

Abstract
Parasympathetic nerves from the pterygopalatine ganglia provide nitroxidergic innervation to forebrain cerebral blood vessels. Disruption of that innervation attenuates cerebral vasodilatation seen during acute hypertension as does systemic administration of a non-selective nitric oxide synthase (NOS) inhibitor. Although such studies suggest that nitric oxide (NO) released from parasympathetic nerves participates in vasodilatation of cerebral vessels during hypertension that hypothesis has not been tested with selective local inhibition of neuronal NOS (nNOS). We tested that hypothesis through these studies performed in anesthetized rats instrumented for continuous measurement of blood pressure, heart rate and pial arterial diameter through a cranial window. We sought to determine if the nNOS inhibitor propyl-L-arginine delivered directly to the outer surface of a pial artery would 1) attenuate changes in pial arterial diameter during acute hypertension and 2) block nNOS mediated dilator effects of N-methyl-D- Aspartate (NMDA) delivered into the window but 3) not block vasodilatation elicited by acetylcholine (ACh) and mediated by endothelial NOS dilator. Without the nNOS inhibitor arterial diameter abruptly increased 70 ± 15% when mean arterial pressure (MAP) reached 183 ± 3 mmHg while with nNOS inhibition diameter increased only 13 ± 10% (p<0.05) even when MAP reached 191 ± 4 (p>0.05). The nNOS inhibitor significantly attenuated vasodilatation induced by NMDA but not ACh delivered into the window. Thus, local nNOS inhibition attenuates breakthrough from autoregulation during hypertension as does complete interruption of the parasympathetic innervation of cerebral vessels. These findings further support the hypothesis that NO released from parasympathetic fibers contributes to cerebral vasodilatation during acute hypertension. Section: Regulatory Systems
Keywords: cerebral blood flow, nitric oxide, nitric oxide synthase, NMDA, parasympathetic
1. INTRODUCTION
Autoregulation of cerebrovascular resistance (CVR)* maintains cerebral blood flow (CBF) near basal levels as arterial blood pressure (AP) increases slowly (Edvinsson, L. et al, 1993). However, when AP exceeds an upper limit, breakthrough of autoregulation occurs, cerebral blood vessels dilate, and CBF increases dramatically (MacKenzie, E. T. et al, 1976; Skinhoj, E. and Strandgaard, S., 1973). We have found that breakthrough may be delayed to much higher blood pressures if the arterial baroreflex is interrupted either peripherally or centrally (Talman, W. T. et al, 1994; Talman, W. T. and Nitschke Dragon, D., 2002) and we recently showed that baroreceptor pathways through the nucleus tractus solitarii project directly to preganglionic parasympathetic neurons that themselves influence CBF (Agassandian, K. et al, 2002; Agassandian, K. et al, 2003). As when the baroreflex is interrupted, breakthrough is also attenuated, and the breakthrough point shifted to higher AP’s, when the nitroxidergic innervation to the cerebral vessels is interrupted (Talman, W. T. and Nitschke Dragon, D., 2000) or when nitric oxide (NO) synthesis is inhibited by systemic administration of a non-selective NO synthase (NOS) inhibitor (Talman, W. T. and Nitschke Dragon, D., 1995a). However, neither of those observations directly tests our hypothesis that neuronal NOS (nNOS) participates in cerebral vasodilatation seen with breakthrough during acute hypertension nor do they directly test whether NO, synthesized by nerves near cerebral vessels, is responsible for that dilatation. In the current study we sought to test that hypothesis by determining if breakthrough occurred when nNOS was selectively locally inhibited. Because systemic administration of an nNOS inhibitor would fail to identify if any effect were mediated at the level of the nerve/vessel interface or at some distant site, we performed the current studies with a pial window that allowed us continuously to assess pial arterial diameter as an index of CBF (Mayhan, W. G., 1989) while changing the local milieu through administration of an nNOS inhibitor directly over an observed vessel.
2. RESULTS
Effects of nNOS inhibition on vasodilatation during hypertension
Neither the basal mean arterial pressure (MAP) nor the maximal MAP (MAPmax) differed between control animals (N = 9) and animals that received the nNOS inhibitor (N = 5; see Table 1). We compared vascular diameters at MAPmax (Fig. 1) with diameters at basal MAP (MAP0) and expressed results as a percent of basal diameters. Propyl-L-arginine (PLA) did not change basal arterial diameters but it significantly attenuated the increase in vascular diameter (13 ± 10%) seen at maximal pressures when compared with the control group (70 ± 15%) (Table 1).
Table 1.
Inhibiting nNOS Attenuates Pial Arterial Dilatation During Acute Hypertension
| Basal MAP (mmHg) | Diam. at Basal MAP (μm) | MAPMAX (mmHg) | Diam. at MAPMax (μm) | |
|---|---|---|---|---|
| Control (9) | 92 ± 3 | 33.2 ± 2.9 | 183 ± 3 | 55.4 ± 5.2* |
| PLA (5) | 86 ± 11 | 53.8 ± 8.5† | 191 ± 4 | 59.4 ± 8NS |
Numbers in parentheses = N;
= p<0.05 compared to basal;
p<0.05 compared with control;
NS = p> 0.05 compared to basal
Figure 1. Inhibiting nNOS Blocks Pial Arterial Vasodilatation Induced by Hypertension.
