

Abstract
Profiling and imaging biological specimens using MALDI mass spectrometry has significant potential to contribute to our understanding and diagnosis of disease. The technique is efficient and high-throughput providing a wealth of data about the biological state of the sample from a very simple and direct experiment. However, in order for these techniques to be put to use for clinical purposes, the approaches used to process and analyze the data must improve. This study examines some of the existing tools to baseline subtract, normalize, align, and remove spectral noise for MALDI data, comparing the advantages of each. A preferred workflow is presented that can be easily implemented for data in ASCII format. The advantages of using such an approach are discussed for both molecular profiling and imaging mass spectrometry.
Keywords: Profiling, Imaging, Mass spectrometry, MALDI, Pre-processing
1. Introduction
Molecular profiling and imaging experiments performed directly on tissue using MALDI mass spectrometry are methods gaining considerable popularity [1-10]. The technique is characterized by a number of valuable and useful features including minimal sample preparation, ease of use, heightened throughput, and cellular specificity. MALDI profiling and imaging has already been successfully applied to samples ranging from individually captured cells [11,12], to intact organs, and whole animal sagittal sections [1]. Although a relatively young method, tissue analysis by imaging mass spectrometry (IMS) has been used to address a number of clinical questions in cancer [13,14], neurodegenerative diseases [15-17], organ development [18] and the study of drug and metabolite distribution [19-21]. Due to the variety of applications and the potential of tissue analysis by MALDI mass spectrometry to offer solutions directed at difficult problems in diagnostic and prognostic medicine, tissue profiling and imaging will continue to be an important topic of research. As the application of these new methodologies mature, so too must the techniques to analyze the data.
Analysis of MALDI data can be separated into two distinct steps: (1) pre-processing and (2) processing or statistical analysis. The purpose of pre-processing is to reduce experimental variance within the data set, conditioning it for subsequent statistical analysis. Raw spectra are conditioned through the removal of background, normalization of intensity, and alignment. The analytical goals of profiling experiments can be two-fold: (1) the classification of samples into two or more classes such as diseased/non-diseased in order to better aid patients’ care [22-24], and (2) the identification of biomarkers characteristic to each class [13,14]. Identification of disease specific proteins could yield mechanistic information as well as potential diagnostic markers or drug targets. The importance in recognizing these points is highlighted in recent comparative analyses of profile spectra [25-31], whereby varied biostatisticians all reported an accuracy >90% in classifying the same profile spectra from either tumor or non-tumor samples. However, in identifying which ions were most significant in determining the classifications there was little to no agreement. Inconsistencies such as these are concerning particularly since these same ions are primary candidates for further identification and study as disease biomarkers. Before undertaking such an extensive study one would expect stronger agreement as to which ions are statistically relevant. We feel that many of these inconsistencies are related more to differences in the pre-processing of data as opposed to the actual statistical analysis imposed. The main point being that the ability to accurately classify, or diagnose samples from profile spectra is an important milestone, and the fact that the accuracy of such studies seems to be, in part, independent of the statistical analysis method is particularly encouraging.
Although a complete profile analysis protocol involves both pre-processing and the statistical analysis, an optimal preprocessing workflow is the key to obtaining reliable statistical analysis of data. Since profile references are not readily available for method development we synthesized a series of samples for use as biological models to test different approaches. This provides us with the ability to independently adjust many sources of variance, providing a level of quantitative judgment of the effectiveness of each algorithm. Further, when these algorithms are applied to IMS data, which is essentially an ordered array of profile spectra, the result is a reduction in the effect of sample preparation on image quality and lower image noise. We have listed a set of guidelines along with a comparative discussion of various methodologies that were examined in devising our present workflow (Fig. 1). Here, we have focused on methods of processing MALDI mass spectrometric data allowing one to compare and determine with statistical confidence subtle expression differences between two or more data sets. Additionally, the same techniques also greatly improve the sensitivity and quality of mass imaging experiments, thereby overcoming to a great extent, current limitations related to tissue specific heterogeneity and sample preparation techniques.
Fig. 1.
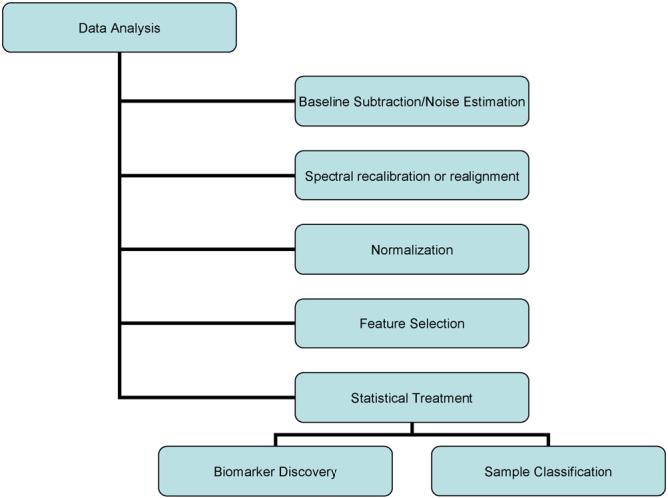
Mass spectra analysis work flow. The mass spectra are treated to processing algorithms responsible for the removal of noise, realignment of the m/z scale, and peak selection and matching. The data is returned to a table, formatted for statistical analysis using a number of established methods. The result of the analysis is a list of biomarker candidates that are subjected to further validation steps.
