

Abstract
Meta-[211At]astatobenzylguanidine ([211At]MABG), an analogue of meta-iodobenzylguanidine (MIBG) labeled with the α-emitter 211At, targets the norepinephrine transporter. Because MABG has been shown to have excellent characteristics in preclinical studies, it has been considered to be a promising targeted radiotherapeutic for the treatment of tumors such as micrometastatic neuroblastoma that over express the norepinephrine transporter. To facilitate clinical evaluation of this agent, a convenient method for the high level synthesis of [211At]MABG that is adaptable for kit formulation has been developed. A tin precursor anchored to a solid-support was treated with a methanolic solution of 211 At in the presence of a mixture of H2O2/HOAc as the oxidant; [211At]MABG was isolated by simple solid-phase extraction. By using C-18 solid-phase extraction, the radiochemical yield from twenty-five batches was 63 ± 13%; however, loss of radioactivity during evaporation of the methanolic solution was a problem. This difficulty was avoided by use of a cation exchange resin cartridge for isolation of [211At]MABG, which resulted in radiochemical yields of 63 ± 9% in a shorter duration of synthesis. The radiochemical purity was more than 90% and no chemical impurity has been detected. The final doses were sterile and apyrogenic. These results demonstrate that [211At]MABG can be prepared via a kit method at radioactivity levels anticipated for initiation of clinical studies.
Keywords: Meta-[211At]astatobenzylguanidine, Meta-iodobenzylguanidine, Neuroblastoma, Astatine-211, Solid-phase synthesis
1. Introduction
The norepinephrine transporter (NET) represents an attractive target for the delivery of therapeutic radiopharmaceuticals to malignancies that arise from the neuroendocrine system. NET is highly over-expressed in neuroendocrine tumors such as neuroblastoma, pheochromocytoma/paraganglioma and carcinoid. Meta-iodobenzylguanidine (MIBG or Iobenguane) is a an avid substrate for NET1, 2 and radioiodinated MIBG has been shown to accumulate in these tumors to such an extent as to be highly useful for both disease detection using nuclear imaging and targeted radiopharmaceutical therapy in humans.3, 4
[131I]MIBG is an active agent in the monotherapy of heavily-pretreated, refractory high-risk neuroblastoma, with 37% responding per International Neuroblastoma Response Criteria (INRC), 27% alive and without disease progression 6 months after [131I]MIBG monotherapy, and with 30% alive at 12 months, including those with progressive disease.5 [131I]MIBG has been used in combination with radiosensitizing agents,6 surgery,7 myeloablative chemotherapy,8 and/or external beam radiotherapy9 in attempts to improve therapeutic response. Similarly, [131I]MIBG is an effective agent in the treatment of malignant pheochromocytoma patients, with 30% showing tumor response and 45% showing hormonal response.10
As [131I]MIBG has demonstrated activity in neuroendocrine cancers, significant data also has demonstrated the ability of other radiohalogenated analogs of MIBG such as 77/76Bromo-, 18Fluoro- and 211Astato- labeled benzylguanidines to accumulate in neuroendocrine tumors.11-13 The ability of this transporter (NET) to recognize and transport these various halogenated benzylguanidines offers several opportunities for medical imaging and radiotherapy applications.
Most intriguing may be the use of the α-particle emitting analogue meta-[211At]astatobenzylguanidine ([211At]MABG)13 for the treatment of neuroendocrine cancers in conjunction with [131I]MIBG, particularly in the treatment of micrometastatic lesions that are often associated with neuroblastoma. Βeta-particles of 131I have a range in tissue that is longer than the dimensions of neuroblastoma metastases. Because of this, the fraction of radiation dose from β-particles that is deposited in these metastatic tumors will be considerably less.14 Alpha particles, on the other hand, have a range in tissue of only a few cell diameters and are more appropriate for the targeted radiotherapy of smaller tumors. Also, because of their high energy and short path length, α-particles are radiations of high linear energy transfer (LET). The LET of 211At α-particles is about 100 keV/μm, a value at which the relative biological effectiveness of ionizing radiation is the highest.15 Astatine-211 has a half-life of 7.2 h and decays by a double-branched pathway, producing one α-particle as a consequence of each decay.16 Its physical half-life is well-matched with the biological half-life of small molecular weight pharmaceuticals such as MIBG. Furthermore, being a heavy halogen, it is often facile to label organic compounds with 211At using aromatic tin intermediates.
