

Abstract
Using a chromatin immunoprecipitation-paired end diTag cloning and sequencing strategy, we mapped estrogen receptor α (ERα) binding sites in MCF-7 breast cancer cells. We identified 1,234 high confidence binding clusters of which 94% are projected to be bona fide ERα binding regions. Only 5% of the mapped estrogen receptor binding sites are located within 5 kb upstream of the transcriptional start sites of adjacent genes, regions containing the proximal promoters, whereas vast majority of the sites are mapped to intronic or distal locations (>5 kb from 5′ and 3′ ends of adjacent transcript), suggesting transcriptional regulatory mechanisms over significant physical distances. Of all the identified sites, 71% harbored putative full estrogen response elements (EREs), 25% bore ERE half sites, and only 4% had no recognizable ERE sequences. Genes in the vicinity of ERα binding sites were enriched for regulation by estradiol in MCF-7 cells, and their expression profiles in patient samples segregate ERα-positive from ERα-negative breast tumors. The expression dynamics of the genes adjacent to ERα binding sites suggest a direct induction of gene expression through binding to ERE-like sequences, whereas transcriptional repression by ERα appears to be through indirect mechanisms. Our analysis also indicates a number of candidate transcription factor binding sites adjacent to occupied EREs at frequencies much greater than by chance, including the previously reported FOXA1 sites, and demonstrate the potential involvement of one such putative adjacent factor, Sp1, in the global regulation of ERα target genes. Unexpectedly, we found that only 22%–24% of the bona fide human ERα binding sites were overlapping conserved regions in whole genome vertebrate alignments, which suggest limited conservation of functional binding sites. Taken together, this genome-scale analysis suggests complex but definable rules governing ERα binding and gene regulation.
Author Summary
Estrogen receptors (ERs) play key roles in facilitating the transcriptional effects of hormone functions in target tissues. To obtain a genome-wide view of ERα binding sites, we applied chromatin immunoprecipitation coupled with a cloning and sequencing strategy using chromatin immunoprecipitation pair end-tagging technology to map ERα binding sites in MCF-7 human breast cancer cells. We identified 1,234 high quality ERα binding sites in the human genome and demonstrated that the binding sites are frequently adjacent to genes significantly associated with breast cancer disease status and outcome. The mapping results also revealed that ERα can influence gene expression across distances of up to 100 kilobases or more, that genes that are induced or repressed utilize sites in different regions relative to the transcript (suggesting different mechanisms of action), and that ERα binding sites are only modestly conserved in evolution. Using computational approaches, we identified potential interactions with other transcription factor binding sites adjacent to the ERα binding elements. Taken together, these findings suggest complex but definable rules governing ERα binding and gene regulation and provide a valuable dataset for mapping the precise control nodes for one of the most important nuclear hormone receptors in breast cancer biology.
Introduction
Cellular transcriptomes are dictated by complex interactions between signal transduction pathways, general and specific transcription factors, chromatin remodeling proteins, and the RNA polymerase complexes. Precise transcriptional responses are achieved in part by targeting transcription factor complexes to the cis-regulatory regions of target genes via specific binding site sequences. The importance of these binding site sequences is reflected in the conservation of sequence motifs in coregulated genes and through evolution.
A number of studies have examined transcriptomic changes to breast cancer cells following estrogen treatment [1–7]. Estrogen receptors (ERs) (specifically ERα and ERβ) are ligand-dependent transcription factors that mediate cellular responses to hormone exposure in vertebrate development, physiological processes, and endocrine-related diseases. ERα, in particular, has been implicated in the etiology of breast cancer and is a major prognostic marker and therapeutic target in disease management [8]. At the molecular level, ERs interact either directly with genomic targets encoded by estrogen response elements (EREs) (5′-GGTCAnnnTGACC-3′) or indirectly by tethering to nuclear proteins, such as AP-1 and Sp1 transcription factors [9–11]. The mechanisms of ER binding site specificity, however, are not clear since these binding site sequence motifs are ubiquitous in the genome, and there is no discernable difference between functional and nonfunctional sites by computational modeling approaches. This ambiguity is likely due to a lack of systemic information on binding site usage and architecture and mechanistic complexity involving additional transcription factors and epigenetic modifications [12,13].