2. Experimental
2.1. Algorithm development and testing
The commercially available software, DataExplorer (Applied Biosystems, Framingham, MA) and ProTS Data (Biodesix, Steamboat Springs, CO.), were used to implement and test baseline subtraction methods. Algorithms for normalization were coded and tested using a combination of Matlab™, and Visual Basic™. Peak alignment was performed using ProTS Data. Processing of image data was performed using custom software (available upon request) integrating automation using all of the previously mentioned processes, and exported into a format suitable for viewing in freely available BioMap (Novartis, Basel, Switzerland).
2.2. Mass spectrometry
All data were collected using Voyager STR (Applied Biosystems, Framingham, MA) and Bruker Ultraflex II (Bruker Daltonics, Billerica, MA) MALDI TOF mass spectrometers. Images were collected using a Bruker Autoflex II equipped with the MALDI Molecular Imager Package. Each instrument was optimized for best resolution on the singly charged CytochromeC molecular ion and operated in automatic acquisition mode. The matrix used for the analysis was sinapinic acid (Fluka) (20 mg/mL in 50% acetonitrile in 0.1% trifluoroacetic acid).
2.3. Protein mixtures
Protein standards were used as provided by Sigma. Mouse liver protein extract was prepared by homogenizing 40 mg of mouse liver in 1 mL of the solvent, 50% acetonitrile in 0.1% trifluoroacetic acid. The insoluble pellet was removed by centrifugation. Aliquots of the liver extract were diluted by addition of equal volumes of a standard protein mixture. The standards were spiked into the mouse liver extract to the final concentrations listed in Table 1 which lists the total amount of each protein standard (pmol) applied to the MALDI target.
Table 1.
Protein standards (pmol) spiked into mouse liver extracts
| Mix 1 | Mix 2 | Mix 3 | Mix 4 | |
|---|---|---|---|---|
| Insulin (porcine) | 0.238 | 0.119 | 0.059 | 0.024 |
| Cytochrome C | 0.95 | 0.475 | 0.238 | 0.095 |
| Apomyoglobin | 2.375 | 1.188 | 0.594 | 0.238 |
Each of the four solutions was measured using MALDI mass spectrometry 10 times. Spectra comprised of 1000 shots were collected.
2.4. MALDI imaging
Brain from a healthy mouse was sectioned to a thickness of 12 μm (bregma, +2.80 mm) and thaw mounted onto a steel MALDI target. The sections were washed using 70% ethanol by immersion for 30 s followed by an additional 30 s in 100% ethanol. The dried samples were then coated with matrix using either a spraying device, (TM Sprayer, LEAP Technologies, Carborro, NC) or an acoustic matrix ejector (LabCyte Inc., CA). For spray coating of matrix, three consecutive passes were made, coating the sample with sinapinic acid matrix (7 mg/mL in 50% acetonitrile in 0.1% trifluoroacetic acid) with each pass. The spray coated section was imaged at a resolution of 75 μm, collecting a 50 shot summed spectra at each raster location. Using the acoustic matrix ejector, discreet matrix deposits (20 mg/mL sinapinic acid in 50% acetonitrile and 0.3% trifluoroacetic acid) were placed in a regular array with a spatial resolution of 150 μm by 150 μm across a rat brain section cut at the level of the frontal cortex (bregma +2.5 mm). Each matrix spot was sampled 20 times, summing up a total of 200 spectra at each location. The matrix was washed off with ethanol after MS acquisition and the section subjected to a heavy Nissl staining (cresyl violet, Sigma-Aldrich). Spectral data were assembled into individual ion images, baselined, and normalized against the total ion current using in-house developed software and ProTSData (Biodesix, Steamboat Springs, CO). Individual interpolated protein images were obtained with the imaging software tool (BioMap, Novartis), where a single ion species was visualized at the width of the peak at half-maximum intensity.
2.5. Statistical analysis
Whole spectrum analysis was performed as previously described with minor modifications as indicated [32]. Briefly, all mass spectra were pre-processed as described above. The processed text files were then imported into a script written in Matlab to be aligned according to a single mass-to-charge (m/z) column. A standard weighted means averaging (WMA) algorithm was then applied [33]. This test is similar to using a non-parametric one-way ANOVA approach (which can also be used), and presents with similar results; however, we prefer WMA due to a robustness observed with the highly variable non-Gaussian data found to be common to MALDI based mass spectra. The weight value (W) is a statistically derived function that approaches significance as the distance between the means for each group increases and the standard deviation decreases using the formula W=(μ1-μ2)/(σ1+σ2). In this way, m/z values were filtered according to the highest weight that best differentiated each group. Finally, only a set of four or more consecutive points were considered to be a true peak. These filtered values were then used to determine a level of confidence for peak detection, and further evaluated by plotting the whole spectra as compared to the difference spectra in Origin 7.0.
3. Results and discussion
Raw MALDI spectra are not amenable to any type of quantitative analysis by direct comparison. Over the course of a typical study, experimental and biological sources of variance can be manifested in the data. One can expect the variability to increase as profiling studies expand to include multiple investigators. The challenge in developing an effective analysis protocol is to adequately distinguish the different sources of variance so that the statistical analysis includes only differences protein expression in the samples. Extending the general ideas of previous profiling studies, we have developed a pre-processing workflow for imaging MS that incorporates procedures for baseline removal, intensity normalization and recalibration/realignment of spectra[34,35].