For these reasons, we have developed a synthesis of [211At]MABG13 and it has been demonstrated that the mechanism for cell uptake and the tissue distribution of [211At]MABG are quite similar to [131I]MIBG but the α-particle emitting analogue is considerably more cytotoxic to human tumor cells that over-express NET.13, 17-26
One of the major impediments to the clinical evaluation of [211At]MABG is that the labeling method developed for this compound is not ideal for large scale production, in part, because it involves an HPLC purification step. Although the method provided [211At]MABG in high yield, only <1 mCi had been synthesized at a time. Because this method would not be practical for the synthesis of [211At]MABG at higher radioactivity levels on a routine basis, a simpler method was sought before getting regulatory approval to initiate clinical evaluation of [211At]MABG as a cancer therapeutic. Our goal was to develop a simple, high yielding procedure that could be adapted to kit formulation in a hospital radiopharmacy.
Solid-phase organic synthesis has become a powerful tool for the syntheses of a variety of chemicals and drug molecules.27 Originally developed by Merrifield for the synthesis of peptides,28 this methodology has several advantages over solution-phase techniques including ease of product isolation via simple filtration, the ability to use large excess of reagents to drive the reaction to completion, and amenability to automation. The speed and simplicity of solid-phase organic synthesis is appealing for radiopharmaceutical applications, especially for those involving high levels of radioactivity.
A very few examples of solid-phase synthesis of radiopharmaceuticals exist. Of particular interest to [211At]MABG synthesis is a work reported for the synthesis of no-carrier-added [*I]MIBG in high yields from poly-(3-{dibutyl[2-(3-and-4-vinylphenyl)ethyl]stannyl}benzylguanidinium acetate)-co-divinylbenzene (tin precursor; Figure 1).29 Herein, we have adapted this strategy for the synthesis of [211At]MABG utilizing a procedure that is amenable to kit formulation. To date, up to 9 mCi of [211At]MABG in a form suitable for patient administration has been produced using this approach.
Figure 1.
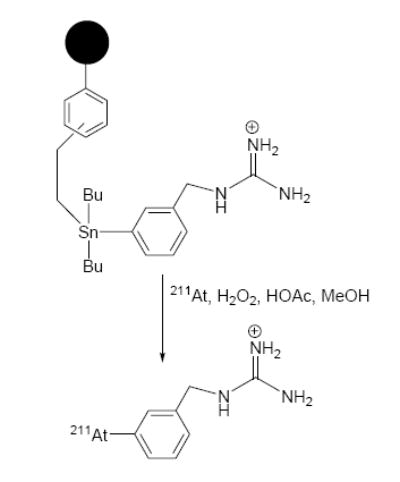
Scheme for the synthesis of [211At]MABG from resin-supported tin precursor.
2. Results and Discussion
The availability of a practical procedure for radiopharmaceutical preparation, especially for molecules tagged with short-lived radionuclides such as 211At at high radioactivity levels, via a kit method should greatly facilitate their clinical evaluation. With this goal in mind, we have evaluated the synthesis of [211At]MABG from a solid-supported tin precursor (Figure 1) that was originally developed for the synthesis of no-carrier-added [*I]MIBG.29
The conditions used by Hunter and Zhu for the synthesis of radioiodinated MIBG involved the use of a mixture of methanol and potassium phosphate as solvent and H2O2/HOAc as the oxidant. The choice of methanol as a solvent should be well suited for 211At because it has been shown that the radiolytic damage of other tin precursors at higher activity levels is relatively low in methanol.30, 31 It should be noted that our original procedure utilized chloroform as the solvent,13 a medium demonstrated to be unsuitable for use at high 211At activity levels due to reaction of radiolytically generated chlorine molecules with the tin precursors.30, 31
For these reasons, all reactions were conducted using methanolic solutions of 211At. Preliminary experiments indicated that use of the buffer, and the large volumes of buffer, methanol, and acetic acid utilized per mg polymer in the Zhu and Hunter procedure were not necessary and might even be counter productive. Initially reactions were conducted using 5 mg of resin per reaction to determine the effect of reaction time on the radiochemical yield. The radiochemical yield was determined by injecting an aliquot of the reaction mixture onto HPLC. As shown in Figure 2, a radiochemical yield of 50.2 ± 6.3% was obtained when the reaction was performed for 5 min. An increased yield of 60.5 ± 6.3% was obtained by prolonging the reaction to 15 min. Increasing the duration of the reaction beyond 15 min did not further increase the radiochemical yield. Although not systematically investigated, from the results of a few reactions, increasing the reaction temperature (up to 70°C) did not improve the radiochemical yield. This is in contrast to the observations of Maresca and Kronague (Molecular Insight Pharmaceuticals Inc.; unpublished results) who found a considerable increase in the yield of [123I]MIBG at 85°C compared to that obtained at 25°C. They conducted the reactions with 10 mg of resin in a mixture of phosphate buffer and ethanol and using the H2O2/HOAc mixture as the oxidant. The HPLC of the reaction mixtures of our temporal effect studies, performed at <1 mCi 211At, indicated the formation of other labeled byproducts that were more polar than [211At]MABG. Frequently, there was a peak eluting with the solvent front, consistent with the presence of astatide or other ionic forms of astatine, and at times, a couple of other peaks with retention times between that of the solvent front peak and that of [211At]MABG.