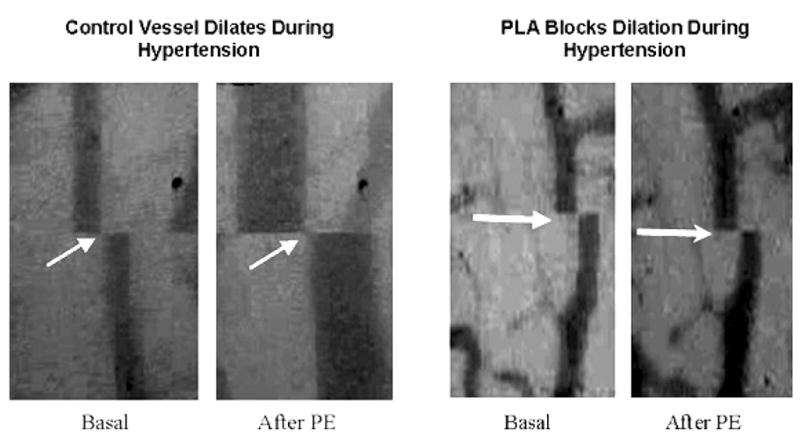
Pial arterial dilatation as a result of hypertension was prominent in a control animal that did not receive PLA (left panels). Between the basal state (far left) when pial arterial diameter was 51.2 μm at an MAP of 89 mmHg and maximal hypertension induced by infusion of phenylephrine (PE; middle left) diameter increased to 123.2 μm with an MAP of 163 mmHg. Pial arterial dilatation was greatly attenuated with administration of PLA (right panels) between the basal state (middle right) when diameter was 48.8 μm and MAP was 85 mmHg and the maximally hypertensive state (far right) when diameter was 54.3 μm and MAP was 188 mmHg.
Because basal diameters in the two groups differed we sought to determine if differences in basal arterial diameter explained differences in dilatation during hypertension. First, we further analyzed data from the 9 control animals by comparing the maximal diameter during hypertension in 4 animals with a basal diameter > 30 μm with the maximal diameter in 5 animals whose basal diameter was < 30 μm. Diameter increased by 81 ± 25.8 % in the former group while in the latter group, with the larger basal diameter, it increased 57 ± 8.0 % (NS). Thus, any influence of diameter on vasodilatation seen with hypertension in controls vs. animals treated with PLA would have been in the opposite direction from what was seen. To further evaluate any possible effect of basal diameter we studied an additional 4 control animals whose basal arterial diameters (52.5 ± 5.2) were matched with the average basal diameter in treated animals. MAP rose from 95 ± 5 to 180 ± 5 mmHg in the former group (change not significantly different from that in animals treated with PLA), but the increase in diameter (102 ± 8%) was significantly greater than in the group treated with PLA (Fig. 1).
Effect of PLA on vasodilator responses to acetylcholine
In 5 animals, infusions of acetylcholine (ACh) into the cranial window prior to PLA led to significant dilatation (7 ± 1 %) of the pial vessel (Fig. 3). Infusion of PLA into the window did not significantly (p >0.05) alter dilator responses (5 ± 2 %) to ACh again infused into the window (Fig. 2). Breakthrough of autoregulation was significantly attenuated (increased arterial diameter of 18 ± 6 %) in animals that received PLA and ACh in this protocol as it was in animals that received PLA alone (N=5; Fig. 3).
Figure 3. Inhibiting nNOS Blocks Breakthrough.
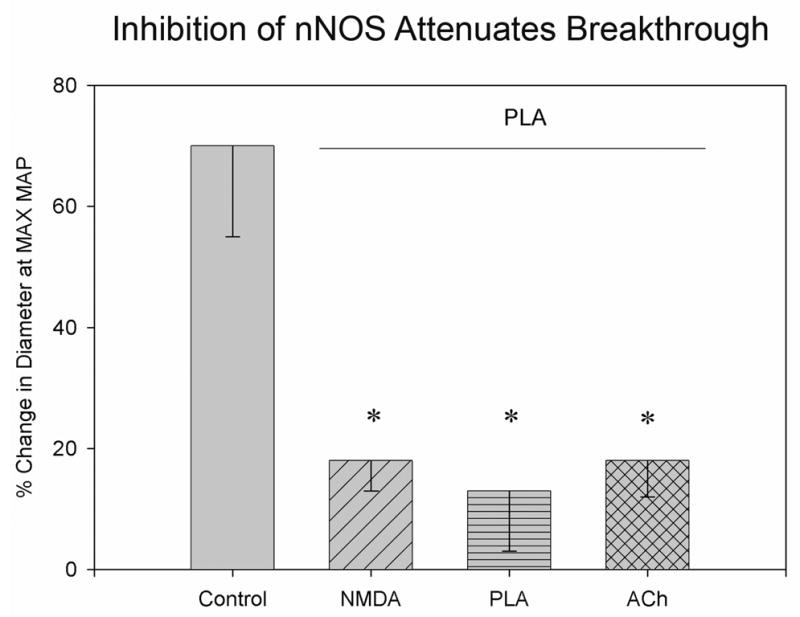
Compared to breakthrough seen in control animals (N = 9), breakthrough was blocked when PLA had blocked vasodilatation induced by NMDA (NMDA; N = 5), when PLA was given alone (PLA; N = 5), and when PLA had failed to block vasodilatory responses to ACh (N = 5). * p<0.05 compared to control.