3.1. Baseline subtraction
Profile spectra typically exhibit an intense and variable chemical noise background that must be quantified before accurate measures of ion intensities can be determined. Example profiles acquired from three different tissues are shown in Fig. 2a. From these one can visually detect global baseline trends. However, development of mathematical algorithms that are equally adept at discerning baselines has not been so straightforward. A number of algorithms have been reported for estimating the baseline [26,36] and many commercial data analysis software packages include baseline subtraction modules. Extending these algorithms to image data requires, a) that the algorithm be capable of accommodating a variety of chemical backgrounds similar to those seen in Fig. 2a with limited or no interaction from the user and b) perform within a reasonable time frame. Perhaps the greatest barrier for anyone developing an algorithm for baselinecorrection is the lack of any quantitative evaluation criteria. In light of this we have been forced as others have done, to compare the results by expert visual inspection. While this introduces a degree of subjectivity it underscores the fact that if it was possible to generate a baseline model adequate for evaluating algorithms then that model in itself would be optimum for baseline correction. The first methods examined are ones available to most investigators, namely the baseline correction functions integrated into the software supplied by the instrument manufacturers, DataExplorer and FlexAnalysis, as well as the imaging standard BioMap. In the case of DataExplorer, the Advanced Baseline Correction function uses three user-set parameters: peak width, flexibility and degree which can be iteratively optimized. We found that one set of parameters are usually not adequate for modeling a baseline over the entire m/z range. Rather, the model generally provides a good estimation of the baseline for only a narrow m/z window of about 10 kDa. In order to extend the usefulness of this algorithm we processed each spectrum multiple times using correction parameters optimized for different m/z regions which were then combined into a final spectrum. While this segmented approach improves the results for a wider m/z range it is somewhat cumbersome to implement and can create anomalous artifacts in the final spectrum particularly if a peak spans two region boundaries. A convex hull option in FlexAnalysis performs well across the full m/z range and requires no user input, but this package currently imports only native data files and as such is not an option for processing data acquired on all instruments. Biomap, the de facto standard for imaging MS, generates baseline models using values from the first and second derivatives of each spectrum that meet certain threshold criteria. While Biomap is optimized for fast processing it also has significant limitations. Among these: the inability to export processed data sets that can be used with external applications and frequent out-of-memory errors when processing large images.
Fig. 2.

MALDI MS profile spectra acquired from three sections from different tissues. (a) Raw spectra; (b) spectra that have been baseline corrected using ProTSData software.
Other techniques for empirically fitting functions to the baseline were examined. For example, monotonic decay functions can be fitted to the spectra and, for a number of spectra, the model produces good results, particularly after removing high-frequency noise. However, the variable slope and intensity of the background for many spectra are not necessarily described by a simple decay function as have been shown [26]. Robust local regression estimation [36] shows very good ability to adapt to variable background signals, but the technique is severely disadvantaged with lengthy computational time to process a profile spectrum of average size, 40-50 s.
Incorporation of the local estimation algorithm in ProTSData (Biodesix, Steamboat Springs, CO) into a batch process has yielded the most widely applicable approach. The user inputs a series of values that establish expected peak resolution at various values along the full m/z range. These values are used as estimators when isolating potential signal components from the baseline. The method can be applied to a variety of spectra acquired at different times provided the spectra have similar resolving power at given m/z values. Fig. 2b presents the results of baseline correction produced by this function. Because it imports data in ASCII format it can be applied to data from different instrument vendors. At first, this may seem an inefficient way of processing the thousands of spectra found in a large image data but here the ASCII file itself is temporary. That is, the batch process sequentially converts each spectrum in the image to an ASCII file that is processed. The processed ASCII spectrum is then converted back to the more compact binary Analyze 7.5 format before being deleted. Once the processing is completed we have only raw and processed image files along with a record of the baseline modeling criteria for archival. This approach is flexible for different baseline shapes and can accommodate large volumes of data without significantly long processing times, typically ∼1-2 s/spectrum.
3.2. Normalization
The next step in spectral pre-processing is to normalize the ion intensities to minimize spectrum-to-spectrum differences in peak intensity. These variations are derived from a number of sources including instrument variation, differences in sample preparation, sample variability, and experimental error. Within the confines of a carefully controlled experiment, one is able to minimize the contribution of each of these sources of error; however, more can be done to further reduce the observed variation. To this end, a number of algorithms were tested for their ability to reduce measurement variation. Normalization is the process of projecting all data onto a common intensity scale to facilitate direct comparisons of spectra. Various normalization routines that have been described in the literature can be categorized according to their methodology which we describe as collective and individual [26,31,34,35,37,38]. In the collective approach all profile spectra in a data set are processed into a singular representation or reference against which each spectrum is then normalized. A basic example of this approach involves normalizing each spectrum to the average of all spectra. Individual normalization differs in that each spectrum is normalized to some reference independent of the collective data set, and an example of this method is the scaling of each spectrum to a constant ion-current.