Figure 2.
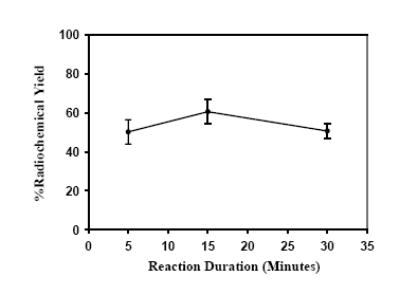
Temporal effect on the radiochemical yield: Resin (5 mg) was treated with a methanolic solution of 211At and a mixture of H2O2/HOAc for 5, 15 and 30 min. Radiochemical yields were determined by reversed-phase HPLC.
Based on the information obtained from these preliminary experiments, reactions using higher radioactivity levels of 211At were performed. Although it may be possible to extract larger amounts of 211At in 50 μl of methanol, a higher volume was desired when higher activities of 211At were used in order to minimize the effect of radiolysis on the labeling reaction.30, 31 However, preliminary studies indicated that the radiochemical yields were considerably lower when more than 100 μl of methanol was used per 5 mg of resin. On the other hand, use of 10 mg of resin and a total volume of 100-200 μl of methanol gave fairly consistent yields.
Initially [211At]MABG was isolated by the C-18 solid-phase cartridge method. Using this method of isolation, twenty-five reactions with relatively high levels of 211At activity under optimized conditions have been performed and the results are summarized in Table 1. These reactions utilized 10 mg of resin, 100-200 μl of methanol and a 10-min reaction time. The range of 211At activity used was 1-23 mCi. The average radiochemical yield, calculated as the percent of radioactivity loaded onto the C-18 cartridge column that eluted in the methanol fractions 2-6, was 63 ± 13%; minimal radioactivity was stuck to the reaction vial.
Table 1.
High level synthesis of [211At]MABG from a tin precursor anchored to a solid support: Isolation by C18 cartridgea.
| Experiment number | Initial 211At activity used (mCi) | Radioactivity eluted in methanol fractions from the C-18 cartridge | Percent of activity loaded |
|---|---|---|---|
| 1 | 1.01 | 0.55 | 57 |
| 2 | 2.71 | 1.66 | 63 |
| 3 | 8.65 | 5.30 | 65 |
| 4 | 7.15 | 2.81 | 41 |
| 5 | 7.41 | 4.33 | 60 |
| 6 | 8.99 | 2.95 | 34 |
| 7 | 2.90 | 1.11 | 39 |
| 8 | 12.63 | 7.61 | 62 |
| 9 | 10.33 | 5.03 | 51 |
| 10 | 4.23 | 2.24 | 55 |
| 11 | 20.47 | 8.56 | 42 |
| 12 | 19.99 | 13.39 | 69 |
| 13 | 4.95 | 3.74 | 80 |
| 14 | 18.35 | 13.87 | 79 |
| 15 | 18.38 | 12.73 | 71 |
| 16 | 13.36 | 10.09 | 77 |
| 17 | 18.75 | 12.71 | 69 |
| 18 | 9.89 | 7.19 | 75 |
| 19 | 20.59 | 13.89 | 70 |
| 20 | 20.22 | 13.71 | 70 |
| 21 | 13.41 | 9.21 | 72 |
| 22 | 20.31 | 14.86 | 75 |
| 23 | 18.42 | 11.60 | 65 |
| 24 | 22.66 | 13.00 | 60 |
|
| |||
| 25 | 18.85 | 13.5 | 72 |
|
| |||
| Mean ± SD | 12.98 ± 6.85 | 8.23 ± 4.89 | 63 ± 13 |
aThe tin precursor resin (10 ± 1 mg) was treated with211At in 100-150 μl of methanol and 20 μl of H2O2/HOAc mixture for 10 min at room temperature. Three different batches of resin were used. For most runs, the loading was ~1 mmol/g; for reaction 6, the resin loading was only 0.7 mmol/g. The resin used for reaction 12-25 had a more powdery consistency; others were clumpy.