Chromatin immunoprecipitation (ChIP) assays have facilitated characterizations of in vivo protein-DNA complexes such as histone modifications and recruitment of transcription factor complexes to specific binding sites [14]. Coupled with microarray technology, the ChIP-on-chip experiments have resulted in more global binding site maps for a number of human transcription factors in proximal promoters and in specific genomic regions. For example, Carroll and colleagues recently mapped ER binding sites first in human Chromosomes 21 and 22 and more recently across the entire human genome [15,16]. In spite of the success of ChIP-on-chip studies, there remain caveats regarding probe specificity and performance, including constraints on probe design in certain genomic regions and potential biases introduced by amplification protocols. Thus, the analysis of ERα binding sites using alternative genome-wide technologies is warranted.
Previously we developed a high throughput cloning and sequencing approach for mapping full-length transcripts. By employing specialized cloning techniques and vectors, paired-end diTags (PETs) from ends of transcripts and this gene identification signature (GIS) can be sequenced and mapped precisely to the genome [17]. The GIS-PET technique increases sequencing efficiency by 30-fold as compared to sequencing the entire transcript insert. We subsequently showed that binding site fragments from ChIP experiments can also be subjected to PET analysis to generate an unbiased whole-genome map of p53 tumor suppressor protein binding sites and demonstrated an association of binding sites and adjacent target genes with p53 functions in patient tumor samples [18]. The PET technology has also been utilized to study OCT4 and Nanog binding sites in stem cell biology [19].
To obtain a global map of ERα binding sites in breast cancer cells, we applied ChIP-PET to generate a library of ERα binding sites in MCF-7 cells following estrogen treatment. We then combined the binding site data with hormone-responsive and breast tumor sample microarray gene expression studies to discern correlations between ER binding, transcriptional activity, and disease status and outcome in patients. We also compared our findings with data from ChIP-on-chip studies to evaluate the performance of each respective technology. Herein, we describe the comprehensive cartographic results and outline the insights they provide into binding site usage and molecular mechanisms of ER transcriptional regulatory functions.
Results
ChIP-PET Analysis Mapped ER Binding Sites across the Human Genome
Hormone-deprived MCF-7 cells were treated with 10 nM estradiol for 45 min, and then DNA-bound receptor complexes were isolated through ChIP using anti-ERα antibodies (HC-20, Santa Cruz Biotechnology, http://www.scbt.com). Prior to generating the PET library for sequencing, we qualified the ChIP products by measuring DNA fragment size and enrichment of known ER binding site in the pS2/TFF1 gene promoter after immunoprecipitation to ensure sample quality. The ChIP DNA fragments ranged from 300 bp to 1 kb, and there was an 80-fold enrichment of ER binding at the known pS2/TFF1 ERE as compared to the irrelevant antibody control. Once ChIP DNA quality had been confirmed, the PET library was constructed and sequenced as described previously [18]. A total of 635,371 PETs were sequenced, of which 361,241 (~56.86%) were unambiguously mapped to unique loci in the human genome (hg17/NCBI build 35) and localized to 136,158 distinct genomic coordinates. One of the first questions we asked was whether the sequencing of these 635,371 clone “equivalents” in the form of pair-end tags provided sufficient representation of the chip library. To assess the degree of saturation of the PET library sequenced (the total number of distinct ChIP DNA fragments that can be captured from the library), we fitted a Hill Function [20] using extrapolated and historical sequencing data (see Figure S1). The degree of saturation of the ER ChIP PET library is estimated at 73.24% (136,158 actual/185,915 expected), suggesting that ~73% of the extrapolated hypothetical limit of coverage by our library was sequenced. Sequencing results are summarized in Figure 