Figure 2. PLA selectively blocks nNOS .
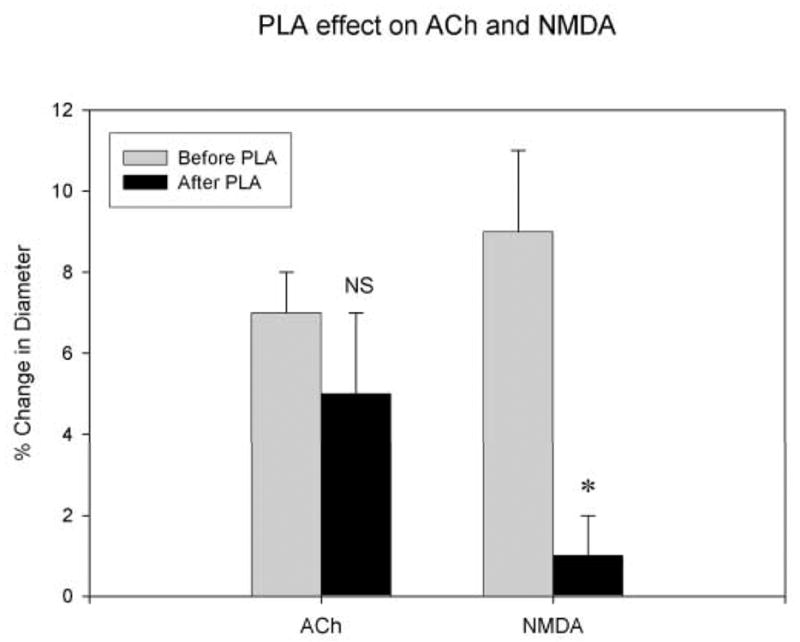
PLA failed to significantly block vasodilatory effects of ACh (10−5 M) infused over cerebral vessels (left) in 5 rats but did significantly block vasodilatory effects of NMDA (3 × 10−5 M) infused over cerebral vessels (right) in 5 other rats. Effects of hypertension on vasodilatation in these groups of animals were compared and contrasted (see figure 3).
Effect of PLA on vasodilator responses to NMDA
We found that infusions of NMDA into the cranial window prior to PLA led to significant dilatation of the pial vessel. In 5 animals infusion of PLA attenuated the dilatation produced by infusion of NMDA (from 9 ± 2 % before PLA to 1 ± 1 % after PLA; Fig. 2) and it significantly attenuated (p<0.02) vasodilation (diameter increased only 18 ± 6 % from basal) seen with breakthrough during hypertension (Fig. 3).
3. DISCUSSION
The data presented here support the hypothesis that nNOS, in close proximity to cerebral vessels, participates in breakthrough of autoregulation during acute hypertension. Numerous studies have shown that NO participates in cerebral vasodilatation; and, while NO derived from endothelial NOS (eNOS) may act as a dilator (Furchgott, R. F. and Vanhoutte, P. M., 1989), NO from neural pathways also directly dilates cerebral vessels (Lee, T. J., 2000). Essentially all of the nitroxidergic innervation of forebrain vessels derives from neurons of the pterygopalatine ganglia (Toda, N. et al, 1993; Yoshida, K. et al, 1993), bilateral ganglia that lie near the floor of the orbit and send ganglionic projections into the cranium through the mesial orbital wall (Hara, H. et al, 1993). Stimulation of those ganglia leads to cerebral arterial dilatation (Seylaz, J. et al, 1988; Toda, N. et al, 2000b; Toda, N. et al, 2000a) and some reports, which indicate that interruption of the ganglia or their innervation leads to cerebral vasoconstriction (Toda, N. et al, 2000b; Toda, N. et al, 2000a), suggest that they exert a tonic vasodilator influence. Stimulation of the superior salivatory nucleus (SSN), the site of preganglionic neurons that project to the pterygopalatine ganglia and thus influence CBF, also leads to vasodilatation of cerebral vessels (Agassandian, K. et al, 2002). Ganglionic neurons of the pterygopalatine ganglia express a number of potentially vasoactive substances including vasoactive intestinal polypeptide, ACh (implied by the presence of choline acetyltransferase), and nNOS, which would synthesize NO (Yu, J. G. et al, 1998). However, it is likely that VIP and ACh may modulate release of NO from ganglionic terminals (Yu, J. G. et al, 1998) and that ACh derived from ganglionic fibers may serve to counter dilator influences by inhibiting release of NO (Kimura, T. et al, 1997). Homeostatic balance between the dilator effects of NO synthesized by neurons and the constrictor effects of ACh could balance changes in CBF under different behavioral conditions.
Because of the close proximity of the nNOS containing parasympathetic nerve terminals and endothelium, from which NO may be generated via eNOS, it was important to establish that the inhibitor used to diminish activity of the one enzyme, nNOS in this case, did not block activity of the other. To that end we sought to determine if vasodilatation induced by infusing into the window ACh, which acts through eNOS to promote release of NO (Faraci, F. M. et al, 1998), was inhibited by PLA and that vasodilatation induced by NMDA, which acts through nNOS to promote release of NO (Kiss, J. P. and Vizi, E. S., 2001), was not. We chose to use PLA in the current studies because it has been shown to be highly selective for nNOS (Cooper, G. R. et al, 2000; Zhang, H. Q. et al, 1997) and because it can be injected in an aqueous solution directly upon pial vessels. Such direct application circumvented problems in interpreting studies with systemic administration of an inhibitor whose site of action could not have been known.