It should be pointed out that more than one batch of precursor resin was used for these high level synthesis reactions. Radiochemical yields of 52 ± 11% were obtained for runs 1-11 in Table 1 and 72 ± 5% for runs 12-25; the batch of resin used for the latter group consisted of finer particles and thus, of higher surface area. In Figure 3, the radiochemical yield as a function of initial 211At activity in the reaction mixture is plotted for both batches of resin. Although there was a small decrease in radiochemical yield as a function of 211At activity added to the reaction mixture, the effect was not statistically significant (r2 = 0.042, first batch; r2 = 0.431, second batch).
Figure 3.
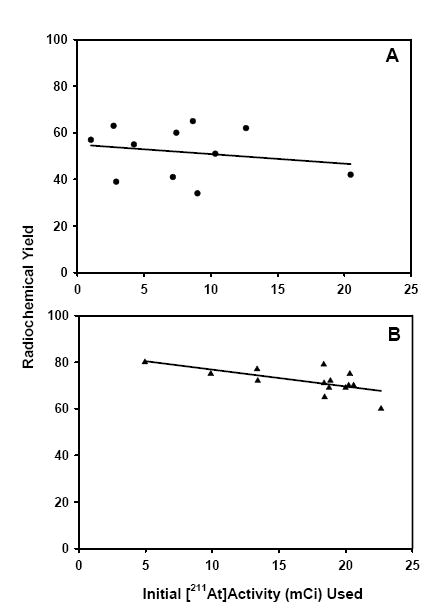
Correlation of initial 211At activity and radiochemical yields for reactions 1-11 (A; r2 = 0.042) and 12-25 (B; r2 = 0.431) of Table 1.
As shown in Figures 4 and 5, there was only one major radioactive peak in both the HPLC and TLC radioactivity profiles of [211At]MABG synthesized using the kit protocol suggesting that any polar byproducts such as those observed in the temporal effect studies, were eliminated during the solid-phase extraction procedure. Based on HPLC and TLC analysis, the radiochemical purity is determined to be more than 90%. No peaks were seen in the HPLC UV profile of the final preparation. Three batches of [211At]MABG prepared in this manner was subjected to sterility and apyrogenicity tests. These batches were found to be sterile and apyrogenic, suggesting the suitability of these methods for preparing [211At]MABG for clinical studies. The maximum amount of [211At]MABG that has been synthesized to date using the C-18 cartridge for isolation is 15 mCi; however, due to loss during evaporation of methanol, the final injectable dose obtained by this method was lower (see below).
Figure 4.
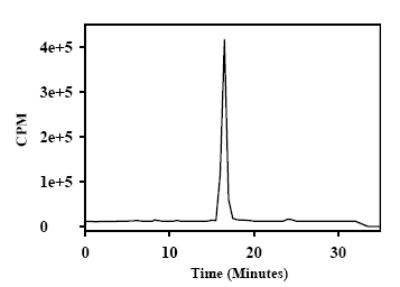
A typical radioactivity profile of reversed-phase HPLC of final [211At]MABG dose obtained by the kit method. For this a Waters' XTerra C18 column was eluted isocratically with 0.1% TFA in 80/20 water/CH3CN at a flow rate of 1 ml/min.
Figure 5.
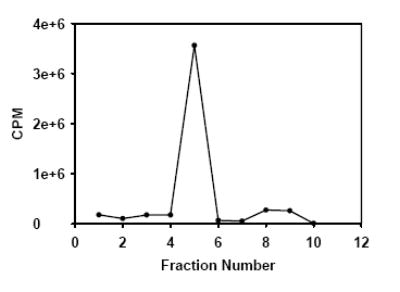
A typical radio TLC of final [211At]MABG dose obtained by the kit method.
If the astatination reaction takes place as expected by breakage of the bond between the tin and the carbon of the aromatic ring substituted with the guanidinomethyl group, the tin atom should stay attached to the resin and should not contaminate the final preparation. Nonetheless, although thermodynamically less favored, there is a possibility of a side reaction whereby the bond between tin and the carbon residing on the polymer can break32 resulting in small molecular weight tin-containing byproducts. As mentioned above, the HPLC UV profile of the final dose did not have any detectable peaks. However, even very small levels of alkyl tin compounds could be extremely toxic. To ensure that the final dose is devoid of any tin impurities, the tin content of a few batches were determined using ICP-MS. The tin content was less than one part per million in all cases.