The task of isolating components of biological variance from components of experimental variance can be challenging given that values of either are unknown and too many profile studies are inadequately designed. Typically sample populations are too small to provide for full statistical treatment of inter- and intra-sample variances and as a result there is always the possibility that experimental differences will be erroneously detected as a biological difference. Clinical samples are difficult to obtain and often in short supply. Realizing this for these cases, the best we can hope for is to estimate the amount of experimental variance present in a data set and use this as threshold for classifying an observed difference as being either greater than or less than what is expected from experimental variance alone. To facilitate these measurements in developing our pre-processing protocol we generated model sample sets with known biological variability to use as test cases.
In a simple example we compared spectra acquired from replicate spots deposited from the same sample to establish an ideal case for experimental variance. An aliquot of the mouse liver homogenate was mixed with sinapinic acid solution and spotted onto 100 sample wells using a 1 μL pipette. Each sample well was analyzed operating the mass spectrometer in automated mode using a constant acquisition method. Under these conditions one would expect that the only source of variance is due to MALDI processes, i.e., variability in matrix crystallization and ion generation. Plotting the log-log of individual spectral intensity versus intensity from average spectra shows distinct misalignment of data from an ideal distribution, i.e., slope = 1. Repeating this plot for each individual spectrum, one can tabulate the estimated slope and R2 for each of the 100 comparisons. The data were then normalized using either of the following: cube root, log, or ln transformations; scaling to a constant total ion current; scaling to a constant noise as determined from the coefficients of wavelet decomposition. The different methods can be compared in this way to determine which of these methods are most effective. The results of this comparison are shown in Fig. 3. When comparing any of the mentioned methods to the case where no normalization was applied, one finds that the average slope for each spectrum is always approximately 1; however, the variation is reduced in cases when a normalization routine is applied. Only two of these methods yield both high correlation and linearity, normalization to total ion current and to constant noise. The other methods involve some form of mathematical transformation such as log, ln, and cube root and are often used for multivariate data such as microarray analysis to reduce variation. However, these data sets are often linear over as many as four orders of magnitude; whereas, MALDI mass spectra are not. For MALDI spectra, data transformation obscures features, causing the sensitivity of the comparison in profiling and the image contrast in IMS data to be poor. The remaining data in this study are processed using the method of total ion current normalization.
Fig. 3.

The effect of normalization on data quality. (a) Single spectra compared against the group average deviate significantly from linearity, whereas (b) the same comparison made after normalization (TIC) results in a linear relationship. (c) This comparison was repeated for each spectrum individually, and the correlation coefficients and slope reported in histogram form. Total ion current (TIC) normalization is the most effective at reducing the deviation from linearity and increasing the correlation coefficient. (Linest and R2∼1.) Linest = regression coefficient of the best fit line (best fit = 1.0). R2 = square of the Pearson product moment correlation coefficient. (proportion of variance in y attributed to variance in x; least variance = 1.0)
3.3. Spectral realignment
At first glance, one might get the impression that realignment of mass spectra onto a common m/z scale is synonymous with calibration. Although these approaches can be the same, and in fact, are carried out in much the same manner, there are important differences. With a properly calibrated instrument, one might expect 100-200 ppm mass accuracy using a linear TOF instrument to measure proteins. While this remains true, considering this amount of variation throughout the course of a number of replicate measurements can cause problems for quantitative comparisons. Further, irregularities within the flatness or thickness of tissue sections magnify the mass measurement variability. Before performing the comparative analysis it becomes necessary to match or bin like peaks into categories to ensure that we are comparing the intensity of the same ions across a number of samples. If spectra have internal calibrants (i.e., landmarks) that can be recognized as such, then calibrating each spectrum to those known peaks can effectively realign the spectra. In many cases a sufficient number of peaks may not be accurately assigned to known m/z. The approach that we have taken instead is to identify a subset of peaks common to most if not all sample spectra and use these as basis for realigning the spectra. Using the criteria that a peak must be found in more than 90% of the spectra, we typically identify 10-20 peaks that are common to a profile data set. It is also important that these common peaks span the entire mass range of interest to prevent extrapolation outside of the chosen alignment points. These common features are assigned a mass arbitrarily chosen as the median value of that peak for the data set, and each spectrum is realigned using a quadratic calibration algorithm. The results of such realignment are shown in Fig. 4. The calibrated, but unaligned data on the top shows a large amount of uncertainty in the m/z dimensions compared to the realigned data. Typically, one observes a 5-10-fold reduction in the range of centroid values reported for a single alignment point. This allows one to more accurately categorize peaks into like groups. While this process facilitates peak comparisons across all spectra it is important to note that the realignment process can produce inaccurate m/z assignments. Once the significant peak differences have been determined from the realigned data an accurate m/z assignment is made from internally calibrated spectra.
Fig. 4.