In the current study we expected the nNOS inhibitor to block vasodilatory responses to NMDA, which is known to elicit such responses through an influence on nNOS (Faraci, F. M. and Breese, K. R., 1993; Faraci, F. M. and Brian, J. E., 1995). PLA significantly inhibited NMDA-mediated vasodilatation and in the same animals markedly attenuated vasodilatation that would have occurred with breakthrough. Our study was designed to provide comparable vasodilatation when ACh, for eNOS activation, or when NMDA, for nNOS activation, was used as the agonist. Thus, PLA, whose dose was kept constant throughout all experiments, was challenged with comparable dilatory events for NMDA and ACh. We adjusted the concentration of NMDA used in this study in order to produce a vasodilatory response that was comparable to that produced by infusion of ACh onto the vessels. The concentration (10−5 M) of ACh used in this study was the same as that used in other studies and the response to ACh (7±1% dilatation) was comparable to that reported previously in studies utilizing rat pial vessels (Fang, Q. et al, 2006). It was beyond the scope of this study to establish full dose-response curves for NMDA and ACh as has been done previously or to derive inhibitory ratios for PLA in challenging NMDA and ACh. We acknowledge the possibility that a significant influence of PLA on responses to ACh may have been missed in our studies, but power analyses indicated that an 80% power would have necessitated studying at least 30–40 animals. Given the readily demonstrated statistical significance of PLA effects on NMDA, such an increase in numbers in the ACh group alone was unjustified.
There has been little evidence to show how the nitroxidergic parasympathetic influence on cerebral blood vessels may participate in physiological processes in the whole animal although its having been found in so many vertebrate species (Goadsby, P. J. et al, 1996; Kimura, T. et al, 1997; Nozaki, K. et al, 1993; Toda, N. et al, 1993; Toda, N. et al, 2000b; Yoshida, K. et al, 1993; Yu, J. G. et al, 1997) certainly suggests that it might serve an important role. In fact, some reports have suggested both pathophysiologic as well as physiologic involvement of the pathway. Specifically, some have shown that the pathway may influence outcomes in stroke in that animals that have sustained interruption of nerve fibers from the pterygopalatine ganglia manifest a significantly larger volume of cerebral damage with ischemia than do intact animals (Kano, M. et al, 1991; Koketsu, N. et al, 1992). Our own laboratory has suggested that the pathway may contribute to cerebral vasodilatation during hypertension and thus extend cerebrovascular autoregulation (Talman, W. T. et al, 1994), a suggestion that has been echoed by others in the field (Toda, N. and Okamura, T., 1992). We have shown that arterial baroreceptors are critical for cerebral vasodilatation when blood pressure has been raised beyond the autoregulatory range (Talman, W. T. et al, 1994; Talman, W. T. and Nitschke Dragon, D., 1995b; Talman, W. T. and Nitschke Dragon, D., 2002); that the NTS, where arterial baroreceptor fibers terminate in the central nervous system (Blessing, W. W. et al, 1999; Ciriello, J., 1983; Kalia, M. and Sullivan, J. M., 1982; Torrealba, F. and Claps, A., 1988), projects directly to the SSN (Agassandian, K. et al, 2002); that interruption of function at the level of SSN or pterygopalatine ganglia extends the autoregulatory range during hypertension as does baroreflex interruption (Agassandian, K. et al, 2003; Talman, W. T. and Nitschke Dragon, D., 2000), and that non-selective inhibition of synthesis of NO released by those ganglionic fibers had the same effect (Talman, W. T. and Nitschke Dragon, D., 1995a). These studies further suggest that nitroxidergic nerves projecting from the pterygopalatine ganglion to cerebral blood vessels play a roll in the increase in CBF seen in breakthrough (Talman, W. T. and Nitschke Dragon, D., 2000). The current study did not directly evaluate CBF but instead monitored changes in cerebrovascular tone in a highly confined area, which alone was treated with the nNOS inhibitor. It is likely that CBF did increase in pial vessels studied here in that arteries more proximal to the site of observation would have themselves dilated in the absence of the nNOS inhibitor. Such anticipated dilatation of proximal vessels likely contributed to what little vasodilation was seen during maximal elevation of arterial pressure.
This study provides another link in the neuronal pathway connecting baroreceptor afferents to cerebral vessels by demonstrating that it is neuronal NOS that is involved in dilatation of cerebral vessels during acute, pharmacologically induced hypertension. We acknowledge, however, that the nNOS inhibitor could have mediated its effects through blockade not only of nNOS in PPG axons but also that in nearby central neurons (Kiss, J. P. and Vizi, E. S., 2001). It is activation of those neurons that has been considered the means for linking cerebral metabolic activity with CBF (Goadsby, P. J. et al, 1992; Iadecola, C., 1993) and it is through those neurons that NMDA has been thought to mediate its nNOS-related vasodilatory action (Faraci, F. M. and Breese, K. R., 1993; Faraci, F. M. and Brian, J. E., 1995). Although NMDA may act through local cerebral neurons to effect release of NO from those neurons, it seems unlikely that the results of this study would support an influence of those neurons on vasomotor tone during autoregulation. Albeit there has been no study to directly test what, if any, role nNOS in cerebral cortical neurons might play in autoregulation during hypertension there is evidence that it could alter, rather than enhance, cerebral vascular hemodynamics in a brain injury model (Armstead, W. M., 2002), an effect that would be opposite what has been shown for parasympathetic NO nerves. Although changes (upregulation) of NMDA receptors may augment vasodilatation with cortical stimulation (Weiss, H. R. et al, 1996), the current study would not be expected to lead to cortical activation in that direct stimulation of parasympathetic nerves, as may occur during intense hypertension, has not led to any change in cerebral metabolism (Goadsby, P. J., 1989). Thus, the current study, together with evidence now from other studies, suggests that it is NO generated by nNOS in parasympathetic nerves from the PPG that is involved in vasodilatation during hypertension. While no studies have suggested that local nitroxidergic cerebral neurons participated otherwise in regulating cerebrovascular tone, further studies will be needed to compare and contrast vasomotor actions of such local neurons with those produced by activation of PPG nitroxidergic neurons.
While the current study demonstrates that nNOS may mediate cerebral vasodilatation, the full physiological implications of cerebrovascular control through these mechanisms remain to be determined. Further study seems warranted in that the parasympathetic nitroxidergic pathway may not only participate in regulating blood flow during acute hypertension but may also provide neural protection during stroke.
4. Experimental Procedure
All methods used in the studies described here were approved by the institutional animal care and use committees both of the Veterans Affairs Health Care System Iowa City and the University of Iowa and adhered to principles defined in the “Guide for the Care and Use of Laboratory Animals” (National Research Council, 1996).
Instrumentation and General Methods: As previously described (Talman, W. T. et al, 1994) adult male Sprague Dawley rats were anesthetized with halothane or isoflurane (2%), instrumented with a femoral arterial cannula for recording AP and an intravenous cannula for delivering drugs, and intubated via the trachea for continuous mechanical ventilation. The femoral arterial cannula was connected to a data acquisition system (Power Lab/8SP, Minneapolis, MN), which acquired AP, mean AP (MAP; calculated as the mean of diastolic and systolic AP’s), and heart rate (HR). Temperature was maintained at 37° Celsius by means of a temperature controller (YSI Model 73A; Yellow Springs Instruments, Yellow Springs, OH) and a rectal thermometer. We modified methods that have previously been described (Paterno, R. et al, 2000) for assessing pial arterial diameter in rats. Animals were placed in a stereotaxic frame and a cranial window was prepared by drilling a 5 mm burr hole over the parietal cortex. The dura was carefully incised and retracted to provide an unobstructed view of the cortical surface. After confirming that we could visualize pial arteries that were of the desired size (approximately 30–60 μm o.d.) we sealed the window with a glass cover slip placed over a ring of melted bone wax that encircled the cranial window. We created an inflow and an outflow port in the bone wax by inserting a blunt 18 and 20 gauge needle, respectively, into the sealed window. Dental resin was placed around the bone wax and needles to fix the window and ports to the skull. We blew air at room temperature over the dental resin for two minutes to offset the thermal reaction created by the resin. The outflow port was attached to a 70mm long tube placed at a 90-degree angle to the port itself. The port and window were then filled with artificial cerebrospinal fluid (aCSF: formula = 0.22 g KCl; 0.132 g MgCl2; 0.22 gCaCl2; 7.71 g NaCl; 0.665 g dextrose; 2 g NaHCO3; and distilled H2O to a total volume of 1 L) to maintain even pressure on the brain surface. Prior to the experiment we flushed the sealed window every 10 minutes with 2 ml of aCSF. Artificial CSF was prepared immediately prior to its use, bubbled with (5% CO2, 5%O2 in N2 balance), and kept at 40°Celsius until it was used in the experiment at which time the temperature was 37° Celsius.
After placing and sealing the window we again confirmed that a pial artery could be visualized clearly through an operating microscope (Olympus) connected to an image-shearing device (Instrumentation for Physiology and Medicine, Inc., San Diego, CA). Inhalation anesthesia was then discontinued and anesthesia maintained for the duration of the study by intravenous (i.v.) infusion of α-chloralose (60 mg/kg loading and 20 mg/kg/hr). We allowed the preparation to equilibrate for at least 30 minutes prior to starting an experiment. Arterial blood gases were assessed immediately prior to each experiment and at regular intervals throughout the study to confirm that arterial PCO2 was maintained within 10 Torr of basal values during protocols for the study.
Protocols
Control experiments
In 13 rats, after identifying the index artery that would be studied, we infused 2 ml of aCSF and 10 minutes later began to gradually (in a “ramp” rather than a “step”) raise mean arterial blood pressure (MAP) over 20–30 minutes from basal values to the maximum achievable value by infusing phenylephrine (0.5 μg/μl i.v. at a rate of 2 μl/min increased slowly to a maximum infusion rate of 120 μl/min or until MAP could no longer be increased or breakthrough had occurred). Breakthrough of autoregulation was recognizable by rapid and pronounced dilatation of the vessel and was seen typically only when MAP exceeded 160 mmHg.
Assessing Effects of an nNOS Inhibitor on Cerebral Arterial Dilatation during hypertension
In 5 animals prior to inducing hypertension, while observing the index artery, we infused 2 ml of a solution containing the selective nNOS inhibitor (Zhang, H. Q. et al, 1997) PLA (Tocris Bioscience, Ellisville, MO) in a 0.3 mM concentration in aCSF and assessed arterial diameter. After 10 minutes we again infused PLA. We then began to induce hypertension as in control experiments and assessed maximal changes in arterial diameter when MAP reached the highest achievable level.
Assessing Effects of the nNOS Inhibitor on eNOS Mediated Vasodilatation
This experimental protocol (n=5) consisted of the following steps: After assessing baseline pial arterial diameter we infused acetylcholine (ACh, 2 ml of a 10−5 M solution in aCSF) into the window and assessed changes in diameter of the index artery. We allowed 10 minutes for vessels to return to their basal diameter, then repeated infusion of ACh, and again assessed arterial diameter to assure stability of the vasodilatation seen with ACh. We proceeded with the protocol only if ACh induced at least a 4% increase in vascular diameter because increases of that magnitude were the minimum that could be clearly defined from basal values and would be consistent with previous studies using ACh as a dilator in vivo (Fang, Q. et al, 2006). We infused 2 ml of a solution of PLA (0.3 mM concentration) as detailed above and again assessed arterial diameter. After 10 minutes we again infused ACh and assessed arterial diameter to determine if ACh still induced vasodilatation. We again infused PLA into the window and assessed effects of hypertension on vascular diameter as described above.
Assessing Effects of the nNOS Inhibitor on nNOS Induced Vasodilatation
In 5 rats the protocol followed the same steps as detailed above except that a 2 ml solution of N-methyl-D-aspartate (NMDA; 0.03 mM) in aCSF was substituted for ACh in the preceding protocol. The concentration of NMDA used in these studies was the concentration that elicited a change in vessel diameter that matched the change elicited by ACh in the above protocol. When the vessel had again returned to its basal diameter we infused PLA into the window and assessed effects of hypertension on vascular diameter as described above.
In each protocol the pial vessel that had been selected was visualized constantly during phenylephrine infusion and data from the image-shearing monitor were stored online for later analysis. MAP and HR were continuously recorded and synchronized with cerebrovascular events as they were recorded from the image-shearing device. If, after administration of the aCSF control or aCSF containing nNOS inhibitor, the window became clouded with proteinaceous material or blood, we flushed the window with aCSF or aCSF containing PLA respectively.
Upon achieving a maximal AP, defined either by breakthrough, dilatation of the pial artery, or an inability to raise MAP further with continued increases in the dose of phenylephrine, a final arterial blood gas was assessed to assure that arterial pCO2 had remained within parameters and the animal was euthanized with concentrated KCl or pentobarbital (150 mg/kg).
Data (expressed as mean ± SEM) generally were analyzed by two-sample t- test or one-way ANOVA and Tukey post hoc comparison as appropriate. We considered a p value ≤0.05 as significant.
Acknowledgments
The authors gratefully acknowledge support provided for these studies through a Merit Review awarded to WTT by the Department of Veterans Affairs and in part through NIH HL R01 59593 (WTT, PI).
- *ACh
acetylcholine
- aCSF
artificial cerebrospinal fluid
- AP
arterial pressure
- CBF
cerebral blood flow
- CVR
cerebrovascular resistance
- Diam
diameter
- eNOS
endothelial nitric oxide synthase
- HR
heart rate
- i.v
intravenous
- MAP
mean AP
- NMDA
N-methyl-D-aspartate
- NO
Nitric oxide
- nNOS
neuronal NOS
- PLA
propyl-L-arginine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agassandian K, Fazan VPS, Adanina V, Talman WT. Direct projections from the cardiovascular nucleus tractus solitarii to pontine preganglionic parasympathetic neurons: a link to cerebrovascular regulation. J Comp Neurol. 2002;452:242–254. doi: 10.1002/cne.10372. [DOI] [PubMed] [Google Scholar]
- Agassandian K, Fazan VPS, Margaryan N, Nitschke Dragon D, Riley J, Talman WT. A novel central pathway links arterial baroreceptors and pontine parasympathetic neurons in cerebrovascular control. CellMolecNeurobiol. 2003;23:463–478. doi: 10.1023/A:1025059710382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstead WM. Age dependent NMDA contribution to impaired hypotensive cerebral hemodynamics following brain injury. Brain ResDevBrain Res. 2002;139:19–28. doi: 10.1016/s0165-3806(02)00511-4. [DOI] [PubMed] [Google Scholar]