A serious problem with the C-18 cartridge method described above for the isolation of [211At]MABG was that a considerable amount of radioactivity (average 50%) volatilized off during the evaporation of methanol from the cartridge elution fractions. Being a polar and ionic molecule, [211At]MABG itself may not volatilize considerably; in fact, we have experienced little or no loss during the evaporation of the solvent from a methanolic solution of [131I]MIBG. We speculate that this observation with [211At]MABG might be due to the formation of some volatile byproduct by the radiolytic decomposition of [211At]MABG during the prolonged evaporation step (about 45-60 min), particularly at the end of the process when the remaining volume was small. Indeed, a TLC analysis of the volatilized radioactivity, which was trapped in another C18 cartridge and eluted with methanol, indicated that only about 10% of it co-eluted with MIBG; most of the radioactivity was in nonpolar fractions. To surmount this problem, a different method, developed at Molecular Insight Pharmaceuticals for no-carrier-added [*I]MIBG purification, was investigated to isolate [211At]MABG. In this method, [211At]MABG was trapped in a cation exchange cartridge, which was subsequently eluted with 0.2 M phosphoric acid and the pH of the eluent was adjusted to the physiological value. This avoided the evaporation step, thereby circumventing the inferred radiolytic decomposition, and helped reducing the total synthesis time by 45-50 min.
The results of the experiments, performed utilizing the cation exchange cartridge, are presented in Table 2. The resin used for these reactions was the finer variety and was of the same batch that was used for reactions 12-25 of Table 1. The average radiochemical yield based on the percent of initial 211At activity that eluted in fractions 2-12 from the cation exchange cartridge was 63 ± 9%. Based on the [211At]MABG activity available for injection, the radiochemical yield was 50 ± 7%. To date, 9 mCi of [211At]MABG that was in a form suitable for patient administration has been prepared. The HPLC and TLC of the final product from these experiments also were similar to those shown in Figure 4 and 5. A few batches of [211At]MABG preparations isolated using the cation cartridge method were analyzed for sterility and apyrogenicity, as well as their tin content determined. They were sterile and apyrogenic and their tin content was less than one part per million.
Table 2.
High level synthesis of [211At]MABG from a tin precursor anchored to a solid support: Isolation by cation exchange cartridge
| Experiment number | Initial 211At activity used (mCi) | [211At]MABG activity (mCi) in cation exchange cartridge fractions | Radiochemical yield (%)a,b | Final sterile filtered [211At]MABG mCi (%) | |
|---|---|---|---|---|---|
| 2 – 12 | 4 – 10 | ||||
| 1 | 14.81 | 10.18 | 9.86 | 69 (67) | 7.71 (52) |
| 2 | 13.16 | 9.67 | 8.69 | 74 (66) | 7.15 (54) |
| 3 | 16.65 | 11.96 | 10.96 | 72 (66) | 9.12 (55) |
| 4 | 2.23 | 1.36 | 1.22 | 61 (55) | 1.08 (49) |
| 5 | 3.35 | 1.59 | 1.51 | 51 (48) | 1.22 (39) |
| 6 | 17.30 | 11.36 | 10.74 | 66 (62) | 9.19 (53) |
| 7 | 11.16 | 7.19 | 6.96 | 64(62) | 5.82 (52) |
| 8 | 0.39 | 0.26 | 0.25 | 68 (64) | 0.24 (62) |
| 9 | 17.80 | 8.63 | 8.12 | 49 (46) | 6.67 (38) |
| 10 | 16.12 | 9.10 | 8.63 | 56 (54) | 7.45 (46) |
|
| |||||
| Mean±SD | 11.30±6.75 | 63±9 (59±8) | (50 ± 7) | ||
aPercent of initial 211At activity that was eluted in all of the fractions; values in parenthesis is based on the activity eluted in just fractions 4-10.
bSterile filtered fractions 4-10 combined.
The original method that we developed for the synthesis of [211At]MABG13 involved a HPLC purification step and thus may not be ideal for high radioactivity level synthesis. The HPLC run and the subsequent removal of volatile solvents from the HPLC fractions take about 30 min. The total time for the synthesis by the kit methods is about 120 min and 70 min for C18 and cation exchange cartridges, respectively; in comparison, the duration was about 150 min for the HPLC method. Because of the 7.2-h half life of 211At, an additional advantage of the cation exchange procedure compared with HPLC procedure is that it will increase the amount of [211At]MABG available for use by about 10%. More importantly, this method provides a convenient way to produce relatively high levels of [211At]MABG that can be readily adapted for use in a hospital radiopharmacy.
