TAT-Bcl-xL peptides protect against sepsis-induced lymphocyte apoptosis in vivo. J Immunol. 2006;176:5471–5477. doi: 10.4049/jimmunol.176.9.5471. [DOI] [PubMed] [Google Scholar]
- 23.Wesche-Soldato DE, Lomas-Neira JL, Perl M, Jones L, Chung CS, Ayala A. The role and regulation of apoptosis in sepsis. J Endotoxin Res. 2005;11:375–382. doi: 10.1179/096805105X76904. [DOI] [PubMed] [Google Scholar]
- 24.Srivastava SK, Ramana KV, Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev. 2005;26:380–392. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- 25.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 26.Chung SS, Chung SK. Aldose reductase in diabetic microvascular complications. Curr Drug Targets. 2005;6:475–486. doi: 10.2174/1389450054021891. [DOI] [PubMed] [Google Scholar]
- 27.Ramana KV, Dixit BL, Srivastava S, Balendiran GK, Srivastava SK, Bhatnagar A. Selective recognition of glutathiolated aldehydes by aldose reductase. Biochemistry. 2000;39:12172–12180. doi: 10.1021/bi000796e. [DOI] [PubMed] [Google Scholar]
- 28.Dixit BL, Balendiran GK, Watowich SJ, Srivastava S, Ramana KV, Petrash JM, Bhatnagar A, Srivastava SK. Kinetic and structural characterization of the glutathione-binding site of aldose reductase. J Biol Chem. 2000;275:21587–21595. doi: 10.1074/jbc.M909235199. [DOI] [PubMed] [Google Scholar]
- 29.Iwata T, Sato S, Jimenez J, McGowan M, Moroni M, Dey A, Ibaraki N, Reddy VN, Carper D. Osmotic response element is required for the induction of aldose reductase by tumor necrosis factor-alpha. J Biol Chem. 1999;274:7993–8001. doi: 10.1074/jbc.274.12.7993. [DOI] [PubMed] [Google Scholar]
- 30.Ko BC, Ruepp B, Bohren KM, Gabbay KH, Chung SS. Identification and characterization of multiple osmotic response sequences in the human aldose reductase gene. J Biol Chem. 1997;272:16431–16437. doi: 10.1074/jbc.272.26.16431. [DOI] [PubMed] [Google Scholar]
- 31.Nishinaka T, Yabe-Nishimura C. EGF receptor-ERK pathway is the major signaling pathway that mediates upregulation of aldose reductase expression under oxidative stress. Free Radic Biol Med. 2001;31:205–216. doi: 10.1016/s0891-5849(01)00571-8. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura J, Kasuya Y, Hamada Y, Nakashima E, Naruse K, Yasuda Y, Kato K, Hotta N. Glucose-induced hyperproliferation of cultured rat aortic smooth muscle cells through polyol pathway hyperactivity. Diabetologia. 2001;44:480–487. doi: 10.1007/s001250051646. [DOI] [PubMed] [Google Scholar]
- 33.Rittner HL, Hafner V, Klimiuk PA, Szweda LI, Goronzy JJ, Weyand CM. Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J Clin Invest. 1999;103:1007–1013. doi: 10.1172/JCI4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naruse K, Nakamura J, Hamada Y, Nakayama M, Chaya S, Komori T, Kato K, Kasuya Y, Miwa K, Hotta N. Aldose reductase inhibition prevents glucose-induced apoptosis in cultured bovine retinal microvascular pericytes. Exp Eye Res. 2000;71:309–315. doi: 10.1006/exer.2000.0882. [DOI] [PubMed] [Google Scholar]
- 35.Mimura K, Umeda F, Yamashita T, Kobayashi K, Hashimoto T, Nawata H. (1995) Effects of glucose and an aldose reductase inhibitor on albumin permeation through a layer of cultured bovine vascular endothelial cells. Horm Metab Res. 1995;27:442–446. doi: 10.1055/s-2007-979998. [DOI] [PubMed] [Google Scholar]
- 36.Tesfamariam B, Palacino JJ, Weisbrod RM, Cohen RA. Aldose reductase inhibition restores endothelial cell function in diabetic rabbit aorta. J Cardiovasc Pharmacol. 1993;21:205–211. doi: 10.1097/00005344-199302000-00004. [DOI] [PubMed] [Google Scholar]