- Blessing WW, Yu YH, Nalivaiko E. Medullary projections of rabbit carotid sinus nerve. Brain Res. 1999;816:405–410. doi: 10.1016/s0006-8993(98)01147-0. [DOI] [PubMed] [Google Scholar]
- Ciriello J. Brainstem projections of aortic baroreceptor afferent fibers in the rat. NeurosciLett. 1983;36:37–42. doi: 10.1016/0304-3940(83)90482-2. [DOI] [PubMed] [Google Scholar]
- Cooper GR, Mialkowski K, Wolff DJ. Cellular and enzymatic studies of N(omega)-propyl-l-arginine and S-ethyl-N-[4-(trifluoromethyl)phenyl]isothiourea as reversible, slowly dissociating inhibitors selective for the neuronal nitric oxide synthase isoform. ArchBiochemBiophys. 2000;375:183–194. doi: 10.1006/abbi.1999.1658. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, MacKenzie ET, McCulloch J. Cerebral blood flow and metabolism. Raven Press; New York: 1993. [Google Scholar]
- Fang Q, Sun H, Arrick DM, Mayhan WG. Inhibition of NADPH oxidase improves impaired reactivity of pial arterioles during chronic exposure to nicotine. JApplPhysiol. 2006;100:631–636. doi: 10.1152/japplphysiol.00975.2005. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Breese KR. Nitric oxide mediates vasodilatation in response to activation of N-methyl-D-aspartate receptors in brain. CircRes. 1993;72:476–480. doi: 10.1161/01.res.72.2.476. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Brian JE. 7-Nitroindazole inhibits brain nitric oxide synthase and cerebral vasodilatation in response to n-methyl-d-aspartate. Stroke. 1995;26:2172–2175. doi: 10.1161/01.str.26.11.2172. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Sigmund CD, Shesely EG, Maeda N, Heistad DD. Responses of carotid artery in mice deficient in expression of the gene for endothelial NO synthase. AmJPhysiol. 1998;274:H564–H570. doi: 10.1152/ajpheart.1998.274.2.H564. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- Goadsby PJ. Effect of stimulation of facial nerve on regional cerebral blood flow and glucose utilization in cats. AmJPhysiol. 1989;257:R517–R521. doi: 10.1152/ajpregu.1989.257.3.R517. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Kaube H, Hoskin KL. Nitric oxide synthesis couples cerebral blood flow and metabolism. Brain Res. 1992;595:167–170. doi: 10.1016/0006-8993(92)91470-y. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Uddman R, Edvinsson L. Cerebral vasodilatation in the cat involves nitric oxide from parasympathetic nerves. Brain Res. 1996;707:110–118. doi: 10.1016/0006-8993(95)01206-0. [DOI] [PubMed] [Google Scholar]
- Hara H, Zhang QJ, Kuroyanagi T, Kobayashi S. Parasympathetic cerebrovascular innervation: an anterograde tracing from the sphenopalatine ganglion in the rat. Neurosurgery. 1993;32:822–827. doi: 10.1227/00006123-199305000-00016. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Regulation of the cerebral microcirculation during neural activity: Is nitric oxide the missing link. Trends Neurosci. 1993;16:206–214. doi: 10.1016/0166-2236(93)90156-g. [DOI] [PubMed] [Google Scholar]
- Kalia M, Sullivan JM. Brainstem projections of sensory and motor components of the vagus nerve in the rat. JCompNeurol. 1982;211:248–264. doi: 10.1002/cne.902110304. [DOI] [PubMed] [Google Scholar]
- Kano M, Moskowitz MA, Yokota M. Parasympathetic denervation of rat pial vessels significantly increases infarction volume following middle cerebral artery occlusion. JCerebBlood Flow Metab. 1991;11:628–637. doi: 10.1038/jcbfm.1991.114. [DOI] [PubMed] [Google Scholar]
- Kimura T, Yu JG, Edvinsson L, Lee TJ. Cholinergic, nitric oxidergic innervation in cerebral arteries of the cat. Brain Res. 1997;773:117–124. doi: 10.1016/s0006-8993(97)00889-5. [DOI] [PubMed] [Google Scholar]
- Kiss JP, Vizi ES. Nitric oxide: a novel link between synaptic and nonsynaptic transmission. Trends Neurosci. 2001;24:211–215. doi: 10.1016/s0166-2236(00)01745-8. [DOI] [PubMed] [Google Scholar]
- Koketsu N, Moskowitz MA, Kontos HA, Yokota M, Shimizu T. Chronic parasympathetic sectioning decreases regional cerebral blood flow during hemorrhagic hypotension and increases infarct size after middle cerebral artery occlusion in spontaneously hypertensive rats. JCerebBlood Flow Metab. 1992;12:613–620. doi: 10.1038/jcbfm.1992.85. [DOI] [PubMed] [Google Scholar]
- Lee TJ. Nitric oxide and the cerebral vascular function. JBiomedSci. 2000;7:16–26. doi: 10.1007/BF02255914. [DOI] [PubMed] [Google Scholar]
- MacKenzie ET, Strandgaard S, Graham DI, Jones JV, Harper AM, Farrar JK. Effects of acutely induced hypertension in cats on pial arteriolar caliber, local cerebral blood flow, and the blood- brain barrier. CircRes. 1976;39:33–41. doi: 10.1161/01.res.39.1.33. [DOI] [PubMed] [Google Scholar]