3. Conclusion
In summary, we have developed a method for the synthesis of relatively high amounts of [211At]MABG in good radiochemical yields from a tin precursor that was anchored to a solid support. This method is adaptable to a kit formulation and the quality control characteristics of the final dose are consistent with those appropriate for clinical studies.
4. Experimental
4.1. General
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. A tin precursor anchored to a polystyrene matrix (1; Figure 1) was synthesized as reported before.29 High-pressure liquid chromatography was performed using a Beckman System Gold HPLC equipped with a Model 126 programmable solvent module, a Model 166 NM variable wavelength detector (a wavelength of 254 nm was used for all runs), a Model 170 radioisotope detector, and a Beckman System Gold remote interface module SS420X; data was acquired using 32 Karat software. For reversed-phase chromatography, a Waters’ XTerra C18 column (4.6 · 250 mm, 5μ) was used. The column was eluted in isocratic mode at 1 ml/min with one of the two mobile phases: 1) 10% THF in 0.2M ammonium dihydrogen phosphate, pH 7.0 or 2) 0.1% TFA in 80:20 water:acetonitrile.
4.2. Astatine-211 Production
Caution! Astatine-211 is an α-emitter of high LET. It should be handled in a well-ventilated fume hood; protective masks should be worn. Ingestion or inhalation of 211 At can cause severe radiotoxicity. The 211At activity was produced on the Duke University Medical Center CS-30 cyclotron via the 209Bi(α, 2n)211At reaction by bombarding natural bismuth metal targets with 28 MeV α-particles using an MIT-1 internal target system (Cyclotron Inc., Napa, CA).33 The radioactivity was condensed into a PEEK (polyetheretherketone) tube34 immersed in a dry ice/ethanol bath and subsequently extracted from the tube into methanol.
4.3. Astatine-211 Quantification
For measurement of 211At activity using the gamma counter, the energy window was set to encompass the 77-92 keV Po K X-rays emitted by its electron capture decay branch. Both the gamma counter and dose calibrator had been cross calibrated for 211At with a germanium semiconductor detector. For the dose calibrator, the 133Xe setting was used and a correction factor of 2.3 was applied.
4.4. [ 211At]MABG Synthesis
A) Temporal effect on the radiochemical yield
To 5 mg of resin 1 (~1 mmol/g) in a Reacti® vial was added 50 μl of a methanolic solution of 211At (200-300 μCi) followed by 10 μl of a 17:10 (v/v) mixture of hydrogen peroxide (30% w/v):HOAc. The suspension was stirred gently at room temperature for various periods of time. The resin from the reaction mixture was removed by filtration through a syringe filter and washed with three 25 μl portions of methanol. Radiochemical yield was determined injecting an aliquot of the filtrate onto reversed-phase HPLC (tR = 23-24 min and ~18 min using HPLC conditions 1 and 2, respectively).
B) High level synthesis—i) C18 cartridge
To 10-12 mg of the resin was added 211At (up to 23 mCi) in 100 μl of methanol followed by 20 μl of hydrogen peroxide-acetic acid mixture. The reaction mixture was stirred gently for 10 min at room temperature and diluted with 10 ml of water. This mixture was passed through an activated C18 solid-phase cartridge (Waters). The cartridge was sequentially washed with 2 H 5 ml water and 0.25 ml 5 mM hexane sulfonic acid, pH 2.5. The [211At]MABG was eluted from the cartridge with 0.25 ml portions of methanol; most of the radioactivity eluting in fractions 2-6. The methanol was evaporated from pooled fractions containing [211At]MABG and the radioactivity was reconstituted in saline and sterile filtered. During the evaporation of methanol, a C-18 solid-phase cartridge was attached to the vial to trap the volatile radioactivity. In some cases, the radioactivity trapped in the cartridge was eluted with methanol to determine the amount of intact [211At]MABG in the volatilized fraction by TLC.
ii) Cation exchange resin cartridge
Astatine-211 (2-18 mCi in 100 – 150 μl MeOH) was added to 10mg of resin in a Reacti® vial. The vial was swirled to ensure uniform coating of radioactivity and 20 μl of a 10:17 (v/v) mixture of glacial acetic acid and hydrogen peroxide (30% - w/v) was added to the vial. A triangular stir bar was placed in the vial and the contents were stirred at room temperature for 10 min. The entire mixture was transferred to a 14-ml Falcon tube with the aid of 10 ml of sterile water for injection (SWFI). A cation exchange cartridge (BondElut – CBA 3ml, 500mg; Varian Inc., Palo Alto, CA) attached with a 3-way stopcock was activated by passing 2.5 ml each of anhydrous MeOH and SWFI . The diluted reaction mixture was added in portions to the activated BondElut cartridge and the effluents were withdrawn using a 10 cc syringe. The Falcon tube was rinsed with an additional 20 ml of SWFI and passed through the cartridge as above. [211At]MABG was eluted from the cartridge with 12 H 1ml of 0.2M phosphoric acid. Generally, the majority of the radioactivity elutes in fractions 4 – 10. The pooled fractions were adjusted to pH 7.0 with the addition of 5N NaOH and then filtered through a 0.2 :m sterile filter (preconditioned by passing 0.25 ml of sterile-filtered 1% HSA in PBS, pH 7.14) into a sterile vial.
C. Quality Control
Purity of the final preparation was determined using both HPLC and TLC. For TLC, silica plates were eluted with 5% HOAc in EtOH, and under these conditions MIBG (and [211At]MABG) eluted with an Rf of 0.4. After developing, the TLC plate was cut into small strips, which were then counted using a gamma counter. The CPM values were plotted against the corresponding strip (fraction) number. Sterility and apyrogenicity testing of a few batches was performed. Sterility was determined using the standard USP sterility test and apyrogenicity by a Limulus amoebocyte lysate (LAL) assay for detection of bacterial endotoxins. The tin content of a few batches of the final [211At]MABG product was determined by inductively coupled plasma mass spectroscopy (ICP-MS; Galbraith Laboratories, Knoxville, TN).
Acknowledgments
This work was supported by Grants CA42324 and CA 93371 from the National Institutes of Health, a Grant DE-FG05-05ER63963 from the Department of Energy, and a Grant from the Pediatric Brain Tumor Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Jaques S, Jr, Tobes MC, Sisson JC. Cancer Res. 1987;47:3920. [PubMed] [Google Scholar]
- 2.Smets LA, Loesberg C, Janssen M, Metwally EA, Huiskamp R. Cancer Res. 1989;49:2941. [PubMed] [Google Scholar]
- 3.Bomanji JB, Wong W, Gaze MN, Cassoni A, Waddington W, Solano J, Ell PJ. Clin Oncol (R Coll Radiol) 2003;15:193. doi: 10.1016/s0936-6555(02)00273-x. [DOI] [PubMed] [Google Scholar]
- 4.Kaltsas G, Rockall A, Papadogias D, Reznek R, Grossman AB. Eur J Endocrinol. 2004;151:15. doi: 10.1530/eje.0.1510015. [DOI] [PubMed] [Google Scholar]
- 5.Matthay KK, DeSantes K, Hasegawa B, Huberty J, Hattner RS, Ablin A, Reynolds CP, Seeger RC, Weinberg VK, Price D. J Clin Oncol. 1998;16:229. doi: 10.1200/JCO.1998.16.1.229. [DOI] [PubMed] [Google Scholar]
- 6.Gaze MN, Chang YC, Flux GD, Mairs RJ, Saran FH, Meller ST. Cancer Biother Radiopharm. 2005;20:195. doi: 10.1089/cbr.2005.20.195. [DOI] [PubMed] [Google Scholar]
- 7.Wangberg B, Muth A, Khorram-Manesh A, Jansson S, Nilsson O, Forssell-Aronsson E, Tisell L, Ahlman H. Ann N Y Acad Sci. 2006;1073:512. doi: 10.1196/annals.1353.054. [DOI] [PubMed] [Google Scholar]
- 8.Matthay KK, Tan JC, Villablanca JG, Yanik GA, Veatch J, Franc B, Twomey E, Horn B, Reynolds CP, Groshen S, Seeger RC, Maris JM. J Clin Oncol. 2006;24:500. doi: 10.1200/JCO.2005.03.6400. [DOI] [PubMed] [Google Scholar]
- 9.Rose B, Matthay KK, Price D, Huberty J, Klencke B, Norton JA, Fitzgerald P. A Cancer. 2003;98:239. doi: 10.1002/cncr.11518. [DOI] [PubMed] [Google Scholar]
- 10.Loh KC, Fitzgerald PA, Matthay KK, Yeo PP, Price DC. J Endocrinol Invest. 1997;20:648. doi: 10.1007/BF03348026. [DOI] [PubMed] [Google Scholar]
- 11.Loc'h C, Mardon K, Valette H, Brutesco C, Merlet P, Syrota A, Maziere B. Nucl Med Biol. 1994;21:49. doi: 10.1016/0969-8051(94)90128-7. [DOI] [PubMed] [Google Scholar]
- 12.Vaidyanathan G, Affleck DJ, Zalutsky MR. J Med Chem. 1994;37:3655. doi: 10.1021/jm00047a022. [DOI] [PubMed] [Google Scholar]
- 13.Vaidyanathan G, Zalutsky MR. Bioconjugate Chem. 1992;3:499. doi: 10.1021/bc00018a006. [DOI] [PubMed] [Google Scholar]
- 14.Humm JLJ. Nucl Med. 1986;27:1490. [PubMed] [Google Scholar]
- 15.Hall EJ. Radiobiology for the Radiobiologist. 4. J.B. Lippincott; Philadelphia: 2000. [Google Scholar]
- 16.Zalutsky MR, Vaidyanathan G. Curr Pharm Des. 2000;6:1433. doi: 10.2174/1381612003399275. [DOI] [PubMed] [Google Scholar]
- 17.Boyd M, Mairs RJ, Keith WN, Ross SC, Welsh P, Akabani G, Owens J, Vaidyanathan G, Carruthers R, Dorrens J, Zalutsky MRJ. Gene Med. 2004;6:937. doi: 10.1002/jgm.578. [DOI] [PubMed] [Google Scholar]
- 18.Boyd M, Ross SC, Dorrens J, Fullerton NE, Tan KW, Zalutsky MR, Mairs RJ. J Nucl Med. 2006;47:1007. [PubMed] [Google Scholar]
- 19.Cunningham SH, Mairs RJ, Wheldon TE, Welsh PC, Vaidyanathan G, Zalutsky MR. Br J Cancer. 1998;77:2061. doi: 10.1038/bjc.1998.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fullerton NE, Boyd M, Ross SC, Pimlott SL, Babich J, Kirk D, Zalutsky MR, Mairs RJ. Med Chem. 2005;1:611. doi: 10.2174/157340605774598090. [DOI] [PubMed] [Google Scholar]
- 21.McCarthy HO, Worthington J, Barrett E, Cosimo E, Boyd M, Mairs RJ, Ward C, McKeown SR, Hirst DG, Robson T. Gene Ther. 2007;14:246. doi: 10.1038/sj.gt.3302871. [DOI] [PubMed] [Google Scholar]
- 22.Strickland DK, Vaidyanathan G, Friedman HS, Zalutsky MR. J Neurooncol. 1995;25:9. doi: 10.1007/BF01054718. [DOI] [PubMed] [Google Scholar]
- 23.Strickland DK, Vaidyanathan G, Zalutsky MR. Cancer Res. 1994;54:5414. [PubMed] [Google Scholar]
- 24.Vaidyanathan G, Friedman HS, Keir ST, Zalutsky MR. Nucl Med Biol. 1996;23:851. doi: 10.1016/0969-8051(96)00115-1. [DOI] [PubMed] [Google Scholar]
- 25.Vaidyanathan G, Strickland DK, Zalutsky MR. Int J Cancer. 1994;57:908. doi: 10.1002/ijc.2910570622. [DOI] [PubMed] [Google Scholar]
- 26.Vaidyanathan G, Zhao XG, Larsen RH, Zalutsky MR. Br J Cancer. 1997;76:226. doi: 10.1038/bjc.1997.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blaney P, Grigg R, Sridharan V. Chem Rev. 2002;102:2607. doi: 10.1021/cr0103827. [DOI] [PubMed] [Google Scholar]
- 28.Merrifield RB. JAm Chem Soc. 1963;85:2149. [Google Scholar]
- 29.Hunter GH, Zhu X. J Labelled Cpd Radiopharm. 1999;42:653. [Google Scholar]
- 30.Pozzi OR, Zalutsky MR. J Nucl Med. 2005;46:1393. [PubMed] [Google Scholar]
- 31.Pozzi OR, Zalutsky MR. J Nucl Med. 2005;46:700. [PubMed] [Google Scholar]
- 32.Zea-Ponce Y, Baldwin RM, Zoghbi SS, Innis RB. Appl Radiat Isot. 1994;45:63. doi: 10.1016/0969-8043(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 33.Zalutsky MR, Zhao XG, Alston KL, Bigner D. J Nucl Med. 2001;42:1508. [PubMed] [Google Scholar]
- 34.Lindegren S, Back T, Jensen HJ. Appl Radiat Isot. 2001;55:157. doi: 10.1016/s0969-8043(01)00044-6. [DOI] [PubMed] [Google Scholar]