- 37.Ruef J, Liu SQ, Bode C, Tocchi M, Srivastava S, Runge MS, Bhatnagar A. Involvement of aldose reductase in vascular smooth muscle cell growth and lesion formation after arterial injury. Arterioscler Thromb Vasc Biol. 2000;20:1745–1752. doi: 10.1161/01.atv.20.7.1745. [DOI] [PubMed] [Google Scholar]
- 38.Ramana KV, Chandra D, Srivastava S, Bhatnagar A, Aggarwal BB, Srivastava SK. Aldose reductase mediates mitogenic signaling in vascular smooth muscle cells. J Biol Chem. 2002;277:32063–32070. doi: 10.1074/jbc.M202126200. [DOI] [PubMed] [Google Scholar]
- 39.Ramana KV, Friedrich B, Bhatnagar A, Srivastava SK. Aldose reductase mediates cytotoxic signals of hyperglycemia and TNF-alpha in human lens epithelial cells. FASEB J. 2003;17:315–317. doi: 10.1096/fj.02-0568fje. [DOI] [PubMed] [Google Scholar]
- 40.Ramana KV, Bhatnagar A, Srivastava SK. Inhibition of aldose reductase attenuates TNF-alpha-induced expression of adhesion molecules in endothelial cells. FASEB J. 2004;18:1209–1218. doi: 10.1096/fj.04-1650com. [DOI] [PubMed] [Google Scholar]
- 41.Ramana KV, Bhatnagar A, Srivastava SK. Aldose reductase regulates TNF-alpha-induced cell signaling and apoptosis in vascular endothelial cells. FEBS Lett. 2004;570:189–194. doi: 10.1016/j.febslet.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 42.Srivastava S, Ramana KV, Tammali R, Srivastava SK, Bhatnagar A. Contribution of aldose reductase to diabetic hyperproliferation of vascular smooth muscle cells. Diabetes. 2006;55:901–910. doi: 10.2337/diabetes.55.04.06.db05-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes. 2004;53:2910–2920. doi: 10.2337/diabetes.53.11.2910. [DOI] [PubMed] [Google Scholar]
- 44.Ramana KV, Bhatnagar A, Srivastava S, Yadav UC, Awasthi S, Awasthi YC, Srivastava SK. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J Biol Chem. 2006;281:17652–17660. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]
- 45.Denis M, Wedlock DN, Buddle BM. IFN-gamma enhances bovine macrophage responsiveness to Mycobacterium bovis: Impact on bacterial replication, cytokine release and macrophage apoptosis. Immunol Cell Biol. 2005;83:643–650. doi: 10.1111/j.1440-1711.2005.01386.x. [DOI] [PubMed] [Google Scholar]
- 46.Thomadaki H, Scorilas A, Hindmarsh JT. BCL2 family of apoptosis-related genes: functions and clinical implications in cancer. Crit Rev Clin Lab Sci. 2006;43:1–67. doi: 10.1080/10408360500295626. [DOI] [PubMed] [Google Scholar]
- 47.Kim R. Unknotting the roles of Bcl-2 and Bcl-xL in cell death. Biochem Biophys Res Commun. 2005;333:336–343. doi: 10.1016/j.bbrc.2005.04.161. [DOI] [PubMed] [Google Scholar]
- 48.Suh SJ, Chung TW, Son MJ, Kim SH, Moon TC, Son KH, Kim HP, Chang HW, Kim CH. The naturally occurring biflavonoid, ochnaflavone, inhibits LPS-induced iNOS expression, which is mediated by ERK1/2 via NF-kappaB regulation, in RAW264.7 cells. Arch Biochem Biophys. 2006;447:136–146. doi: 10.1016/j.abb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 49.Kim JH, Kim DH, Baek SH, Lee HJ, Kim MR, Kwon HJ, Lee CH. Rengyolone inhibits inducible nitric oxide synthase expression and nitric oxide production by down-regulation of NF-kappaB and p38 MAP kinase activity in LPS-stimulated RAW 264.7 cells. Biochem Pharmacol. 2006;71:1198–1205. doi: 10.1016/j.bcp.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 50.Vicente S, Perez-Rodriguez R, Olivan AM, Martinez Palacian A, Gonzalez MP, Oset-Gasque MJ. Nitric oxide and peroxynitrite induce cellular death in bovine chromaffin cells: Evidence for a mixed necrotic and apoptotic mechanism with caspases activation. J Neurosci Res. 2006;84:78–96. doi: 10.1002/jnr.20853. [DOI] [PubMed] [Google Scholar]
- 51.Chan SH, Wu KL, Wang LL, Chan JY. Nitric oxide- and superoxide-dependent mitochondrial signaling in endotoxin-induced apoptosis in the rostral ventrolateral medulla of rats. Free Radic Biol Med. 2005;39:603–618. doi: 10.1016/j.freeradbiomed.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 52.Yu LC, Flynn AN, Turner JR, Buret AG. SGLT-1-mediated glucose uptake protects intestinal epithelial cells against LPS-induced apoptosis and barrier defects: a novel cellular rescue mechanism. FASEB J. 2005;19:1822–1835. doi: 10.1096/fj.05-4226com. [DOI] [PubMed] [Google Scholar]
- 53.Huang J, Tatsumi T, Pizzoferrato E, Vujanovic N, Storkus WJ. Nitric oxide sensitizes tumor cells to dendritic cell-mediated apoptosis, uptake, and cross-presentation. Cancer Res. 2005;65:8461–8470. doi: 10.1158/0008-5472.CAN-05-0654. [DOI] [PubMed] [Google Scholar]
- 54.Hoffmann J, Haendeler J, Aicher A, Rossig L, Vasa M, Zeiher AM, Dimmeler S. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res. 2001;89:709–715. doi: 10.1161/hh2001.097796. [DOI] [PubMed] [Google Scholar]
- 55.Callsen D, Brune B. Role of mitogen-activated protein kinases in S-nitrosoglutathione-induced macrophage apoptosis. Biochemistry. 1999;38:2279–2286. doi: 10.1021/bi982292a. [DOI] [PubMed] [Google Scholar]
- 56.Nolan Y, Vereker E, Lynch AM, Lynch MA. Evidence that lipopolysaccharide-induced cell death is mediated by accumulation of reactive oxygen species and activation of p38 in rat cortex and hippocampus. Exp Neurol. 2003;184:794–804. doi: 10.1016/S0014-4886(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 57.Boggs SE, McCormick TS, Lapetina EG. Glutathione levels determine apoptosis in macrophages. Biochem Biophys Res Commun. 1998;247:229–233. doi: 10.1006/bbrc.1998.8765. [DOI] [PubMed] [Google Scholar]
- 58.Murakami Y, Shoji M, Hirata A, Tanaka S, Yokoe I, Fujisawa S. Dehydrodiisoeugenol, an isoeugenol dimer, inhibits lipopolysaccharide-stimulated nuclear factor kappa B activation and cyclooxygenase-2 expression in macrophages. Arch Biochem Biophys. 2005;434:326–332. doi: 10.1016/j.abb.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 59.Lee SJ, Bai SK, Lee KS, Namkoong S, Na HJ, Ha KS, Han JA, Yim SV, Chang K, Kwon YG, Lee SK, Kim YM. Astaxanthin inhibits nitric oxide production and inflammatory gene expression by suppressing I(kappa)B kinase-dependent NF-kappaB activation. Mol Cells. 2003;16:97–105. [PubMed] [Google Scholar]
- 60.Chen JC, Ho FM, Pei-Dawn Lee Chao Chen CP, Jeng KC, Hsu HB, Lee ST, Wen Tung Wu Lin WW. Inhibition of iNOS gene expression by quercetin is mediated by the inhibition of IkappaB kinase, nuclear factor-kappa B and STAT1, and depends on heme oxygenase-1 induction in mouse BV-2 microglia. Eur J Pharmacol. 2005;521:9–20. doi: 10.1016/j.ejphar.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 61.Hsu TC, Young MR, Cmarik J, Colburn NH. Activator protein 1 (AP-1)- and nuclear factor kappaB (NF-kappaB)-dependent transcriptional events in carcinogenesis. Free Radic Biol Med. 2000;28:1338–1348. doi: 10.1016/s0891-5849(00)00220-3. [DOI] [PubMed] [Google Scholar]
- 62.Bai J, Cederbaum AI. Overexpression of catalase in the mitochondrial or cytosolic compartment increases sensitivity of HepG2 cells to tumor necrosis factor-alpha-induced apoptosis. J Biol Chem. 2000;275:19241–19249. doi: 10.1074/jbc.M000438200. [DOI] [PubMed] [Google Scholar]
- 63.Manna SK, Zhang HJ, Yan T, Oberley LW, Aggarwal BB. Overexpression of manganese superoxide dismutase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappaB and activated protein-1. J Biol Chem. 1998;273:13245–13254. doi: 10.1074/jbc.273.21.13245. [DOI] [PubMed] [Google Scholar]
- 64.Djavaheri-Mergny M, Accaoui MJ, Rouillard D, Wietzerbin J. Gamma-glutamyl transpeptidase activity mediates NF-kappaB activation through lipid peroxidation in human leukemia U937 cells. Mol Cell Biochem. 2002;232:103–111. doi: 10.1023/a:1014834315936. [DOI] [PubMed] [Google Scholar]
- 65.Abou-Elella AM, Siendones E, Padillo J, Montero JL, De la Mata M, Muntane Relat J. Tumour necrosis factor-alpha and nitric oxide mediate apoptosis by D-galactosamine in a primary culture of rat hepatocytes: exacerbation of cell death by cocultured Kupffer cells. Can J Gastroenterol. 2002;16:791–799. doi: 10.1155/2002/986305. [DOI] [PubMed] [Google Scholar]
- 66.Nakagawa T, Yamaguchi M. Overexpression of regucalcin suppresses apoptotic cell death in cloned normal rat kidney proximal tubular epithelial NRK52E cells: change in apoptosis-related gene expression. J Cell Biochem. 2005;96:1274–1285. doi: 10.1002/jcb.20617. [DOI] [PubMed] [Google Scholar]
- 67.Satake K, Matsuyama Y, Kamiya M, Kawakami H, Iwata H, Adachi K, Kiuchi K. Nitric oxide via macrophage iNOS induces apoptosis following traumatic spinal cord injury. Brain Res Mol Brain Res. 2000;85:114–122. doi: 10.1016/s0169-328x(00)00253-9. [DOI] [PubMed] [Google Scholar]
- 68.Price SA, Agthong S, Middlemas AB, Tomlinson DR. Mitogen-activated protein kinase p38 mediates reduced nerve conduction velocity in experimental diabetic neuropathy: interactions with aldose reductase. Diabetes. 2004;53:1851–1856. doi: 10.2337/diabetes.53.7.1851. [DOI] [PubMed] [Google Scholar]
- 69.Galvez AS, Ulloa JA, Chiong M, Criollo A, Eisner V, Barros LF, Lavandero S. Aldose reductase induced by hyperosmotic stress mediates cardiomyocyte apoptosis: differential effects of sorbitol and mannitol. J Biol Chem. 2003;278:38484–38494. doi: 10.1074/jbc.M211824200. [DOI] [PubMed] [Google Scholar]
- 70.Jiang T, Che Q, Lin Y, Li H, Zhang N. Aldose reductase regulates TGF-beta1-induced production of fibronectin and type IV collagen in cultured rat mesangial cells. Nephrology (Carlton) 2006;11:105–112. doi: 10.1111/j.1440-1797.2006.00553.x. [DOI] [PubMed] [Google Scholar]
- 71.Irie T, Muta T, Takeshige K. TAK1 mediates an activation signal from toll-like receptor(s) to nuclear factor-kappaB in lipopolysaccharide-stimulated macrophages. FEBS Lett. 2000;467:160–164. doi: 10.1016/s0014-5793(00)01146-7. [DOI] [PubMed] [Google Scholar]
- 72.Chen CC, Wang JK, Lin SB. Antisense oligonucleotides targeting protein kinase C-alpha, -beta I, or -delta but not -eta inhibit lipopolysaccharide-induced nitric oxide synthase expression in RAW 264.7 macrophages: involvement of a nuclear factor kappa B-dependent mechanism. J Immunol. 1998;161:6206–6214. [PubMed] [Google Scholar]
- 73.Ramana KV, Friedrich B, Tammali R, West MB, Bhatnagar A, Srivastava SK. Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes. 2005;54:818–829. doi: 10.2337/diabetes.54.3.818. [DOI] [PubMed] [Google Scholar]
- 74.Singh R, White MA, Ramana KV, Petrash JM, Watowich SJ, Bhatnagar A, Srivastava SK. Structure of a glutathione conjugate bound to the active site of aldose reductase. Proteins. 2006;64:101–110. doi: 10.1002/prot.20988. [DOI] [PubMed] [Google Scholar]