- Mayhan WG. Impairment of endothelium-dependent dilatation of cerebral arterioles during diabetes mellitus. AmJPhysiolHeart CircPhysiol. 1989;256:H621–H625. doi: 10.1152/ajpheart.1989.256.3.H621. [DOI] [PubMed] [Google Scholar]
- Nozaki K, Moskowitz MA, Maynard KI, Koketsu N, Dawson TM, Bredt DS, Snyder SH. Possible origins and distribution of immunoreactive nitric oxide synthase-containing nerve fibers in cerebral arteries. JCerebBlood Flow Metab. 1993;13:70–79. doi: 10.1038/jcbfm.1993.9. [DOI] [PubMed] [Google Scholar]
- Paterno R, Heistad DD, Faraci FM. Potassium channels modulate cerebral autoregulation during acute hypertension. AmJPhysiolHeart CircPhysiol. 2000;278:H2003–H2007. doi: 10.1152/ajpheart.2000.278.6.H2003. [DOI] [PubMed] [Google Scholar]
- Seylaz J, Hara H, Pinard E, Mraovitch S, MacKenzie ET, Edvinsson L. Effect of stimulation of the sphenopalatine ganglion on cortical blood flow in the rat. JCerebBlood Flow Metab. 1988;8:875–878. doi: 10.1038/jcbfm.1988.145. [DOI] [PubMed] [Google Scholar]
- Skinhoj E, Strandgaard S. Pathogenesis of hypertensive encephalopathy. Lancet. 1973;i:461–462. doi: 10.1016/s0140-6736(73)91884-9. [DOI] [PubMed] [Google Scholar]
- Talman WT, Nitschke Dragon D. Inhibition of nitric oxide synthesis extends cerebrovascular autoregulation during hypertension. Brain Res. 1995a;672:48–54. doi: 10.1016/0006-8993(94)01381-q. [DOI] [PubMed] [Google Scholar]
- Talman WT, Nitschke Dragon D. Mechanisms for preserved cerebrovascular autoregulation during hypertension in rats after sinoaortic denervation. ClinExpPharmacolPhysiol. 1995b;22(Supp 1):S77–S79. doi: 10.1111/j.1440-1681.1995.tb02978.x. [DOI] [PubMed] [Google Scholar]
- Talman WT, Nitschke Dragon D. Parasympathetic nerves influence cerebral blood flow during hypertension in rat. Brain Res. 2000;873:145–148. doi: 10.1016/s0006-8993(00)02490-2. [DOI] [PubMed] [Google Scholar]
- Talman WT, Nitschke Dragon D. Inhibiting the nucleus tractus solitarii extends cerebrovascular autoregulation during hypertension. Brain Res. 2002;931:92–95. doi: 10.1016/s0006-8993(02)02264-3. [DOI] [PubMed] [Google Scholar]
- Talman WT, Nitschke Dragon D, Ohta H. Baroreflexes influence autoregulation of cerebral blood flow during hypertension. AmJPhysiolHeart CircPhysiol. 1994;267:H1183–H1189. doi: 10.1152/ajpheart.1994.267.3.H1183. [DOI] [PubMed] [Google Scholar]
- Toda N, Ayajiki K, Tanaka T, Okamura T. Preganglionic and postganglionic neurons responsible for cerebral vasodilation mediated by nitric oxide in anesthetized dogs. JCerebBlood Flow Metab. 2000a;20:700–708. doi: 10.1097/00004647-200004000-00007. [DOI] [PubMed] [Google Scholar]
- Toda N, Ayajiki K, Yoshida K, Kimura H, Okamura T. Impairment by damage of the pterygopalatine ganglion of nitroxidergic vasodilator nerve function in canine cerebral and retinal arteries. CircRes. 1993;72:206–213. doi: 10.1161/01.res.72.1.206. [DOI] [PubMed] [Google Scholar]
- Toda N, Okamura T. Regulation by nitroxidergic nerve of arterial tone. NIPS. 1992;7:148–152. [Google Scholar]
- Toda N, Tanaka T, Ayajiki K, Okamura T. Cerebral vasodilatation induced by stimulation of the pterygopalatine ganglion and greater petrosal nerve in anesthetized monkeys. Neurosci. 2000b;96:393–398. doi: 10.1016/s0306-4522(99)00557-6. [DOI] [PubMed] [Google Scholar]
- Torrealba F, Claps A. The carotid sinus connections: A WGA-HRP study in the cat. Brain Res. 1988;455:134–143. doi: 10.1016/0006-8993(88)90122-9. [DOI] [PubMed] [Google Scholar]
- Weiss HR, Sinha AK, Lu X. Effect of up-regulation of NMDA receptors on cerebral O2 consumption and blood flow in rat. Brain Res. 1996;730:193–198. doi: 10.1016/0006-8993(96)00446-5. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Okamura T, Kimura H, Bredt DS, Snyder SH, Toda N. Nitric oxide synthase-immunoreactive nerve fibers in dog cerebral and peripheral arteries. Brain Res. 1993;629:67–72. doi: 10.1016/0006-8993(93)90482-3. [DOI] [PubMed] [Google Scholar]
- Yu JG, Kimura T, Chang XF, Lee TJ. Segregation of VIPergic-nitric oxidergic and cholinergic-nitric oxidergic innervation in porcine middle cerebral arteries. Brain Res. 1998;801:78–87. doi: 10.1016/s0006-8993(98)00548-4. [DOI] [PubMed] [Google Scholar]
- Yu JG, O’Brien WE, Lee TJ. Morphologic evidence for L-citrulline conversion to L-arginine via the argininosuccinate pathway in porcine cerebral perivascular nerves. JCerebBlood Flow Metab. 1997;17:884–893. doi: 10.1097/00004647-199708000-00007. [DOI] [PubMed] [Google Scholar]
- Zhang HQ, Fast W, Marletta MA, Martasek P, Silverman RB. Potent and selective inhibition of neuronal nitric oxide synthase by N omega-propyl-L-arginine. JMedChem. 1997;40:3869–3870. doi: 10.1021/jm970550g. [DOI] [PubMed] [Google Scholar]