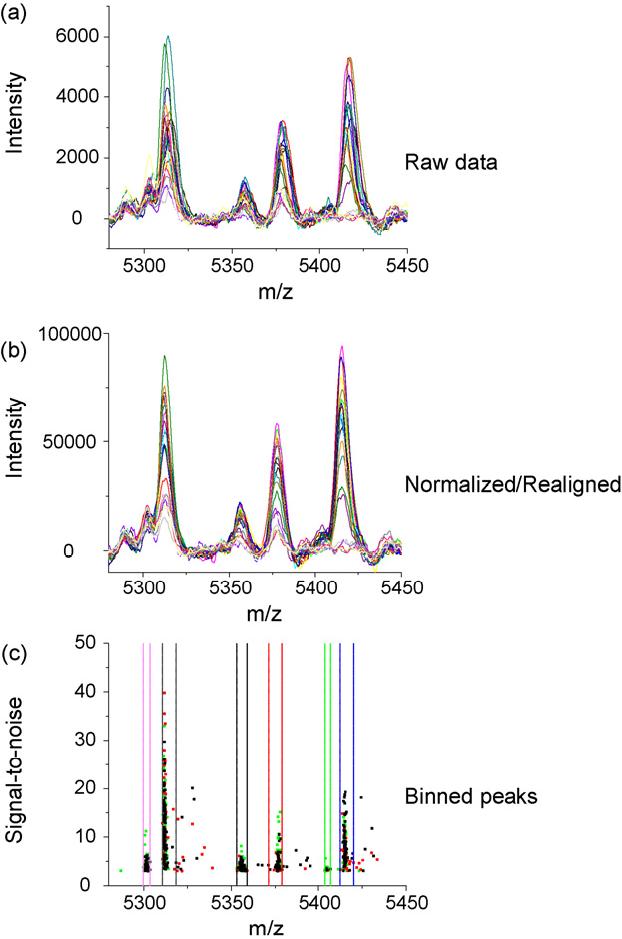
Baseline correction and peak binning. A portion of the data before (a), and after (b) spectral alignment. Spectral alignment is performed using common peaks across the set of spectra. Peaks are selected that are common to 90% of the data collected. These features are used as arbitrary calibrants for a quadratic calibration of the data. (c) The selected peaks from the data set are categorized into bins, grouping peaks originating from the same ion.
There are other methods for the aligning the m/z scale of MALDI mass spectra. For example, Matlab provides an alignment algorithm, msalign, that iteratively shifts the spectrum and computes a cross correlation of the spectrum with a theoretical spectrum generated using user provided alignment points. It was found that this algorithm provides very robust alignment; however, the computation time for each spectrum ranges from 10-60 s, depending on the amount of data per spectrum. Although this method works well and is widely available, it cannot practically be applied to image size data sets.
3.4. Effect of pre-processing on data quality
Before blindly applying these processing steps to real-world data, we wanted to insure that the effect produced on the data made sense both from mass spectrometry and biological points of view. We synthesized a sample set having known differences in the levels of known proteins to determine a practical limit at which we could expect to determine protein differences. Standard proteins were added to the liver extract mentioned previously. These proteins were chosen because they are commonly used MALDI calibrants and therefore sensitivity is not an issue and because they did not overlap with ions endogenous to the liver extract. These ions were added in increasing concentrations, ranging over one order of magnitude. It was found that higher or lower concentrations of spiked proteins could not be added without significantly affecting the sensitivity of the extract or the spiked standards because of ion suppression. The spiked standards can only be measured simultaneously with the liver extract across a 10-fold dynamic range. Each of these spiked mixtures was measured 10 times using linear MALDI TOF MS. The resulting response is shown in Fig. 5. Operator bias, variation in dried droplet sample preparation, and instrumental variations, produced strong ion intensity differences that depended on sample and the location within the matrix spot being interrogated using the laser. These differences manifest themselves in individual spectra as variations in overall peaks intensity and the amount of chemical noise observed in the low molecular weight region. Although these effects can be tolerated in a qualitative study such as peptide mass fingerprinting, in MALDI profiling and imaging, the quantitative information relates directly to the sensitivity of the assays and the quality of the images being generated. The ion intensity measured as a function of protein concentration is displayed in Fig. 5 both before spectral pre processing and after. The raw data deviates from linearity for each of the proteins measured, and the experimental error is quite large in the uncorrected data. In contrast, after pre-processing the data using the approach outline in Fig. 1, the ion intensity is more reproducible. This results in lower experimental error and the ion intensity approaches a linear correlation with protein concentration. Data having a more predictable response as a function of concentration yields more reliable predictive results.
Fig. 5.

(a) Comparison of the relationship between protein concentration and instrument response for processed and unprocessed data. The unprocessed data shows a marked deviation from the expected relationship to protein concentration. However, processing the data according to the proposed scheme restores data quality comparable to the ideal case. The factors that contribute to this deviation include manual sample preparation, MALDI target spotting, and sample acquisition. (b) Effect of baseline/noise subtraction to remove chemical noise and the effect of normalization on spectral quality. Spectra were normalized using TIC. Spectral features were extracted using a minimum S/N threshold of 3. Each spectrum was comprised of 5-7 ions originating from the spiked standard proteins and approximately 110-120 ions originating from the liver extract. The error bars represent 95% confidence intervals for the intensity.
3.5. Processing of imaging mass spectrometry data
Previous studies have highlighted the need to consider sample preparation as a critical component in developing MALDI IMS applications [4,8,39]. However, in spite of the effort to exert control over environmental factors affecting matrix applications, instrument variation during the course of data collection, and inherent cellular heterogeneity within the samples themselves, there is inevitably some contribution of each of these effects to the image. In much the same way as with profile spectra, this experimental variability has direct bearing on image quality and overall ability to visually observe biological changes in protein levels. Comparing two single spectra comprising two pixels of the image, it is often difficult to estimate the amount of variation between the two; however, when examining a single mass plotted as an image the differences become obvious.Fig. 6 shows selected ion images collected from two different regions of mouse brain. The images on the left are unprocessed, and represents purely the integrated ion intensity from each spectrum over the peak at m/z 6219 (Fig. 6a) and m/z 14,575 (Fig. 6c), respectively. Gross structural features within the tissue are clearly distinguishable; however, they are obscured by a high background signal. In this mass range, unprocessed spectra can have a high amount of background signal due to the contributions of chemical noise. This effect is for example visible in the upper corners of the unprocessed image in Fig. 6a away from the tissue edge. The background signal is additionally affected by any irregularities in the sample preparation. In the same image, this is evidenced by the bright stripe of high intensity (white) running horizontal across the center of the image. Moreover, some of the bright areas in Fig. 6a do not follow the contours of any of the tissue features, but rather an area of matrix that is of greater density due to overlapping passes of the matrix sprayer. Similar effects are observed in Fig. 6c where tissue features in the left brain hemisphere are obscured due to similar sample preparation effects.