1A. Overlapping uniquely mapped PETs that form PET clusters (see Figure 1B) have been shown to be highly enriched for “true” binding sites [18]. We previously set the selection parameter (i.e., number of PETs within a given cluster) for high probability binding regions by using the goodness-of-fit analysis employing a mixture of two standard Pareto distributions to model the signal and noise within the dataset [18]. MCF-7 cells, however, pose a special analytical challenge in that regions of gene amplification in the cell line also appeared to amplify cluster PET numbers from low quality binding sites (Figure S2). Therefore, we devised an alternative strategy that normalizes regions of gene amplification so that all chromosomal regions can be directly compared. Using this “adaptive maximum overlap PET (moPET) threshold” approach (unpublished data) and setting the false-positive rate of <0.01, 1,234 moPET3+ ER binding site clusters were then defined as high quality binding regions and were used for all subsequent analysis. Among the high quality binding regions, 45% are moPET3 clusters, another 20% are moPET4s, and the remaining 35% have five or more PETs in overlap regions within the clusters (see Figure 1C). An indication that the ChIP-PET experiment and the analytical methods were properly executed is that many known ER binding sites, including the pS2/TFF1, GREB1, ADORA1, and CYP1B1 EREs are present in the defined set of regions [6,21,22]. The complete list of 1,234 high quality binding regions and their chromosomal location can be found in Table S1.
Figure 1. ChIP-PET Analysis Identified 1,234 High Confidence ER Binding Sites in MCF-7 Cells.

(A) A total of 635,371 PETs were sequenced and resolved into 1,234 binding site clusters after filtering for ambiguous and redundant mapping and local noise in amplified regions of the genome.
(B) This diagram illustrates binding site mapping by a cluster of ChIP-PETs. ER binding site is situated in the region of overlap between the ChIP fragments and their PETs.
(C) All of the 1,234 high confidence clusters have at least three PETs in their region of overlap (moPET3+) with the largest number of clusters being moPET3s.
ERα binding regions defined by ChIP-PET are located in every chromosome in the human genome, with the exception of the Y chromosome, which is not present in MCF-7 cells derived from a female breast cancer patient (see Figure S3A and S3B). When regions of gene amplification are accounted for, the frequency of binding clusters per chromosome generally corresponds to the size and gene density of the chromosome, and ER does not appear to localize to specific chromosomal regions within the genome. Figure S3C shows ER binding clusters distribution relative to the nearest University of California Santa Cruz (UCSC) Known Genes (KG) (see Materials and Methods). Binding regions were mapped to the precise positions relative to the 5′ and 3′ ends of transcripts in the UCSC KG database. Only 5% of the regions map to the proximal promoter regions, defined as 0–5 kb upstream of the transcriptional start site (TSS), where the vast majority of current known EREs have been identified and characterized thus far. The largest fraction (38%) of binding regions map to intragenic regions of transcripts and are localized to introns, whereas 23% are within 100 kb from the 5′ start sites, and 19% are within 100 kb of 3′ polyadenylation sites. Only 20% of the ER binding regions are located in gene deserts where the nearest KG is >100 kb away. These findings initially suggest that DNA-bound ER can interact with the transcriptional machinery through both proximal- and distal-acting mechanisms, and these interactions are not likely to be limited by binding site orientation (5′ or 3′) relative to the TSSs. Intriguingly, functional ER binding sites were rarely in exons and when in exons were in probable untranslated regions. We did not detect any binding regions that mapped to a protein-coding domain of a transcriptional unit. These observations further suggest a dynamic selection of ER binding sites that excludes exonic regions and raise the possibility that transcription factor binding sites (TFBSs) in exons may undergo negative selection during evolution.
The ERE Sequence Motif Is Enriched in Validated ChIP-PET Binding Sites
As a preliminary assessment on the fidelity of the 1,234 high quality binding regions, we considered presence of putative ERE motif as a proxy for a real binding event. A total of 13 base pair sites that were at most two Hamming distance away (i.e., two base deviation) from the consensus ERE (GGTCA-nnn-TGACC) were called putative EREs. Upon scanning the 1,234 binding regions, we discovered that 884 (~71%) binding regions contained at least one ERE-like sequence, 25% encoded putative half-ERE sites, and the remaining 4% bore no ERE sequence motifs whatsoever (Figure 2). To further confirm the validity of the discovered binding regions, we selected 107 out of the 1,234 high quality clusters for further ChIP-qPCR validation (Figure 3A). All clusters containing a full putative ERE (47 sites) showed significant (>2-fold over control) enrichment, and based on this 100% success rate the 884 genomic loci of ER binding containing consensus ERE motifs are highly likely to encompass true ER binding sites. We also tested 37 sites with half EREs and validated ER binding in 84% (31 of 37) of selected sites. A similar success rate was found for the non-ERE ChIP-PET clusters, as 19 out of 23 tested sites (83%) showed binding. The median fold enrichment of the validated sites containing a full ERE was 81-fold, which was considerably higher than the median fold enrichment observed for half- and non-ERE-containing PET clusters (36- and 51-fold, respectively). These results support the idea that EREs tend to encode high affinity ER binding sites whereas the half- and non-ERE binding likely support moderate affinity binding, perhaps through indirect tethering mechanisms. This is further supported by the enrichment of EREs as the number of PETs in the moPET clusters increase, corresponding to higher ChIP efficiency and potentially reflecting the higher affinity binding. The positive gradient of the curve supports the notion that higher moPET clusters are more likely to contain full ERE-like sequences (p = 3.204e−8) (Figure 3B). Thus, the canonical ERE sequences appears to be a hallmark of ER control on a genome-wide scale.
Figure 2. The ERE Sequence Motif Is Enriched in ER Binding Sites.

Overall, 71.6% of the 1,234 high confidence ChIP-PET clusters encode at least one ERE motif, and 3.73% contained no ERE or half site motif.
Figure 3. Putatively Higher Affinity Binding by ER Is Associated with the ERE.

(A) ChIP-PET ER binding sites are validated by conventional ChIP followed by quantitative PCR. Data shown represent average of duplicate experiments. Binding sites are considered validated if the binding ratio (ER ChIP/irrelevant antibody control) ≥ 2. Validated sites are grouped by presence of EREs (allowing for up to two base deviation from consensus), half ERE sites, and no ERE motifs.
(B) The frequency of ERE motifs increase as the size of binding site clusters increase. ERE motifs are only present in 7.05% similarly sized genomic fragments shown as the background.
Out of the ten clusters that failed validation, eight of the loci that were misclassified as true binders belonged to the moPET 3 category, which would be considered in the borderline confident range; one was a moPET 4 cluster, whereas a false positive in a moPET7 was located in an amplified region of the MCF-7 genome on Chromosome 20 (which we have shown overestimates the binding efficiency of a DNA fragment to ER). When adjusted for the frequency of full, half, and no ERE motifs in the PET-defined binding loci, our validation rate for binding calls is 94%. These validation results are in line with our previous whole-genome ChIP-PET analysis of p53, Oct4, and Nanog binding sites [18,19] and compares favorably with other genome-wide technologies such as ChIP-on-chip.
To examine whether ER ChIP-PET binding sites containing putative full EREs harbor transcriptional enhancer activities, we PCR amplified 11 binding sites from MCF-7 genomic DNA and cloned them upstream of the pGL4-TATA luciferase reporter. For negative and positive controls we used pGL4-TATA and pGL4-2ERE-TATA, respectively. These constructs were transiently transfected into hormone-depleted MCF-7 cells, treated with either ethanol or E2 for 18–24 hours, and then assayed for luciferase activity. A total of eight out of 11 ER ChIP-PET binding site reporter constructs tested were E2 responsive (Figure 4A). To show that the transcriptional enhancer activities of the ER ChIP-PET binding sites are mediated via EREs, we mutated the putative ERE motifs in the eight E2 responsive constructs and transfected them into MCF-7 cells. Destroying the putative EREs resulted in either significant reduction or complete loss of estrogen response, thus demonstrating the EREs of the ER ChIP-PET binding sites are responsible for their enhancer properties (Figure 4B).
Figure 4. ERE Sequences in ER ChIP-PET Binding Sites Are Functional Transcriptional Enhancers.

(A) ER ChIP-PET binding sites were cloned into the pGL4-TATA luciferase reporter construct and transfected into MCF-7 cells that have been grown in hormone depleted medium for at least three days. The cells were treated with either ethanol or 10 nM estradiol for 18–24 h before harvesting for luciferase activity. pGL4-TATA and pGL4-2ERE-TATA (two copies of the vitellogenin ERE cloned upstream of TATA box of pGL4-TATA) were used as negative and positive controls, respectively. The cells were also cotransfected with the HSV-TK renilla vector as an internal control for transfection efficiency. The data represent the average of three individual experiments ± standard error of mean. The binding site coordinates and their adjacent genes are: ERE1 and ESR1, Chromosome 6: 152029288–152029705; ERE2 and ESR1, Chromosome 6: 152071268–152071889; ERE3 and FOXA1, Chromosome 14: 37189409–37189699; ERE4 and GREB1, Chromosome 2: 11589053–11589737; ERE5 and GREB1, Chromosome 2: 11621762–11622024; ERE6 and GREB1, Chromosome 2: 11622967–11623504; ERE7 and GREB1, Chromosome 2: 11630097–11630780; ERE8 and PGR, Chromosome 11: 100554271–100554807; ERE9 and PGR, Chromosome 11: 100712072–100712428; ERE10 and CA12, Chromosome 15: 61467060–61467460; ERE11 and TFF1, Chromosome 21: 42669273–42670075.
(B) Putative ERE motifs in ER ChIP-PET binding sites were mutated and examined in transient transfection studies as described in (A).
Comparative Analysis of ChIP-PET and ChIP-on-Chip Data Reveals Differential Binding Site Discovery by These Technologies
Other technologies have been used to map ER binding sites on a genomic scale. Carroll et al. using ChIP and human genome tiling arrays have mapped ER binding sites in Chromosomes 21 and 22 and across the human genome in MCF-7 cells [15,16]. We first compared our mapped ER binding sites to the previously published results in Chromosomes 21 and 22 [15]. In the 1,234 binding sites mapped by ChIP-PET, we detected 36 ER binding sites in these chromosomes and found that 20 binding sites were identified by both techniques (Figure 5A) and 16 sites were unique to ChIP-PET. We then tested ER binding to technology-specific and overlapping sites by conventional ChIP to determine the validity of the mapped sites. We selected 25 sites detected by one technique or by both for further validation by ChIP and quantitative PCR (Figure 5B). The six sites identified by both technologies were validated as bona fide ER binding sites and had the greatest fold enrichment (median ≈ 300×). All nine of the sites discovered only by ChIP-PET were validated compared to nine of the ten selected sites discovered by ChIP-on-chip alone. The median fold enrichment of the sites identified solely by the ChIP-PET approach was higher than that of the ChIP-on-chip (medians ~45× versus ~22×). A total of four of the ten sites discovered only by ChIP-on-chip were also associated with ChIP-PET clusters but not deemed high probability sites since three had two PETs in their cluster, and one site was a one PET singleton (unpublished data). Moreover, these four ChIP-on-chip sites overlapping with lower probability PET clusters had higher fold enrichment for ER binding than the remaining sites with only ChIP-on-chip supporting evidence (~37× versus ~12× median), suggesting that conjoint assignment of sites by the two technologies even at suboptimal thresholds may identify higher quality ER binding sites. To assess the two technology platforms across the entire genome, we also compared the 1,234 ChIP-PET ER binding sites identified in this study to the recently published whole-genome ChIP-on-chip ER binding site map of 3,665 binding sites [16] and found 624 (50.6%) ChIP-PET sites in common with the ChIP-on-chip data and 610 (49.4%) sites unique to the PET technology (see Figure 5C). These results are consistent with the data of Chromosomes 21 and 22 where 44.4% (16/36) of the ChIP-PET sites are unique (see Figure 5A).
Figure 5. Comparison of ER Binding Sites Discovered by ChIP-PET and Published ChIP-on-Chip Experiments Indicate Sites Common to Both Technologies and Platform-Form Specific Sites.

(A) In human Chromosome 21 and 22 studies, 57 ER binding sites were discovered by Carroll et al. [15], and 36 sites were identified in this study. There is an overlap of 20 sites between the two studies.
(B) Validation of select binding sites from both studies by ChIP and qPCR suggest that the common sites (both) are high affinity sites, whereas sites unique to each technology tend to have more moderate affinity for ER.
(C) Venn diagram of the overlap between the 3,665 sites discovered by Carroll et al. and the 1,234 sites identified in this study. The 624 binding sites identified in this study actually correspond to 633 binding regions reported by Carroll et al. [16].
It is likely that the difference between the two platforms are due to lower affinity ER binding sites being more susceptible to constraints and limitations inherent in the detection technologies and to possible differences in biological handling of cell lines in each study. Moreover, there appears to be content differences between the discovery capacity of the two technologies. An inherent disadvantage of the ChIP-on-chip approach is that arrays mask sites that contain repetitive sequences, whereas the output of the ChIP-PET technology is completely unbiased in regard to the presence or absence of repetitive sequences. To this point, we found that ~27.9% of the base pairs in bona fide binding sites discovered by the pair-end tag approach were associated with repetitive sequences, whereas 5.3% of those in Carroll et al. bore repeats. Taken together, these results further suggest that the ChIP-PET and the ChIP-on-chip are complementary methods for the identification of TFBSs in a whole genome manner.
Integration of Binding Site and Gene Expression Data Indicates Diversity in Regulatory Region Architecture and Transcriptional Response
The binding site map denotes ER transcriptional regulatory potential for a large number of genes. To determine the specific transcriptional responses corresponding to estrogen treatment and ER recruitment to cis-regulatory sites in MCF-7 cells, we performed gene expression profiling experiments using Affymetrix U133 microarrays on a time course following estradiol exposure. Differentially expressed genes were selected based on a q-value cut-off of less than 2% using a stringent significance analysis of microarrays analysis algorithm. We identified 802 probe sets, representing 544 unique named genes, whose expression levels were up-regulated in response to 10 nM E2 treatment for 12, 24, or 48 h, and 1,168 probe sets corresponding to 704 unique named genes, were down-regulated following hormone treatment. When combined with the ER binding site mapping data (within 100 kb of and closest to high quality binding regions identified by ChIP-PET), 171 up-regulated genes and 116 down-regulated genes were associated with high confidence ER binding sites. Table S2 contains the complete listing of estrogen responsive genes with adjacent ER binding sites identified in this study.
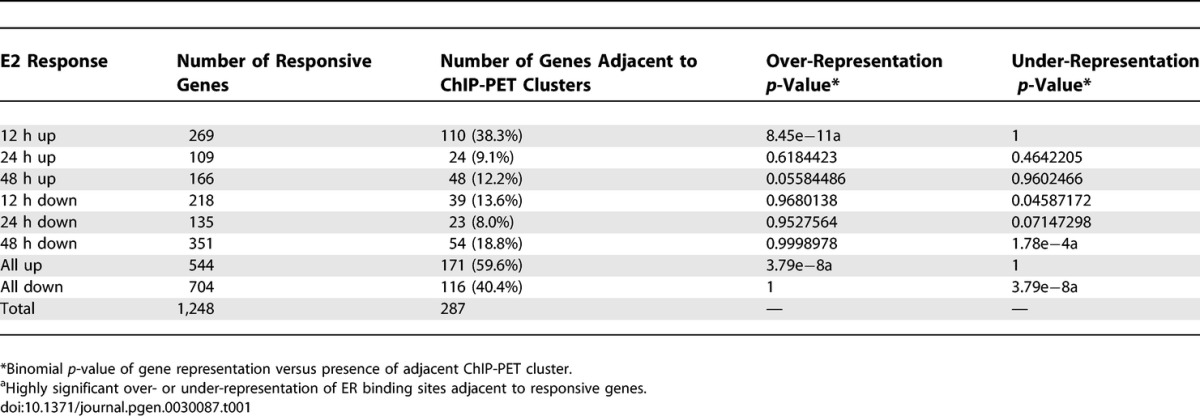
We next examined whether there was a preference for positioning of the ER binding sites in up-regulated versus down-regulated genes. Our analysis revealed that there was a statistically significant association between the presence of an ER ChIP-PET cluster near an up-regulated gene and an under-representation of ER ChIP-PET clusters associated with down-regulated genes (p = 8.379e−08) (see Table 1). The over-representation of ER ChIP-PET clusters that can be associated with E2-up-regulated genes is particularly noticeable at the early 12-h time point. That more binding site clusters are associated with E2 up-regulated genes (60%) compared with down-regulated genes (40%) suggests that the ER protein more frequently is directly involved in the transcriptional regulation of E2 up-regulated genes as compared to down-regulated genes. This finding is in concordance with a previous study we conducted in T-47D human mammary carcinoma cells, where we found that out of 89 genes identified as direct target genes (i.e., E2-responsive and cycloheximide insensitive), 59 (66.3%) were up-regulated and only 30 (33.7%) were down-regulated by E2 treatment [6].
Table 1.
Statistics Showing Enrichment of ER Binding Sites Adjacent to Up-Regulated E2 Responsive Genes
To further confirm the observed association of binding sites with the transcriptional response, we examined the association of the 107 sites validated by conventional ChIP qPCR to E2 regulated gene expression data. Of the 107 sites tested, 22 sites were found to be associated with an E2-regulated probe from the Affymetrix dataset. Out of these 22 sites, 16 were up-regulated by E2 whereas six were down-regulated by E2-treatment. The 16 sites associated with E2 up-regulated probes all showed high levels of enrichment (~25–473×) when analyzed by ChIP qPCR, whereas the six sites associated with down-regulated genes showed lower levels of enrichment (~1–25×) (Figure 6A). These data suggested that some potential mechanistic association exist between high affinity ER binding sites and induction of gene expression by ER.
Figure 6. Comparative Analysis of Binding Site Affinity and Location Adjacent to E2 Up-Regulated Genes Versus Down-Regulated Genes.

(A) Binding site affinity measured by ChIP and qPCR for 22 ChIP-PET sites within 100 kb of E2 responsive genes detected in the microarray studies. Up-regulated genes are denoted in red bars and down-regulated genes in green bars. Each binding site is furthered characterized for the presence of EREs, half EREs, or no EREs.
(B) Locations of binding sites adjacent to up- (blue line) and down- (red line) regulated genes are mapped relative to the transcripts. Relative location to a random set of genes from the UCSC KGs database is included as a reference.
Exploring the association between ER binding sites and the directionality of the transcriptional response further, we mapped the locations of the binding sites relative to the start and termination sites of E2 up- and down-regulated genes as assessed by time course studies using Affymetrix expression arrays. As shown in Figure 6B, binding clusters associated with E2-induced genes are found, significantly above background, 50 kb upstream of the TSS, 50 kb downstream of the TSS within the early introns, and within 25 kb downstream of the termination site of their associated genes. Intriguingly, approximately 35 ER binding sites were found within 5 kb of the transcriptional initiation sites of up-regulated genes (Figure 6B). No such enrichment was detected with down-regulated genes or with genes randomly selected from the UCSC KG database. Taken together, our results suggest that ER binds to many sites in the genome, most bearing a discernable ERE-like sequences, and that genes induced by estrogen are significantly more likely to have an ER binding site within 50 kb of the TSS. Manual analysis of the intronic binding sites showed no evidence for internal alternative TSSs. Genes down-regulated by estrogen show no such positional enrichment and appear to be associated with lower ChIP efficiency ER binding sites. Moreover, genes repressed by estrogen are usually down-regulated later than those induced (48 h versus 12 h; as also observed elsewhere [3,16]). This suggests that genes repressed by ER may require further synthesis and recruitment of other factors to the ER binding sites and that the mechanism of gene repression is topographically distinct from that of gene induction. Supporting this is our observation that binding sites for up-regulated genes have higher moPET counts than binding sites for down-regulated genes (p = 0.0005). When sampled for validation by quantitative PCR, ER binding sites associated with induced genes had much higher fold enrichment for ER occupancy than repressed genes (see Figure 6A).
Genes Adjacent to ER Binding Sites Are Associated with Breast Cancer Biology
We posited that the genes adjacent to the ER binding sites identified by ChIP-PET are putative ER target genes and should reflect ER function in vivo. To assess this possibility, we examined whether the behavior of the collection of adjacent genes could determine the ER status of human breast cancers. All genes within 100 kb of an ER binding site or with an ER binding cluster within an intron were used to cluster 251 breast tumors from a cohort from Uppsala, Sweden previously analyzed for gene expression using Affymetrix U133 A and B arrays [23].
Our results showed that this proximate gene list could easily segregate ER-positive and ER-negative breast tumors (see Figure 7A), whereas a random gene list from the U133 A and B gene set could not (unpublished data). Statistical analysis using the Fisher's exact test showed a highly significant separation based on ER-status with p = 3.914e−12. Moreover, patients with ER-positive like expression profiles, based on the ER-associated genes, have better disease-specific survival over ten years of follow-up as compared to those with the ER-negative like profiles (p = 0.0057) (Figure 7B). This is consistent with all current knowledge of the impact of ER-status in breast cancer prognosis. These results provide strong evidence that the ERα binding sites identified using ChIP-PET enrich for ER responsive genes that are associated with the biology of human breast cancers.
Figure 7. ER Binding Sites Are Adjacent to Genes Associated with ER-Status and Disease Outcome in Breast Cancer Patients.

(A) Expression profiles of genes adjacent to the 1,234 ER binding sites (<100 kb) cluster 260 breast cancer patients into ER+ and ER− groups. ER status is indicated by blue (ER+) and orange (ER−) bars beneath each patient sample.
(B) Kaplan-Meier analysis of disease outcome indicates significantly longer survival for patients with the ER+ profile (red) and compared to those in the ER− cluster (black).
There Is Limited Conservation of ER Binding Site Sequences and Specific ERE-Like Motifs
We have previously shown that ERE sequences in promoter regions of putative ER target genes are not highly conserved, even though both conserved and nonconserved sites appear to be involved in ER binding [6]. Carroll et al., however, indicated conservation of ER binding sites based on their analysis of binding sites discovered in Chromosomes 21 and 22 [15]. To resolve this apparent difference in our observations, we performed comparative analysis of binding site regions and ERE motifs across species using this genome-wide dataset. The overall conservation of a binding region is measured using the base-by-base conservation score and the presence of conserved elements (PhastCons score and PhastCons Conserved Elements [24]), (see Materials and Methods). Using this approach, although a clear conservation signal is visible, as compared to a randomly generated set of regions, the actual proportion of binding regions that are conserved is hidden. To analyze this conservation further, we examined the presence of PhastCons Elements in the ER binding sites. