

Abstract
Whether environmental toxicants impact an individual woman’s risk for developing endometriosis remains uncertain. Although the growth of endometrial glands and stroma at extra-uterine sites is associated with retrograde menstruation, our studies suggest that reduced responsiveness to progesterone may increase the invasive capacity of endometrial tissue in women with endometriosis. Interestingly, our recent studies using isolated human endometrial cells in short-term culture suggest that experimental exposure to the environmental contaminant 2,3,7,8-tetracholorodibenzo-p-dioxin (TCDD) can alter the expression of progesterone receptor isotypes. Compared to adult exposure, toxicant exposure during development can exert a significantly greater biological impact, potentially affecting the incidence of endometriosis in adults. To address this possibility, we exposed mice to TCDD at critical developmental time points and subsequently examined uterine progesterone receptor expression and steroid responsive transforming growth factor-β2 expression in adult animals. We find that the uterine phenotype of toxicant-exposed mice is markedly similarly to the endometrial phenotype of women with endometriosis.
Keywords: Dioxin, TCDD, Progesterone, Progesterone receptor, TGF-β2, Endometrium, Endometriosis, Development, Fetal origin
1.Introduction
Polychlorinated dibenzo-p-dioxins, generally called dioxins, are a family of chlorinated aromatic hydrocarbons that accumulate as ubiquitous contaminants in our environment. In human populations, ingestion of contaminated food is the primary source of dioxin exposure [1-3]. These chemicals are resistant to degradation and, due to their lipophilic nature, bioaccumulate and biomagnify at higher levels within the food chain [4]. Among the numerous dioxin-like environmental contaminants, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is often considered to be the prototypical disruptor of steroid action within endocrine sensitive tissues, affecting steroid receptor levels as well as steroid metabolism and serum transport [5-7]. In addition to dramatically affecting the endocrine system, TCDD exposure also impacts the expression of multiple cellular signaling systems that regulate key elements of the immune system. For example, TCDD exposure can activate local inflammation through increased expression of tumor necrosis factor-α, interleukin-1β (IL-1β) and IL-6 [8,9]. Within either the endocrine or immune system, exposure to TCDD affects individual cell behavior by initially binding to the aryl hydrocarbon receptor (AhR; [10]) which rapidly forms a heterodimeric complex with ARNT (AhR nuclear translocator). The TCDD/AhR/ARNT complex can associate with dioxin response elements within the promoter of certain genes, subsequently exerting a wide variety of effects. The biological activity of various families of chemical toxicants on human health is diverse; however, toxicant exposure is generally classified into two models. The organizational model refers to chemical exposure of the embryo/fetus at very early stages of development, leading to both structural and functional changes in tissue development, whereas the activational model denotes activation of specific genes in adult tissue following acute or chronic exposure to environmental toxicants (reviewed by [11]). Although environmental toxicants have traditionally been classified separately as either endocrine-disruptors or immune-disruptors, a significant degree of cross-talk occurs between these two systems and chronic inflammation has recently been associated with the development of numerous human diseases affecting multiple organ systems [12-14].
Not surprisingly, animal and human research has suggested that exposure to environmental agents may adversely affect the development and function of the reproductive system (reviewed by [15]). Notably, human exposure to TCDD has been suggested to negatively impact endometrial function during pregnancy, increasing the risk of spontaneous abortion [16]. More recently, evidence has begun to accumulate which suggests that TCDD exposure promotes the establishment of endometriosis [17,18], a disease resulting from the ectopic invasion of endometrial tissue, usually within the peritoneal cavity. Endometriosis is a disease of menstruating species; only humans and other primates exhibit naturally occurring endometriosis. Establishment of ectopic sites of endometrial growth can develop following retrograde menstruation due to the capacity of endometrial fragments to undergo a cancer-like invasive process. Although well-vascularized sites of ectopic endometrial growth can be extremely difficult to eliminate therapeutically using either surgical or medical approaches, endometriosis is estrogen dependent and rarely persists post-menopausally in the absence of ovarian steroid production.
In opposition to the role of estrogen as a risk factor for endometriosis, progesterone may play a critical role in protecting a woman from the development and progression of this disease. For example, progesterone exposure during pregnancy, or exogenous therapy with various progestins, has been associated with disease regression in some women [19]. Nevertheless, reduced progesterone responsiveness has been noted in the eutopic endometrium of women with endometriosis, correlating with an altered expression of progesterone receptor (PR) isotypes [20]. In the endometrium of endometriosis patients, reduced levels of PR-B expression relative to PR-A likely explains the altered expression of progesterone-responsive genes that we and others have recently described during endometrial differentiation [20,21]. Additionally, diminished endometrial PR-B expression may be related to an increased sensitivity of stromal cells to locally produced proinflammatory cytokines [22,23] since this PR isoform has been shown to completely disappear within the inflammatory-like microenvironment that exists at ectopic sites of endometrial tissue growth [24]. As noted above, the eutopic endometrium of women with endometriosis exhibits alterations in the expression of a number of progesterone-regulated genes and proteins, including growth factors, cytokines and retinoid signaling proteins that are known to be critical for matrix metalloproteinase (MMP) regulation [25]. For example, the ability of progesterone or all-trans retinoic acid to down-regulate MMP expression in human endometrial tissue fragments, thereby reducing the invasive establishment of ectopic growth in nude mice, requires transforming growth factor-β (TGF-β) signaling [26-28]. Thus, due to reduced responsiveness to progesterone, blocking the establishment of experimental disease by endometrial tissues acquired from the endometrium of women with endometriosis requires TGF-β treatments in addition to progesterone and all-trans retinoic acid [27,29]. These results strongly suggest that reduced endometrial expression of progesterone-responsive genes and proteins, due to decreased expression of PR-B, may be an important contributing factor to an individual’s overall risk for developing endometriosis. Although the cellular mechanism(s) associated with the development of reduced endometrial responsiveness to progesterone among endometriosis patients is not completely understood, our experimental data suggests that acute exposure of human tissue or cells to TCDD can dramatically reduce both PR-B expression and progesterone-mediated TGF-β2 expression, contributing to a more invasive endometrial phenotype in our experimental endometriosis model [20,27-29].
A possible role of environmental contaminants with dioxinlike activity in the development of endometriosis emerged with the demonstration that the incidence and severity of spontaneous endometriosis in rhesus monkeys was increased following dietary exposure to TCDD [30]. Although two recent epidemiologic studies demonstrated an increased level of dioxin-like compounds in the serum of women with endometriosis compared to disease-free women [31,32], other epidemiologic examinations have been less definitive [33,34]. Since human and animal exposures actually begin in utero, when toxicant sensitivity is greatest, developing a clearer understanding of the potential effects of TCDD and other environmental contaminants will likely require examining the impact of early, developmental exposure. In the current study, we provide further evidence, supporting our previously published work, of reduced endometrial responsiveness to progesterone in women with endometriosis. Additionally, we have examined the adult murine uterus following developmental exposure to TCDD and determined the impact of additional pre-pubertal and pubertal exposures to this toxicant, singly or in combination. We find that the altered expression of progesterone receptors and TGF-β2 that we observed in the endometrium of women with endometriosis occurs in the uterus of adult mice following TCDD exposure during critical periods of reproductive tract development and function.
Materials and methods
2.1. Acquisition of human tissues
Control endometrial tissues were acquired by Pipelle® (Unimar Inc., Wilton, CT) biopsy during the proliferative (days 9-12; n = 8) or secretory phase (days 13-17; n = 7) of the menstrual cycle from a donor population (age 18-45) exhibiting normal menstrual cycles and no history of endometriosis. Endometrial tissue from women with surgically confirmed endometriosis was also obtained by biopsy during the proliferative (n = 4) and secretory (n =4) phases. Serum progesterone levels were assessed in order to confirm the cycle stage (proliferative ≤ 1.5 ng/mL; secretory > 1.6). Individuals with a recent (≤3 months) history of hormone therapy (i.e., oral contraceptives) were excluded. Biopsies were washed in prewarmed, phenol-red free Dulbecco’s modified eagles medium/Ham’s F-12 Medium (DME/F-12) (Sigma) to remove residual blood and mucous prior to culturing. Informed consent was obtained prior to biopsy and the use of human tissues was approved by Vanderbilt University’s Institutional Review Board and Committee for the Protection of Human Subjects. Additional archived samples of formalin-fixed, paraffin-embedded endometrial tissues from women with surgically confirmed endometriosis were obtained from Vanderbilt University Medical Center’s Histopathology Tissue Core.
2.2. Organ cultures of human tissues
For organ cultures, endometrial biopsies were dissected into small cubes (∼1mm × 1mm × 1 mm) and 8-10 pieces of tissue per treatment group were suspended in tissue culture inserts (Millipore; Bedford, MA) as previously described [26]. Prior to establishing experimental conditions, proliferative phase organ cultures were maintained 24 h under serum-free conditions in DME/F-12 supplemented with 1% Insulin-Transferrin-Selenium (ITS+; Collaborative Biomedical; Bedford, MA), 0.1% Excyte (Miles Scientific, Kankakee, IL) and 17β-estradiol (E, 1 nM). Experimental conditions were established by providing fresh media containing E (1 nM) or E plus progesterone (EP, 1 and 500 nM, respectively). Experimental conditions were maintained for 48 h; the total culture time for all samples was 72 h. All cultures were incubated at 37 °C in a humidified chamber with 5% CO2.
2.3. Animals
Virgin female and timed-pregnant C57BL/6 mice were purchased from Harlan Sprague-Dawley (Indianapolis, IN). Animals were housed in the Animal Care Facility according to the National Institutes of Health institutional guidelines for laboratory animals. All animals received low phytoestrogen rodent chow (Picolab 5VO2, Purina) and water ad libitum. Animal rooms were maintained at a temperature of 22-24 °C and a relative humidity of 40-50% on a 12-h light:dark schedule. Experiments described herein were approved by Vanderbilt University Institutional Animal Care and Use Committee in accordance with the Animal Welfare Act.
2.4. Chemicals
TCDD in nonane solution was obtained from Cambridge Isotope Laboratories (Andover, MA). 17β-Estradiol, progesterone, corn oil, and all other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO).
2.5. Animal TCDD exposure
Female mice (5-6 in each group) received no treatment (unexposed) or were exposed to TCDD or vehicle corn oil at different developmental stages of life (Table 1). Singly exposed animals received TCDD (10 μg/kg) by gavage as follows: (1) pregnant mice on gestation day (GD) 15 (when organogenesis is completed) resulting in utero plus lactational exposure (i.e., perinatal exposure). Note: vaginal plug = day 0 of gestation, (2) pre-pubertal virgin females at 4 weeks of age or (3) pubertal virgin females at the age of 9 weeks [35]. Some mice were exposed to TCDD (10 μg/kg) at two subsequent developmental stages: (1) first exposure to TCDD through pregnant dam on GD 15 followed by a second exposure at 4 weeks (pre-pubertal) or (2) first exposure of TCDD to virgin females at 4 weeks (pre-pubertal) followed by the second exposure at 9 weeks (pubertal). Finally, some animals were exposed to TCDD (10 μg/kg) at all three developmental stages (in utero, pre-pubertal and pubertal). The dose of TCDD used in these studies reflects the more rapid clearance of this toxicant in mice compared to humans and is well below the LD50 for adult mice of this strain (230 μg/kg) [36]. To allow for uniform lactational TCDD exposure within the treatment groups, litter sizes were standardized on postnatal day (PND) 1 (PND 0 = day of birth) to eight pups each by removing and/or adding offspring from another litter within the same treatment group [37]. Dams were euthanized when offspring were weaned on PND 21. Note: data from all endpoints demonstrated no difference between unexposed (no treatment) and corn oil exposed mice, therefore, only the results of the corn oil exposed control mice are shown.
Table 1.
The treatment groups of mice
| Treatment |
||||||||
|---|---|---|---|---|---|---|---|---|
| 1 (No treatment) | 2 (Corn oil) | 3 (TCDD) | 4 (TCDD) | 5 (TCDD) | 6 (TCDD) | 7 (TCDD) | 8 (TCDD) | |
| In utero (GD15)a | x | X | X | X | ||||
| Pre-pubertal (4 weeks) | x | X | X | X | X | |||
| Pubertal (9 weeks) | x | X | X | X | ||||
X = 10 μg/kg TCDD in corn oil by gavage at indicated time. x: equal volume of corn oil.
Pregnant dam only.
2.6. Ovariectomy and ovarian steroid hormone treatments
As described by Brasted et al. [38] in order to mimic many of the events of the adult endometrial cycle in the murine uterus, all female offspring were ovariectomized at 12 weeks of age (young adult) under isoflourane anesthesia and a slow-release estradiol capsule (8 mg) inserted subcutaneously along the dorsal surface of each mouse. Six days later, 50% of the mice in each group additionally received slow-release progesterone capsules (15 mg). Preparation of the silastic implants has been previously described [29]. Four days after placement of the second capsule, all the mice were euthanized by cervical dislocation under anesthesia and uterine and liver tissues were collected. Tissues were flash frozen and stored at -80 °C for subsequent analysis. A portion of each uterus was formalin-fixed, paraffin-embedded for immunohistochemical analysis. The sequence of experimental steps is shown diagrammatically in Fig. 1.
Fig. 1.
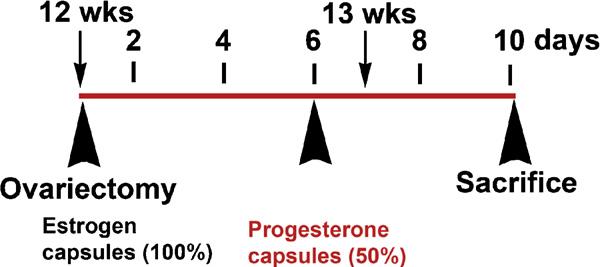
Schematic of exogenous steroid treatment of mice. Regardless of exposure status, all female offspring of pregnant dams were subjected to ovariectomy at 12 weeks of age. Mice were immediately implanted with a slow-release estradiol capsule (8 μg estradiol and 17 μg cholesterol). Six days after placement of the estradiol capsule, half of all mice were additionally implanted with a slow-release silastic capsule containing progesterone (25 μg). All mice were sacrificed 4 days after placement of the second capsule.
2.7. Evaluation of hydronephrotic response
TCDD is a specific and potent inducer of hydronephrosis in both fetal and neonatal mice, with males being more severely affected [39]. Therefore, as an indicator of TCDD exposure, a representative sample of perinatal toxicant exposed and vehicle exposed male pups were sacrificed on postnatal day (PND) 26 and the kidneys examined for hydronephrosis by assessing gross morphology and wet weight.
2.8. Preparation of whole tissue extracts and Western blotting
As described previously [20], proteins were extracted from frozen tissue samples by homogenization in buffer containing 50 mM Tris (pH 7.4), 1 mM EDTA, 150 mM NaCl, and proteinase inhibitors (1 μg/ml phenylmethylsulfonylfluoride, 10 μg/ml aprotinin, and 1 μg/ml leupeptin). The homogenates were centrifuged at 2000 × g for 15 min at 4 °C. The supernatants collected and protein concentration determined using the BCA protein assay reagent kit (Pierce, Rockford, IL). Proteins (20 μg) were electrophoresed by 10% SDS-PAGE under reducing conditions and transferred onto PVDF membranes. Nonspecific binding was blocked by incubation with phosphate-buffered saline containing 0.5% Tween-20 and 5% nonfat dry milk for 1 h at room temperature. For uterine samples (human and mouse), the membranes were incubated with 200 ng/ml of primary antibodies diluted in blocking buffer for PR (catalog No. RB-1492-P1, Neomarker). The PR antibody recognizes both PR-A (approximately 83 kDa) and PR-B (approximately 115 kDa) isoforms by Western blot. Membranes were stripped as previously described [20] and probed for TGF-β2 (catalog No sc-90, Santa Cruz Biotechnology). As a loading control, blots were also probed for the housekeeping protein, β-actin (catalog No sc-1616, Santa Cruz Biotechnology) overnight at 4 °C. Liver samples were similarly incubated with Cyp1A1 (catalog no sc-9828, Santa Cruz Biotechnology) followed by stripping and probing with β-actin. Following incubation with the primary antibody, membranes were washed (PBS/Tween) and incubated in horseradish peroxidase conjugated secondary antibodies (rabbit anti-goat IgG (Santa Cruz Biotechnology) for Cyp1A1 and β-actin (40 ng/ml) and goat anti-rabbit IgG (Santa Cruz Biotechnology) for PR and TGF-β2 (20 ng/ml)) in blocking buffer for 1 h followed by ECL detection (Amersham Biosciences Inc., Piscataway, NJ).
2.9. Immunohistochemistry
Universal DAKO LSAB® + Kit (labeled streptavidin-biotin), HRP (Dak-Coytomation California Inc., Carpinteria, CA), were used for immunohistochemical staining according to the manufacturer’s instruction using the same PR and TGF-β2 antibodies used above. In brief, formalin-fixed paraffin-embedded uterine sections (6 μM) were deparaffinized, hydrated, and microwave irradiated for 10 min in 10 mM citrate buffer solution (pH 6.0) for antigen retrieval. Endogenous peroxidase activity was quenched by immersing in 3% H2O2 for 12 min followed by blocking of nonspecific reaction by incubation in 1% bovine serum albumin in phosphate-buffered saline containing 0.5% Tween-20 for 20 min. Sections were then incubated with primary antibody (2 μg/ml) diluted with blocking solution for 1 h at room temperature followed by incubation with biotinylated MultiLink antibodies (anti-goat, anti-mouse and anti-rabbit immunoglobulins) for 30 min. After rinsing with phosphate-buffered saline containing 0.5% Tween-20, the sections were reacted with labeled streptavidin horseradish peroxidase complex. The reaction was visualized using 3-3 diaminobenzidene (DAB) chromagen solution resulting in brown precipitate. Slides using anti-TGF-β2 antibody were counterstained with Gill’s hematoxylin (Fisher Scientific, Pittsburgh, PA) and slides incubated with anti-PR antibody were counterstained with eosin (Volu-sol Inc. Salt Lake City, Utah; human) or hematoxylin (mouse). The negative control specimens were incubated only with secondary antibody to verify the specificity of immunostaining (data not shown).
2.10. Staining assessment
For calculation of H-score of slides subjected to immunohistochemistry, 10 fields (human samples) or 5 fields (mouse samples) were selected at random and photographed at 400× magnification. Staining intensity within endometrial glands and stroma was independently assessed by two investigators and scored as 0, 1, 2 or 3 corresponding to the presence of negative, weak, intermediate or intense staining, respectively. The average of percent positive cells at each level of intensity was determined, and the H-score calculated based on the formula: H-score=(% of cells stained at level 1 intensity×1) + (% of cells stained at level 2 intensity × 2) + (% of cells stained at level 3 intensity×3). An H-score of between 0 and 300 was obtained where a score of 300 is equivalent to 100% of cells maximally stained.
3. Results
3.1. PR expression in human endometrium from women with and without endometriosis
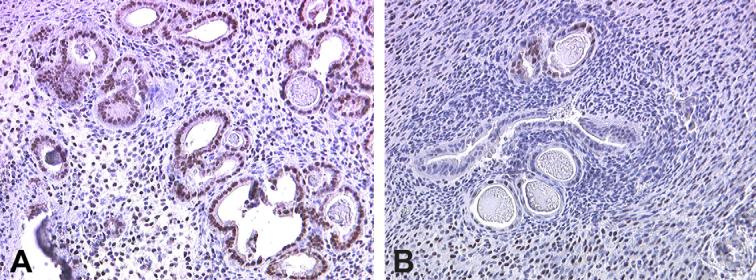
Endometrial progesterone resistance has been described in women with endometriosis resulting in an altered ability of this steroid to regulate a number of genes and proteins during the progesterone-dominated secretory phase (reviewed by [22]). Endometrial PR expression is induced by estradiol and is highly expressed in the late proliferative and early secretory phase of the normal menstrual cycle [40]. As shown in Fig.2 and Table 2, imunohistochemical localization of PR in samples of proliferative phase endometrium from women with and without endometriosis reveals abundant PR expression in control samples, whereas a reduced level of expression of PR is apparent in samples obtained from women with endometriosis. These results are consistent with our published data demonstrating that whole tissue extracts from late proliferative phase samples obtained from women with endometriosis exhibits a significant reduction in PR-B expression compared to late proliferative samples from disease-free women [20]. Using Western analysis of whole tissue protein extracts, we now demonstrate that both PR-A and PR-B are normally highly expressed in the endometrium during the early secretory phase, but PR-B expression is specifically reduced in samples obtained from women with endometriosis (Fig. 3).
Fig. 2.
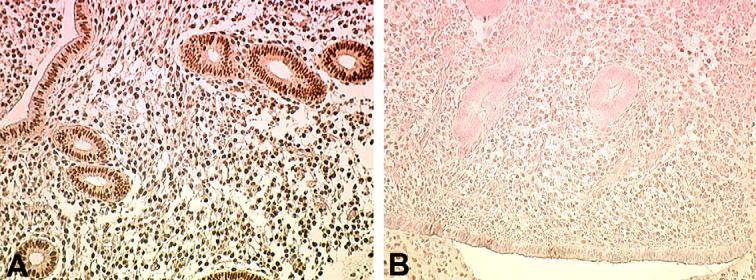
Immunohistochemical localization of PR in human proliferative phase endometrium from women with and without endometriosis. Proliferative phase endometrium from a woman with no history of endometriosis (A) demonstrates abundant expression of PR in both endometrial glands and stroma. Proliferative phase endometrium from a woman with endometriosis reveals limited expression of PR in either cell type (B). A minimum of three specimens from each patient group was examined; typical results are shown. Original magnification is 200×.
Table 2.
H-scores of staining intensity from samples analyzed by immunohistochemistry
| Sample type | Antibody | Cell type | Mean (median) | Range |
|---|---|---|---|---|
| Human endometrium (Control tissue donors) | α-PR | Epithelial | 287 (240) | 90-300 |
| Stromal | 240 (210) | 60-300 | ||
| α-TGF-β2 | Epithelial | 212 (200) | 120-300 | |
| Stromal | 156 (145) | 80-280 | ||
| Human endometrium (Endometriosis patients) | α-PR | Epithelial | 45 (40) | 0-150 |
| Stromal | 35 (30) | 0-120 | ||
| α-TGF-β2 | Epithelial | 0 | n/a | |
| Stromal | 3a | 0-20 | ||
| Mouse endometrium (Corn oil exposed animals) | α-PR | Epithelial | 190 (175) | 120-275 |
| Stromal | 206 (180) | 110-300 | ||
| α-TGF-β2 | Epithelial | 150 (140) | 90-200 | |
| Stromal | 125 (115) | 70-200 | ||
| Mouse endometrium (TCDD exposed)b | α-PR | Epithelial | 1.75a | 0-25 |
| Stromal | 12 (10) | 0-100 | ||
| α-TGF-β2 | Epithelial | 0.75a | 0-10 | |
| Stromal | 4.5a | 0-25 |
n/a = not applicable.
Majority of samples received a score of 0.
Tissues from maximally exposed animals.
Fig. 3.
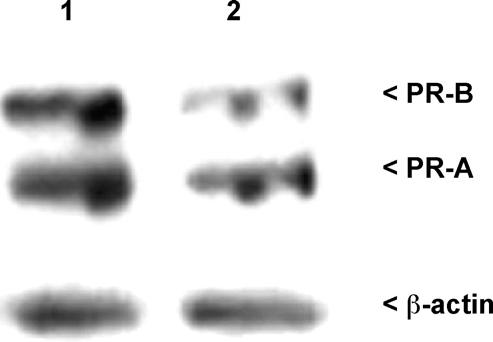
Western analysis of human PR-A and PR-B expression in secretory phase whole tissues from control women (1) and women with endometriosis (2). Total protein was extracted from samples and subjected to Western analysis of PR-A and PR-B expression. Equal loading of sample proteins were confirmed by stripping and reprobing the same blot for β-actin. A minimum of three specimens from each patient group was examined; results from a representative experiment are shown.
3.2. TGF-β2 expression in human endometrium from women with and without endometriosis
This laboratory has previously demonstrated that endometrial TGF-β2 expression is closely linked to progesterone associated differentiation [26]. Taken with the data presented above indicating a decreased endometrial PR-B expression in women with endometriosis, it is likely that expression of TGF-β2 may also be compromised in secretory endometrium from these patients. Indeed, immunohistochemical localization reveals abundant TGF-β2 in stromal cells and glands in samples obtained during the secretory phase of control women, but a greatly reduced expression of this growth factor is noted in secretory endometrium from women with endometriosis (Fig. 4 and Table 2). To further assess whether or not a reduced endometrial response to progesterone plays a role in altered TGF-β2 expression, we established proliferative phase organ cultures using endometrial tissues acquired from women with and without endometriosis. As shown in Fig. 5, progesterone exposure readily induces expression of this growth factor in control tisues; however, induction in TGF-β2 expression is not observed in progesterone-treated tissues from women with endometriosis, consistent with decreased endometrial progesterone responsiveness in these patients.
Fig. 4.
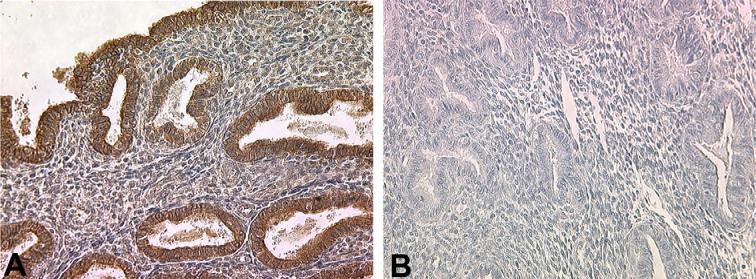
Immunohistochemical localization of TGF-β2 in human secretory phase endometrium from women with and without endometriosis. Secretory phase endometrium from a woman with no history of endometriosis (A) demonstrates expression of TGF-β2 in both endometrial glands and stroma, with localization more prominent within the glands. Secretory phase endometrium from a woman with endometriosis reveals very limited expression of TGF-β2 in either cell type (B). A minimum of three specimens from each patient group was examined; typical results are shown. Original magnification is 200×.
Fig. 5.
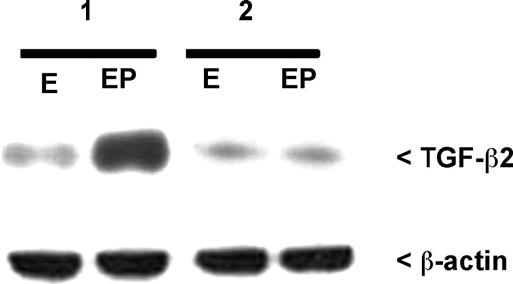
Western analysis of secreted human TGF-β2 in proliferative phase organ cultures from control women (1) and from women with endometriosis (2). Conditioned media were examined for the presence of TGF-β2 following 48 h in culture with estradiol (1 nM) or estradiol plus progesterone (1 nM and 500 nM, respectively). Equal loading of sample proteins was confirmed by stripping and reprobing the same blot for β-actin. A minimum of three specimens from each patient group was examined; results from a representative experiment are shown.
3.3. Assessment of TCDD exposure
In order to determine the possible influence of early TCDD exposure on adult endometrial responsiveness to progesterone, we have treated mice with TCDD at various developmental stages and assessed expression of PR and TGF-β2 in adult uteri. However, prior to examining these endpoints, we assessed expression of CYP1A1 in livers of control and TCDD exposed mice. CYP1A1 is induced by a variety of toxicants, including TCDD. The incidence of hydronephrosis, which is specifically linked to in utero TCDD exposure, was also assessed.
3.3.1. CYP1A1 expression
Induction of cytochrome P4501A1 (CYP1A1) gene is frequently used as a marker of AhR activation upon binding of ligands such as TCDD [41]. Therefore, hepatic expression of CYP1A1 subsequent to TCDD exposure was evaluated in female mice exposed at various timepoints. Prior to sacrifice, adult mice were ovariectomized and treated with estrogen or estrogen plus progesterone as described in methods. As shown in Fig. 6, Western analysis revealed increased expression of CYP1A1 protein in the liver of all TCDD exposed mice except the perinatally exposed group (in utero/lactational exposure to TCDD), which demonstrated faint expression similar to that of the control animals.
Fig. 6.
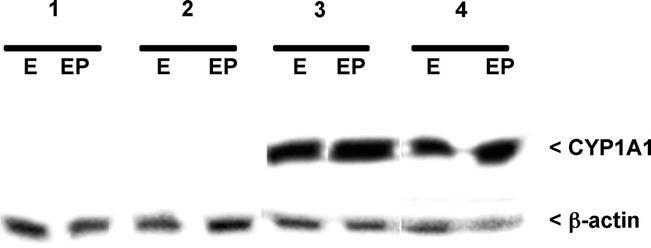
Western analysis of murine CYP1A1 expression in whole liver extracts from adult mice exposed to corn oil or TCDD during development. As described in methods, prior to sacrifice mice were ovariectomized and treated exogenously with estradiol (E) or estradiol plus progesterone (EP). Exposure groups included: (1) corn oil exposure; (2) in utero TCDD; (3) in utero TCDD plus pre-pubertal TCDD and (4) in utero TCDD, pre-pubertal TCDD and pubertal TCDD. Blots were stripped and reprobed with β-actin to assure equal loading. A minimum of five mice per group was examined; typical results are shown.
3.3.2. Hydronephrosis
Hydronephrosis is a characteristic response to TCDD in the developing embryo [42,43] with the greatest manifestation detected in 26-day-old male pups [39]. Thus, we used this parameter to assess the exposure to TCDD in perinatally exposed male mice compared with the corn oil treated male mice. The majority (80%) of male offspring exposed in utero to TCDD exhibited moderate to severe hydronephrosis on PND 26 (Table 3). Consistent with previous reports, the severity of hydronephrosis was greatest in the right kidney (data not shown). None of the corn oil exposed pups exhibited hydronephrosis. A total of 15 mice from 4 different TCDD-exposed dams and 8 mice from 3 corn oil exposed dams were examined; typical results are shown (Table 3).
Table 3.
Wet weight of kidneys from 26-day-old male mice exposed in utero to corn oil (control) or to TCDD
| In utero exposure | Total wet weight (both kidneys) mean (median) (mg) | Range (mg) | Weight of mice mean (median) (g) | Number of mice | Pa |
|---|---|---|---|---|---|
| Corn oil | 119.96 (121.0) | 116.3-131.7 | 9.86 (9.9) | 5 | |
| TCDD | 158.5 (150.2) | 138.1-199.6 | 10.16 (10.1) | 5 | 0.024 |
Compared to corn oil exposed mice. Unpaired t-test due to small sample size.
3.4. Endometrial PR expression in adult mice subsequent to developmental TCDD exposure
After confirming the toxicant exposure of mice, we next examined PR expression in uteri of adult animals following TCDD exposure at different developmental stages. The PR expression in the young adult uteri of the TCDD-exposed and corn oil exposed ovariectomized mice treated with estradiol or estradiol plus progesterone at 13 weeks of age were analyzed by Western blot (Fig. 7) and immunohistochemistry (Fig. 8). As shown in Fig. 7, abundant expression of both PR-B and PR-A was observed in the corn oil-treated mice, regardless of exogenous steroid treatment. Within the TCDD exposed groups, we found a progressive loss of both proteins as the number of exposures increased. PR expression in mice exposed singly at prepuberty or puberty or at both prepuberty and puberty demonstrated a similar expression pattern as the singly and doubly exposed mice in Fig. 7 (data not shown).
Fig. 7.
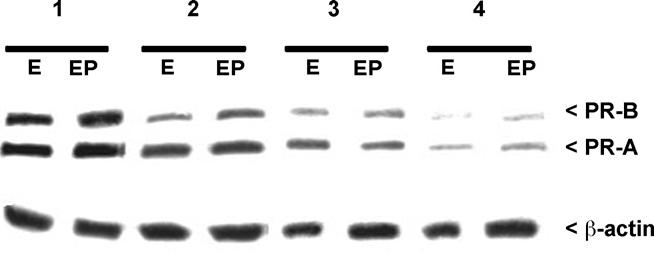
Western analysis of murine PR-A and PR-B expression in whole uterine extracts from adult mice exposed to corn oil or TCDD during development. As described in methods, prior to sacrifice mice were ovariectomized and treated exogenously with estradiol (E) or estradiol plus progesterone (EP). Exposure groups included: (1) corn oil exposure; (2) in utero TCDD; (3) in utero TCDD plus pre-pubertal TCDD and (4) in utero TCDD, pre-pubertal TCDD and pubertal TCDD. Blots were stripped and reprobed with β-actin to assure equal loading. A minimum of five mice per group was examined; typical results are shown.
Fig. 8.
Immunohistochemical localization of PR in uteri of mice exposed to corn oil or TCDD during development. Formalin-fixed, paraffin-embedded uteri from adult mice exposed during development to corn oil (A) demonstrates abundant expression of PR in both endometrial glands and stroma. Fixed tissues from mice exposed to TCDD in utero, pre-pubertally and at puberty demonstrate greatly reduced expression of PR protein. A minimum of five mice per group was examined; typical results are shown. Original magnification is 200×.
Immunohistochemical analysis revealed a similar loss of PR expression following three developmental exposures to TCDD. As shown in Fig. 8 and Table 2, corn oil exposed mice, treated with estrogen, exhibit staining for PR in both glands and stromal cells while mice exposed at all developmental stages (perinatally, prebertally and at puberty) demonstrate a near complete loss of staining. Although PR was detected by immunohistochemistry in singly and doubly exposed mice, expression was decreased compared to control mice (data not shown).
3.5. Endometrial TGF-β2 expression in adult mice subsequent to developmental TCDD exposure
Although the role of progesterone in regulating endometrial TGF-β2 expression in the primate is well-established [26,44,45], expression of this protein in the non-pregnant mouse uterus in response to progesterone has not been described. Nevertheless, successful pregnancy, which is reliant on progesterone in either species, is also known to be dependent upon appropriate expression of decidual TGF-β2 in mice [46-48]. Therefore, in the current study, we further explored whether the reduced PR expression observed in adult mice uteri subsequent to in vivo TCDD exposure impacted TGF-β2 expression in response to either estradiol alone or estradiol plus progesterone. TGF-β2 expression was evaluated in the young adult uterus at the age of 13 weeks from the TCDD-exposed and corn oil-exposed ovariectomized mice following exogenous estrogen or estrogen plus progesterone supplementation. Western analysis of whole tissue protein extracts revealed a high level of expression of TGF-β2 in corn oil exposed mice following treatment with either estrogen or progesterone, but a marked decrease in expression following multiple exposures to TCDD (Fig. 9). Singly and doubly TCDD-exposed animals at other developmental stages also exhibited slightly decreased TGF-β2 expression compared to corn oil exposed mice (data not shown).
Fig. 9.
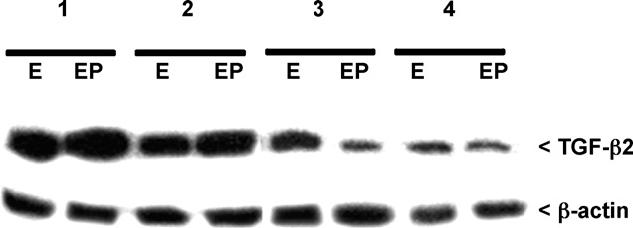
Western analysis of secreted human TGF-β2 expression in whole uterine extracts from adult mice exposed to corn oil or TCDD during development. Proteins were subjected to Western analysis for expression of TGF-β2. As described in methods, prior to sacrifice mice were ovariectomized and treated exogenously with estradiol (E) or estradiol plus progesterone (EP). Exposure groups included: (1) corn oil exposure; (2) in utero TCDD; (3) in utero TCDD plus pre-pubertal TCDD and (4) in utero TCDD, pre-pubertal TCDD and pubertal TCDD. Blots were stripped and reprobed for β-actin. A minimum of five mice per group was examined; typical results are shown.
Immunohistochemical analysis of uteri also revealed abundant expression of TGF-β2 in control animals treated exogenously with progesterone; staining was most prominent within the surface and glandular epithelium (Fig.10A and Table 2). A dramatic decrease in expression of TGF-β2 was observed in the uteri from progesterone treated mice maximally exposed to TCDD, although some stromal cell staining remained (Fig.10B and Table 2).
Fig. 10.
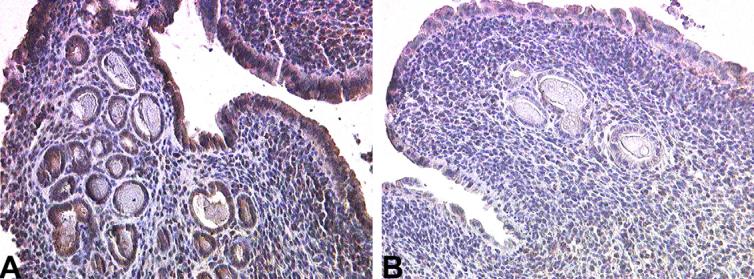
Immunohistochemical localization of TGF-β2 in uteri of mice exposed to corn oil or TCDD during development. Formalin-fixed, paraffin-embedded uteri from adult mice exposed during development to corn oil and exogenously treated with estradiol and progesterone during adulthood (A) demonstrates abundant expression of TGF-β2 in both endometrial glands and stroma. Fixed tissues from estradiol and progesterone treated adult mice previously exposed to TCDD in utero, pre-pubertally and at puberty demonstrate greatly reduced expression of TGF-β2 protein. A minimum of five mice per group was examined; typical results are shown. Original magnification is 200×.
It is important to note that although progesterone has been found to increase expression of TGF-β2 in the human endometrium (i.e., [26]), we found no apparent difference in uterine expression of TGF-β2 in estradiol-treated mice compared to mice receiving both estradiol and progesterone (immunohistochemical data not shown, see Western analysis in Fig. 9). Specifically, this protein was highly expressed in the endometrium of both estradiol treated and estradiol plus progesterone treated mice. Nevertheless, a dramatic reduction of TGF-β2 was observed in TCDD exposed estradiol-treated animals, similar to the reduction observed in estradiol plus progesterone treated mice exposed developmentally to TCDD (Fig. 9).
4. Discussion
There is growing concern that human exposure to man-made chemicals in our environment may impact the pathophysiology of disease. Although human populations are constantly exposed to complex mixtures of environmental toxicants, beginning in utero, the consequences of developmental versus adult toxicant exposure to the risk for adult onset disease remains an issue of much debate. Numerous environmental contaminants structurally related to TCDD are known to disrupt steroid action, not only impacting the actions of steroids during reproductive processes, but also potentially disrupting reproductive tract development. Rier et al. [30] were the first to report a possible association between TCDD exposure and the development of severe, spontaneous endometriosis in a colony of macaque monkeys. Establishment of endometriosis is an invasive event that requires degradation of the extracellular matrix, a process that is associated with the expression and action of numerous members of the MMP family [25,49,50]. Although endometriosis is not normally a life-threatening condition, the severity of disease, including multiple organ invasion, noted in the TCDD-exposed primate colony described by Rier et al. [30] suggested to us that TCDD may disrupt the regulation of the endometrial MMP system. Indeed, we found that following short-term exposure of control human endometrium to TCDD, the capacity of endometrial fragments to invade the peritoneal wall of nude mice in our experimental endometriosis model was greatly enhanced [28]. Since a woman’s exposure to progesterone is recognized as a negative risk factor for the development of endometriosis, it is important to determine whether TCDD may act to disrupt key elements of progesterone action in the reproductive tract.
Traditionally, TCDD has been viewed as a toxic compound with both estrogenic and antiestrogenic activity [51,52]; consequently, the potential that TCDD exposure may disrupt progesterone action has received much less attention. Nevertheless, our recent research suggests that, in the endometrium of women with endometriosis, a loss of responsiveness to progesterone may represent a key element of endometrial dysfunction associated with the invasive pathophysiology of this disease (reviewed by [22]). Progesterone action, mediated by specific progesterone receptor (PR) isotypes, is essential for the function of the female reproductive tract of all mammals. Using a PR null mouse strain, uterine stromal cells are implicated as the sole mediators of the inhibitory effect of progesterone on estrogen-induced proliferation of epithelial cells [53] Thus, mice lacking PR exhibit epithelial cell hyperplasia, related to unopposed exposure to estrogen without the modulating influence of progesterone [54].
In addition to PR expression, our early studies of the human endometrium demonstrated that progesterone-mediated stromal-epithelial communication through TGF-β signaling was also critical for down-regulation of MMP-7 expression by epithelial cells [26,55]. Not unexpectedly, we found that the ability of TCDD exposure to specifically reduce progesteronemediated expression of TGF-β2 not only inhibited the downregulation of MMPs by both stromal and epithelial cells but also promoted the capacity of human endometrial tissue fragments to establish ectopic sites of growth in nude mice [28]. Therefore, TCDD-mediated disruption of steroid-regulated TGF-β2 expression could partially explain the ability of this toxicant to disrupt normal patterns of stromal-epithelial cell communication related to the invasive establishment of endometriosis [20,14,28]. Recently, we have shown that short-term exposure of endometrial stromal and epithelial co-cultures to TCDD can further disrupt progesterone-mediated stromal-epithelial communication by reducing stromal cell PR-B expression [20]. As noted above, stromal cell responsiveness to progesterone has been shown to be key to the action of progesterone on the growth of adjacent epithelial cells [53] as well as the expression of MMPs [26]. Therefore, the ability of TCDD exposure to reduce PR expression in stromal cells and increase MMP expression by both stromal and epithelial cells [22] could explain the aggressive endometriosis phenotype observed in the primate colony by Rier et al. [30].
A careful study of Belgian patients presenting with not only peritoneal endometriotic lesions, but also deep infiltrating lesions found an increase in TCDD-like toxicants in the blood of women with endometriosis compared to disease-free control patients [31]. Although not all studies of adult human populations agree on the potential role of environmental toxicants in the pathophysiology of reproductive tract disease (reviewed by [15]), a recent Italian study also found an increase in both TCDD-like and non-TCDD-like PCBs in patients with this disease [32]. It is unlikely that women and children living in any developed country will be able to avoid significant exposure to environmental toxicants; therefore the potential developmental impact of endocrine-disrupting chemicals likely to be present in the maternal circulation during pregnancy must be better understood. In the current study, we found that female mice exposed to TCDD in utero, and again during the development of reproductive potential demonstrate a progressive loss of both PR-A and PR-B expression. Since in vitro exposure of adult endometrial tissue and cells to TCDD blocks progesterone-induced TGF-β2 expression in humans [28], we were not surprised to find that the expression of this growth factor was also greatly reduced in toxicant-exposed mice. Although mice exposed at any single time point exhibit only moderately reduced expression of PR and TGF-β2, our results suggest a cumulative effect of adult toxicant exposure following an initial exposure at a critical timepoint during in utero development. Importantly, our results demonstrate that the uterine phenotype of toxicant-exposed mice is markedly similarly to the endometrial phenotype we have described in women with endometriosis. Our murine findings support the possibility that exposure of children to TCDD during critical periods (i.e., in utero development, perinatally, or as young adults) could play a cumulative role in disrupting adult endometrial function, potentially predisposing some women to the development of endometriosis. The studies presented herein examined the impact of a relatively high dose (10 μg/kg) of TCDD compared to human background levels. However, the dose of TCDD used in the present study was similar to that reported by other investigators [56-58] and was chosen to insure that we could identify appropriate endpoints in offspring animals. Importantly, humans are exposed to complex mixtures of toxicants which are collectively capable of interacting with the AhR [59]. Nevertheless, it will be important to determine the impact of lower doses of TCDD in future murine studies in order to better determine the potential risks to human populations.
Over the past several years, data has begun to accumulate which suggests that many diseases and conditions of adults may be rooted in the fetal environment [60]. This concept, “the developmental basis of health and disease” is still theory, but requires that we begin to examine the potential role of fetal/neonatal programming on adult susceptibility to disease. Importantly, fetal programming can be impacted by many factors, i.e., maternal stress, nutrition and exposure to toxicants. Although there is strong data to support a link between the in utero environment and obesity, heart disease and cancer [61-63], endometriosis has not been examined in this regard. While our data may be the first to link developmental exposure to TCDD to adult uterine dysfunction, it will be important to continue to follow accidentally exposed human populations in order to determine if children exposed at early developmental timepoints demonstrate an increased sensitivity to additional adult toxicant exposures and whether repeat exposures are necessary for the development of adult onset diseases such as endometriosis.
Acknowledgements
The authors would like to express their grateful appreciation to all their donors of endometrium, without whom this work could not have been achieved. We also express our grateful appreciation to Esther Eisenberg, MD, for performing the endometrial biopsies and to Ms. Santhi Gladson for expert technical assistance. This work was supported by grants from NIH (NIEHS R21ES12298 (KGO), NIH K12HD043483 (KB-T)) and by The Endometriosis Association.
References
- [1].Harrad S, Wang Y, Sandaradura S, Leeds A. Human dietary intake and excretion of dioxin-like compounds. J Environ Monit. 2003;5:224–8. doi: 10.1039/b211406b. [DOI] [PubMed] [Google Scholar]
- [2].Pompa G, Caloni F, Fracchiolla ML. Dioxin and PCB contamination of fish and shellfish: assessment of human exposure. Review of the international situation. Vet Res Commun. 2003;27(Suppl 1):159–67. doi: 10.1023/b:verc.0000014134.23782.10. [DOI] [PubMed] [Google Scholar]
- [3].Schecter A, Wallace D, Pavuk M, Piskac A, Papke O. Dioxins in commercial United States baby food. J Toxicol Environ Health A. 2002;65:1937–43. doi: 10.1080/00984100290071450. [DOI] [PubMed] [Google Scholar]
- [4].Birnbaum LS. The mechanism of dioxin toxicity: relationship to risk assessment. Environ Health Perspect. 1994;102(Suppl 9):157–67. doi: 10.1289/ehp.94102s9157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Heimler I, Rawlins RG, Owen H, Hutz RJ. Dioxin perturbs, in a dose- and time-dependent fashion, steroid secretion, and induces apoptosis of human luteinized granulosa cells. Endocrinology. 1998;139:4373–9. doi: 10.1210/endo.139.10.6264. [DOI] [PubMed] [Google Scholar]
- [6].Moran FM, VandeVoort CA, Overstreet JW, Lasley BL, Conley AJ. Molecular target of endocrine disruption in human luteinizing granulosa cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin: inhibition of estradiol secretion due to decreased 17alpha-hydroxylase/17,20-lyase cytochrome P450 expression. Endocrinology. 2003;144:467–73. doi: 10.1210/en.2002-220813. [DOI] [PubMed] [Google Scholar]
- [7].Pocar P, Fischer B, Klonisch T, Hombach-Klonisch S. Molecular interactions of the aryl hydrocarbon receptor and its biological and toxicological relevance for reproduction. Reproduction. 2005;129:379–89. doi: 10.1530/rep.1.00294. [DOI] [PubMed] [Google Scholar]
- [8].Lai ZW, Hundeiker C, Gleichmann E, Esser C. Cytokine gene expression during ontogeny in murine thymus on activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol Pharmacol. 1997;52:30–7. doi: 10.1124/mol.52.1.30. [DOI] [PubMed] [Google Scholar]
- [9].Lai ZW, Pineau T, Esser C. Identification of dioxin-responsive elements (DREs) in the 5 regions of putative dioxin-inducible genes. Chem Biol Interact. 1996;100:97–112. doi: 10.1016/0009-2797(96)03691-5. [DOI] [PubMed] [Google Scholar]
- [10].Mimura J, Fujii-Kuriyama Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys Acta. 2003;1619:263–8. doi: 10.1016/s0304-4165(02)00485-3. [DOI] [PubMed] [Google Scholar]
- [11].Osteen KG, Sierra-Rivera E. Does disruption of immune and endocrine systems by environmental toxicants contribute to development of endometriosis. Semin Reprod Endocrinol. 1997;15:301–8. doi: 10.1055/s-2008-1068760. [DOI] [PubMed] [Google Scholar]
- [12].Jackson L, Evers BM. Chronic inflammation and pathogenesis of GI and pancreatic cancers. Cancer Treat Res. 2006;130:39–65. doi: 10.1007/0-387-26283-0_2. [DOI] [PubMed] [Google Scholar]
- [13].Kao PC, Shiesh SC, Wu TJ. Serum C-reactive protein as a marker for wellness assessment. Ann Clin Lab Sci. 2006;36:163–9. [PubMed] [Google Scholar]
- [14].Shi YL, Luo XZ, Zhu XY, Hua KQ, Zhu Y, Li DJ. Effects of combined 17beta-estradiol with TCDD on secretion of chemokine IL-8 and expression of its receptor CXCR1 in endometriotic focus-associated cells in co-culture. Hum Reprod. 2006;21:870–9. doi: 10.1093/humrep/dei414. [DOI] [PubMed] [Google Scholar]
- [15].Miller KP, Borgeest C, Greenfeld C, Tomic D, Flaws JA. In utero effects of chemicals on reproductive tissues in females. Toxicol Appl Pharmacol. 2004;198:111–31. doi: 10.1016/j.taap.2003.07.016. [DOI] [PubMed] [Google Scholar]
- [16].Le TN, Johansson A. Impact of chemical warfare with agent orange on women’s reproductive lives in Vietnam: a pilot study. Reprod Health Matter. 2001;9:156–64. doi: 10.1016/s0968-8080(01)90102-8. [DOI] [PubMed] [Google Scholar]
- [17].Birnbaum LS, Cummings AM. Dioxins and endometriosis: a plausible hypothesis. Environ Health Perspect. 2002;110:15–21. doi: 10.1289/ehp.0211015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rier S, Foster WG. Environmental dioxins and endometriosis. Semin Reprod Med. 2003;21:145–54. doi: 10.1055/s-2003-41321. [DOI] [PubMed] [Google Scholar]
- [19].Olive DL, Pritts EA. The treatment of endometriosis: a review of the evidence. Ann NY Acad Sci. 2002;955:360–72. doi: 10.1111/j.1749-6632.2002.tb02797.x. [DOI] [PubMed] [Google Scholar]
- [20].Igarashi TM, Bruner-Tran KL, Yeaman GR, et al. Reduced expression of progesterone receptor-B in the endometrium of women with endometriosis and in cocultures of endometrial cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fertil Steril. 2005;84:67–74. doi: 10.1016/j.fertnstert.2005.01.113. [DOI] [PubMed] [Google Scholar]
- [21].Kao LC, Germeyer A, Tulac S, et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology. 2003;144:2870–81. doi: 10.1210/en.2003-0043. [DOI] [PubMed] [Google Scholar]
- [22].Osteen KG, Bruner-Tran KL, Eisenberg E. Reduced progesterone action during endometrial maturation: a potential risk factor for the development of endometriosis. Fertil Steril. 2005;83:529–37. doi: 10.1016/j.fertnstert.2004.11.026. [DOI] [PubMed] [Google Scholar]
- [23].Tseng JF, Ryan IP, Milam TD, et al. Interleukin-6 secretion in vitro is upregulated in ectopic and eutopic endometrial stromal cells from women with endometriosis. J Clin Endocrinol Metab. 1996;81:1118–22. doi: 10.1210/jcem.81.3.8772585. [DOI] [PubMed] [Google Scholar]
- [24].Attia GR, Zeitoun K, Edwards D, Johns A, Carr BR, Bulun SE. Progesterone receptor isoform A but not B is expressed in endometriosis. J Clin Endocrinol Metab. 2000;85:2897–902. doi: 10.1210/jcem.85.8.6739. [DOI] [PubMed] [Google Scholar]
- [25].Curry TE, Jr, Osteen KG. The matrix metalloproteinase system: changes, regulation, and impact throughout the ovarian and uterine reproductive cycle. Endocr Rev. 2003;24:428–65. doi: 10.1210/er.2002-0005. [DOI] [PubMed] [Google Scholar]
- [26].Bruner KL, Rodgers WH, Gold LI, et al. Transforming growth factor beta mediates the progesterone suppression of an epithelial metalloproteinase by adjacent stroma in the human endometrium. Proc Natl Acad Sci USA. 1995;92:7362–6. doi: 10.1073/pnas.92.16.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bruner-Tran KL, Eisenberg E, Yeaman GR, Anderson TA, McBean J, Osteen KG. Steroid and cytokine regulation of matrix metalloproteinase expression in endometriosis and the establishment of experimental endometriosis in nude mice. J Clin Endocrinol Metab. 2002;87:4782–91. doi: 10.1210/jc.2002-020418. [DOI] [PubMed] [Google Scholar]
- [28].Bruner-Tran KL, Rier SE, Eisenberg E, Osteen KG. The potential role of environmental toxicants in the pathophysiology of endometriosis. Gynecol Obstet Invest. 1999;48(Suppl 1):45–56. doi: 10.1159/000052868. [DOI] [PubMed] [Google Scholar]
- [29].Bruner-Tran KL, Zhang Z, Eisenberg E, Winneker RC, Osteen KG. Downregulation of endometrial matrix metalloproteinase-3 and -7 expression in vitro and therapeutic regression of experimental endometriosis in vivo by a novel nonsteroidal progesterone receptor agonist, tanaproget. J Clin Endocrinol Metab. 2006;91:1554–60. doi: 10.1210/jc.2005-2024. [DOI] [PubMed] [Google Scholar]
- [30].Rier SE, Martin DC, Bowman RE, Dmowski WP, Becker JL. Endometriosis in rhesus monkeys (Macaca mulatta) following chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam Appl Toxicol. 1993;21:433–41. doi: 10.1006/faat.1993.1119. [DOI] [PubMed] [Google Scholar]
- [31].Heilier JF, Nackers F, Verougstraete V, Tonglet R, Lison D, Donnez J. Increased dioxin-like compounds in the serum of women with peritoneal endometriosis and deep endometriotic (adenomyotic) nodules. Fertil Steril. 2005;84:305–12. doi: 10.1016/j.fertnstert.2005.04.001. [DOI] [PubMed] [Google Scholar]
- [32].Porpora MG, Ingelido AM, di Domenico A, et al. Increased levels of polychlorobiphenyls in Italian women with endometriosis. Chemosphere. 2006;63:1361–7. doi: 10.1016/j.chemosphere.2005.09.022. [DOI] [PubMed] [Google Scholar]
- [33].Fierens S, Mairesse H, Heilier JF, et al. Dioxin/polychlorinated biphenyl body burden, diabetes and endometriosis: findings in a population-based study in Belgium. Biomarkers. 2003;8:529–34. doi: 10.1080/1354750032000158420. [DOI] [PubMed] [Google Scholar]
- [34].Pauwels A, Schepens PJ, D’Hooghe T, et al. The risk of endometriosis and exposure to dioxins and polychlorinated biphenyls: a case-control study of infertile women. Hum Reprod. 2001;16:2050–5. doi: 10.1093/humrep/16.10.2050. [DOI] [PubMed] [Google Scholar]
- [35].Nomura M, Durbak L, Chan J, et al. Genotype/age interactions on aggressive behavior in gonadally intact estrogen receptor beta knockout (betaERKO) male mice. Horm Behav. 2002;41:288–96. doi: 10.1006/hbeh.2002.1773. [DOI] [PubMed] [Google Scholar]
- [36].Vogel CF, Zhao Y, Wong P, Young NF, Matsumura F. The use of c-src knockout mice for the identification of the main toxic signaling pathway of TCDD to induce wasting syndrome. J Biochem Mol Toxicol. 2003;17:305–15. doi: 10.1002/jbt.10096. [DOI] [PubMed] [Google Scholar]
- [37].Mably TA, Moore RW, Peterson RE. In utero and lactational exposure of male rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Effects on androgenic status. Toxicol Appl Pharmacol. 1992;114:97–107. doi: 10.1016/0041-008x(92)90101-w. [DOI] [PubMed] [Google Scholar]
- [38].Brasted M, White CA, Kennedy TG, Salamonsen LA. Mimicking the events of menstruation in the murine uterus. Biol Reprod. 2003;69:1273–80. doi: 10.1095/biolreprod.103.016550. [DOI] [PubMed] [Google Scholar]
- [39].Theobald HM, Peterson RE. In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-rho-dioxin: effects on development of the male and female reproductive system of the mouse. Toxicol Appl Pharmacol. 1997;145:124–35. doi: 10.1006/taap.1997.8173. [DOI] [PubMed] [Google Scholar]
- [40].Lessey BA, Killam AP, Metzger DA, Haney AF, Greene GL, McCarty KS., Jr Immunohistochemical analysis of human uterine estrogen and progesterone receptors throughout the menstrual cycle. J Clin Endocrinol Metab. 1988;67:334–40. doi: 10.1210/jcem-67-2-334. [DOI] [PubMed] [Google Scholar]
- [41].Nebert DW, Jones JE. Regulation of the mammalian cytochrome P1-450 (CYP1A1) gene. Int J Biochem. 1989;21:243–52. doi: 10.1016/0020-711x(89)90182-1. [DOI] [PubMed] [Google Scholar]
- [42].Couture LA, Abbott BD, Birnbaum LS. A critical review of the developmental toxicity and teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin: recent advances toward understanding the mechanism. Teratology. 1990;42:619–27. doi: 10.1002/tera.1420420606. [DOI] [PubMed] [Google Scholar]
- [43].Couture-Haws L, Harris MW, McDonald MM, Lockhart AC, Birnbaum LS. Hydronephrosis in mice exposed to TCDD-contaminated breast milk: identification of the peak period of sensitivity and assessment of potential recovery. Toxicol Appl Pharmacol. 1991;107:413–28. doi: 10.1016/0041-008x(91)90305-x. [DOI] [PubMed] [Google Scholar]
- [44].Ace CI, Okulicz WC. Differential gene regulation by estrogen and progesterone in the primate endometrium. Mol Cell Endocrinol. 1995;115:95–103. doi: 10.1016/0303-7207(95)03674-v. [DOI] [PubMed] [Google Scholar]
- [45].Sachdeva G, Patil V, Katkam RR, Manjramkar DD, Kholkute SD, Puri CP. Expression profiles of endometrial leukemia inhibitory factor, transforming growth factor beta2 (TGFbeta2), and TGFbeta2 receptor in infertile bonnet monkeys. Biol Reprod. 2001;65:1–8. doi: 10.1095/biolreprod65.1.1. [DOI] [PubMed] [Google Scholar]
- [46].Clark DA, Merali FS, Hoskin DW, et al. Decidua-associated suppressor cells in abortion-prone DBA/2-mated CBA/J mice that release bioactive transforming growth factor beta2-related immunosuppressive molecules express a bone marrow-derived natural suppressor cell marker and gamma delta T-cell receptor. Biol Reprod. 1997;56:1351–60. doi: 10.1095/biolreprod56.5.1351. [DOI] [PubMed] [Google Scholar]
- [47].Fein A, Magid N, Savion S, et al. Diabetes teratogenicity in mice is accompanied with distorted expression of TGF-beta2 in the uterus. Teratog Carcinog Mutagen. 2002;22:59–71. doi: 10.1002/tcm.1039. [DOI] [PubMed] [Google Scholar]
- [48].Gorivodsky M, Torchinsky A, Zemliak I, Savion S, Fein A, Toder V. TGF beta 2 mRNA expression and pregnancy failure in mice. Am J Reprod Immunol. 1999;42:124–33. [PubMed] [Google Scholar]
- [49].Bruner KL, Matrisian LM, Rodgers WH, Gorstein F, Osteen KG. Suppression of matrix metalloproteinases inhibits establishment of ectopic lesions by human endometrium in nude mice. J Clin Invest. 1997;99:2851–7. doi: 10.1172/JCI119478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Spuijbroek MD, Dunselman GA, Menheere PP, Evers JL. Early endometriosis invades the extracellular matrix. Fertil Steril. 1992;58:929–33. doi: 10.1016/s0015-0282(16)55437-5. [DOI] [PubMed] [Google Scholar]
- [51].Grochowalski A, Chrzaszcz R, Pieklo R, Gregoraszczuk EL. Estroenic and antiestrogenic effect of in vitro treatment of follicular cells with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chemosphere. 2001;43:823–7. doi: 10.1016/s0045-6535(00)00440-9. [DOI] [PubMed] [Google Scholar]
- [52].Safe S, Wormke M, Samudio I. Mechanisms of inhibitory aryl hydrocarbon receptor-estrogen receptor crosstalk in human breast cancer cells. J Mammary Gland Biol Neoplasia. 2000;5:295–306. doi: 10.1023/a:1009550912337. [DOI] [PubMed] [Google Scholar]
- [53].Kurita T, Young P, Brody JR, Lydon JP, O’Malley BW, Cunha GR. Stromal progesterone receptors mediate the inhibitory effects of progesterone on estrogen-induced uterine epithelial cell deoxyribonucleic acid synthesis. Endocrinology. 1998;139:4708–13. doi: 10.1210/endo.139.11.6317. [DOI] [PubMed] [Google Scholar]
- [54].Lydon JP, DeMayo FJ, Conneely OM, O’Malley BW. Reproductive phenotpes of the progesterone receptor null mutant mouse. J Steroid Biochem Mol Biol. 1996;56:67–77. doi: 10.1016/0960-0760(95)00254-5. [DOI] [PubMed] [Google Scholar]
- [55].Osteen KG, Rodgers WH, Gaire M, Hargrove JT, Gorstein F, Matrisian LM. Stromal-epithelial interaction mediates steroidal regulation of metalloproteinase expression in human endometrium. Proc Natl Acad Sci USA. 1994;91:10129–33. doi: 10.1073/pnas.91.21.10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nishimura N, Yonemoto J, Miyabara Y, Fujii-Kuriyama Y, Tohyama C. Altered thyroxin and retinoid metabolic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin in aryl hydrocarbon receptor-null mice. Arch Toxicol. 2005;79:260–7. doi: 10.1007/s00204-004-0626-4. [DOI] [PubMed] [Google Scholar]
- [57].Ishida T, Oshimo T, Nishimura A, et al. Reduction of the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice using an antiulcer drug, geranylgeranylacetone. Biol Pharm Bull. 2004;27:1397–402. doi: 10.1248/bpb.27.1397. [DOI] [PubMed] [Google Scholar]
- [58].Couture LA, Abbott BD, Birnbaum LS. A critical review of the developmental toxicity and teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin: recent advances toward understanding the mechanism. Teratology. 1990;42:619–27. doi: 10.1002/tera.1420420606. [DOI] [PubMed] [Google Scholar]
- [59].DeVito MJ, Birnbaum LS, Farland WH, Gasiewicz TA. Comparisons of estimated human body burdens of dioxinlike chemicals and TCDD body burdens in experimentally exposed animals. Environ Health Perspect. 1995;103:820–31. doi: 10.1289/ehp.95103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Heindel JJ. Role of exposure to environmental chemicals in the developmental basis of reproductive disease and dysfunction. Semin Reprod Med. 2006;24:168–77. doi: 10.1055/s-2006-944423. [DOI] [PubMed] [Google Scholar]
- [61].Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. 1971;284:878–81. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- [62].Lau C, Rogers JM. Embryonic and fetal programming of physiological disorders in adulthood. Birth Defects Res C Embryo Today. 2004;72:300–12. doi: 10.1002/bdrc.20029. [DOI] [PubMed] [Google Scholar]
- [63].McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]