Fig. 6.
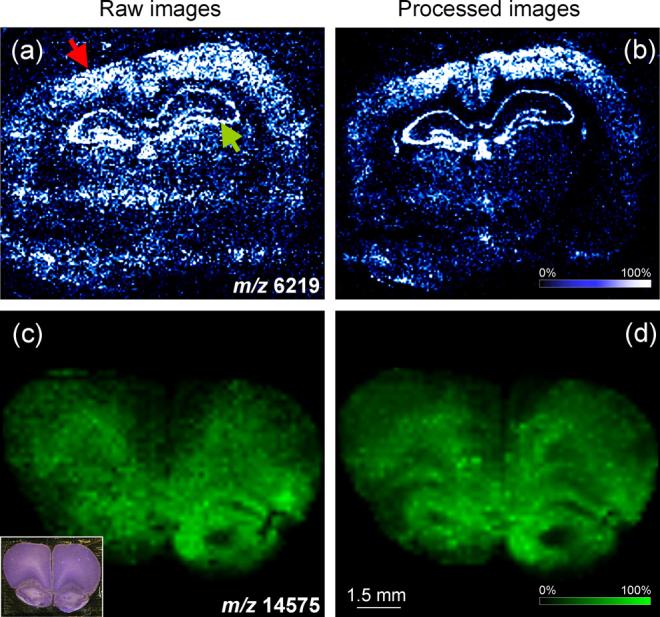
Baseline subtraction and normalization improve image quality. Two selected ion images obtained from mouse brain are chosen to highlight the advantages of spectral processing on image quality. Above are compared raw image data (a and c) and data processed (b and d) to remove baseline and normalized using total ion current. The inset in panel (c) presents a photomicrograph of the section used for IMS after matrix removal and Nissl staining. Red arrow, cortex; green arrow, hippocampus.
Since image data are nothing more than ordered arrays of profile spectra we can apply the pre-processing steps outline in Fig. 1 to markedly increase the quality of images as we have done in the right panels of Fig. 6 (b and d, respectively). First, the image contrast is increased due to the decrease in chemical noise and its contribution to the background signal. In Fig. 6b, it becomes much more apparent that there is a distinct border of the outside edge of the tissue. Additionally, two distinct areas of the tissue, the cortex and the hippocampus, stand out as having higher amounts of this protein. Furthermore, the prominent horizontal bands of brighter pixels seen in Fig. 6a image are not as prominent in the corresponding processed image. There is noticeable improvement in Fig. 6d as well. First, the darker, low intensity pixels present in the raw image, Fig. 6c, have been enhanced by the processing to yield a more expected pixel-to-pixel transition. As a result, features that were only faintly visible before processing are now clearly distinguishable due to the improved image contrast.
The individual mass spectra of mass spectrometric images of tissue having highly compartmentalized and varied protein expression are not always possible to align and normalize. When comparing one part of the tissue to an adjacent position on the tissue composed of different cell types, reduced numbers of common peaks are available as alignment points and changes in the ion currents may be observed. One basic assumption in adopting total ion current as a method for normalizing spectra is that the identity and abundance of ions across the data set are similar in each spectrum. For diverse tissue samples this in not always the case and for these samples normalizing to total ion current shows only modest improvement. Further development of sample preparation techniques would allow for the inclusion of calibration standards in the matrix used for coating tissues. This would allow for images of such samples to be aligned more reliably. In spite of these drawbacks, these methods provide obvious benefits to data quality and have been routinely applied to images of major organs, such as brain, liver and kidney, with success.
3.6. Data quality and sample quality
Although it is beyond the scope of our present discussion it is important to note that the acquisition and analysis of mass spectra are but one component of profiling and imaging studies of tissue sections by MALDI. Before reaching the mass spectrometry lab clinical samples have been collected and handled by people in conditions that are often varied and beyond the control of the mass spectrometry analyst. A well designed profiling study should include protocols for these portions of a study to avoid introducing systematic bias into the profile spectra which may be attributed to biological differences when in fact they fall within experimental error. Profiling studies to determine postischemic variations of the proteome with time are limited and as such every effort and consideration should be made to insure these are minimized as much as possible. Within the relatively small scale of previous profiling studies we can assume that a limited number of individuals are in the chain of custody and that minimal variations exist within sample collection and handling. However, this will most likely not be the case as the future studies expand to include multiple phases or institutions. No amount of processing has demonstrated to compensate for high-quality data from samples appropriate to address the clinical question of interest.
Likewise, it is also important to prepare samples for imaging with the same rigorous standards. Some of the traditional methods for sample preparation do not yield the best matrix coating for tissues, and lack of attention in the way data are collected and instrument performance can negatively affect the outcome of the experiment. New and improved methods for the coating of samples for imaging which are more automated and standardized will go far to increase the quality and usefulness of the data generated with such techniques. This paper is intended as a starting point for the furthering of IMS using data processing approaches.
4. Concluding remarks
In summary, we have presented a data processing workflow that we have used to successfully increase the amount of useful information that can be derived from mass spectrometry profiling and imaging of biological specimens. The steps necessary to obtain such improvements were found to be baseline subtraction, normalization, and spectral realignment/recalibration. For the purposes of protein profiling, these methods allow for a more accurate determination of the relevant changes in protein expression. Furthermore, the application of these methods to the processing of MALDI imaging data allows for improvements in the contrast of the mass spectrometric image and compensates for variations in the quality of the matrix coating. The methods that were highlighted offer some significant improvements, allowing progress that would not otherwise be possible; however, there were limitations on the amount of time and the type of expertise that could be brought to bear on the problem. For each of the steps highlighted, and each method that was tested, there are numerous other algorithms that went untried due to lack of resources and time. Also, the data available for testing is ever increasing, so the availability of data derived from different samples and different instruments continues to grow. Likely, the approach we outlined will need further refinement for some of these special conditions. It is our hope that future extensions of this work will expand upon the basic framework presented here for continued development of the fields of molecular profiling and imaging mass spectrometry.
Acknowledgements
The authors would like to acknowledge Anna Crecelius, Ph.D. (Lugwig Maximilians University Munich) for her help preparing and analyzing the protein extracts and Heinrich Röder, D.Phil. (Biodesix, Inc.) for helpful discussions and software support. The authors also acknowledge support from the NIH (GM 58008-08), the NCI (CA 86243-03 and CA 116123-01) and the DOD (W81XWH-05-1-0179).
References
- [1].Rohner TC, Staab D, Stoeckli M. Mech. Ageing Dev. 2005;126:177. doi: 10.1016/j.mad.2004.09.032. [DOI] [PubMed] [Google Scholar]
- [2].Reyzer ML, Caprioli RM. J. Proteome Res. 2005;4:1138. doi: 10.1021/pr050095+. [DOI] [PubMed] [Google Scholar]
- [3].Crossman L, McHugh NA, Hsieh YS, Korfmacher WA, Chen JW. Rapid Commun. Mass Spectrom. 2006;20:284. doi: 10.1002/rcm.2259. [DOI] [PubMed] [Google Scholar]
- [4].Altelaar AFM, Klinkert I, Jalink K, de Lange RPJ, Adan RAH, Heeren RMA, Piersma SR. Anal. Chem. 2006;78:734. doi: 10.1021/ac0513111. [DOI] [PubMed] [Google Scholar]
- [5].DeKeyser SS, Li LJ. Analyst. 2006;131:281. doi: 10.1039/b510831d. [DOI] [PubMed] [Google Scholar]
- [6].Garrett TJ, Yost RA. Anal. Chem. 2006;78:2465. doi: 10.1021/ac0522761. [DOI] [PubMed] [Google Scholar]
- [7].Jones JJ, Borgmann S, Wilkins CL, O’Brien RM. Anal. Chem. 2006;78:3062. doi: 10.1021/ac0600858. [DOI] [PubMed] [Google Scholar]
- [8].Lemaire R, Tabet JC, Ducoroy P, Hendra JB, Salzet M, Fournier I. Anal. Chem. 2006;78:809. doi: 10.1021/ac0514669. [DOI] [PubMed] [Google Scholar]
- [9].Stemmler EA, Gardner NP, Guiney ME, Bruns EA, Dickinson PS. J. Mass Spectrom. 2006;41:295. doi: 10.1002/jms.989. [DOI] [PubMed] [Google Scholar]
- [10].Woods AS, Ugarov M, Jackson SN, Egan T, Wang HYJ, Murray KK, Schultz JA. J. Proteome Res. 2006;5:1484. doi: 10.1021/pr060055l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guo JZ, Colgan TJ, DeSouza LV, Rodrigues MJ, Romaschin AD, Siu KWM. Rapid Commun. Mass Spectrom. 2005;19:2762. doi: 10.1002/rcm.2119. [DOI] [PubMed] [Google Scholar]
- [12].Xu BJ, Shyr Y, Liang XB, Ma LJ, Donnert EM, Roberts JD, Zhang XQ, Kon V, Brown NJ, Caprioli RM, Fogo AB. J. Am. Soc. Nephrol. 2005;16:2967. doi: 10.1681/ASN.2005030262. [DOI] [PubMed] [Google Scholar]
- [13].Yanagisawa K, Shyr Y, Xu BGJ, Massion PP, Larsen PH, White BC, Roberts JR, Edgerton M, Gonzalez A, Nadaf S, Moore JH, Caprioli RM, Carbone DP. Lancet. 2003;362:433. doi: 10.1016/S0140-6736(03)14068-8. [DOI] [PubMed] [Google Scholar]
- [14].Schwartz SA, Weil RJ, Thompson RC, Shyr Y, Moore JH, Toms SA, Johnson MD, Caprioli RM. Cancer Res. 2005;65:7674. doi: 10.1158/0008-5472.CAN-04-3016. [DOI] [PubMed] [Google Scholar]
- [15].Pierson J, Norris JL, Aerni HR, Svenningsson P, Caprioli RM, Andren PE. J. Proteome Res. 2004;3:289. doi: 10.1021/pr0499747. [DOI] [PubMed] [Google Scholar]
- [16].Laurent C, Levinson DF, Schwartz SA, Harrington PB, Markey SP, Caprioli RM, Levitt P. J. Neurosci. Res. 2005;81:613. doi: 10.1002/jnr.20590. [DOI] [PubMed] [Google Scholar]
- [17].Pierson J, Svenningsson P, Caprioli RM, Andren PE. J. Proteome Res. 2005;4:223. doi: 10.1021/pr049836h. [DOI] [PubMed] [Google Scholar]
- [18].Chaurand P, Fouchecourt S, DaGue BB, Xu BGJ, Reyzer ML, Orgebin-Crist MC, Caprioli RM. Proteomics. 2003;3:2221. doi: 10.1002/pmic.200300474. [DOI] [PubMed] [Google Scholar]
- [19].Hsieh Y, Casale R, Fukuda E, Chen JW, Knemeyer I, Wingate J, Morrison R, Korfmacher W. Rapid Commun. Mass Spectrom. 2006;20:965. doi: 10.1002/rcm.2397. [DOI] [PubMed] [Google Scholar]
- [20].Troendle FJ, Reddick CD, Yost RA. J. Am. Soc. Mass Spectrom. 1999;10:1315. [Google Scholar]
- [21].Reyzer ML, Caldwell RL, Dugger TC, Forbes JT, Ritter CA, Guix M, Arteaga CL, Caprioli RM. Cancer Res. 2004;64:9093. doi: 10.1158/0008-5472.CAN-04-2231. [DOI] [PubMed] [Google Scholar]
- [22].Chaurand P, Schwartz SA, Caprioli RM. J. Proteome Res. 2004;3:245. doi: 10.1021/pr0341282. [DOI] [PubMed] [Google Scholar]
- [23].Chaurand P, Sanders ME, Jensen RA, Caprioli RM. Am. J. Pathol. 2004;165:1057. doi: 10.1016/S0002-9440(10)63367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Caprioli RM. Cancer Res. 2005;65:10642. doi: 10.1158/0008-5472.CAN-04-3581. [DOI] [PubMed] [Google Scholar]
- [25].Tatay JW, Feng X, Sobczak N, Jiang H, Chen CF, Kirova R, Struble C, Wang NJ, Tonellato PJ. Proteomics. 2003;3:1704. doi: 10.1002/pmic.200300512. [DOI] [PubMed] [Google Scholar]
- [26].Wagner M, Naik D, Pothen A. Proteomics. 2003;3:1692. doi: 10.1002/pmic.200300519. [DOI] [PubMed] [Google Scholar]
- [27].Slotta DJ, Heath LS, Ramakrishnan N, Helm R, Potts M. Proteomics. 2003;3:1687. doi: 10.1002/pmic.200300517. [DOI] [PubMed] [Google Scholar]
- [28].Lee KR, Lin XW, Park DC, Eslava S. Proteomics. 2003;3:1680. doi: 10.1002/pmic.200300515. [DOI] [PubMed] [Google Scholar]
- [29].Markey MK, Tourassi GD, Floyd CE. Proteomics. 2003;3:1678. doi: 10.1002/pmic.200300521. [DOI] [PubMed] [Google Scholar]
- [30].Zhu HT, Yu CY, Zhang HP. Proteomics. 2003;3:1673. doi: 10.1002/pmic.200300520. [DOI] [PubMed] [Google Scholar]
- [31].Baggerly KA, Morris JS, Wang J, Gold D, Xiao LC, Coombes KR. Proteomics. 2003;3:1667. doi: 10.1002/pmic.200300522. [DOI] [PubMed] [Google Scholar]
- [32].Mobley JA, Lam YW, Lau KM, Pais VM, L’Esperance JO, Steadman B, Fuster LMB, Blute RD, Taplin ME, Ho SM. J. Urol. 2004;172:331. doi: 10.1097/01.ju.0000132355.97888.50. [DOI] [PubMed] [Google Scholar]
- [33].Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloom-field CD, Lander ES. Science. 1999;286:531. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- [34].Morris JS, Coombes KR, Koomen J, Baggerly KA, Kobayashi R. Bioinformatics. 2005;21:1764. doi: 10.1093/bioinformatics/bti254. [DOI] [PubMed] [Google Scholar]
- [35].Villanueva J, Philip J, Chaparro CA, Li Y, Crow-toledo R, DeNoyer L, Fleisher M, Robbins RJ, Tempst P. J. Proteome Res. 2005;4:1060. doi: 10.1021/pr050034b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cleveland WS. J. Am. Stat. Assoc. 1979;74:829. [Google Scholar]
- [37].Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. Anal. Chem. 2006;78:779. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- [38].Wu BL, Abbott T, Fishman D, McMurray W, Mor G, Stone K, Ward D, Williams K, Zhao HY. Bioinformatics. 2003;19:1636. doi: 10.1093/bioinformatics/btg210. [DOI] [PubMed] [Google Scholar]
- [39].Schwartz SA, Reyzer ML, Caprioli RM. J. Mass Spectrom. 2003;38:699. doi: 10.1002/jms.505. [DOI] [PubMed] [Google Scholar]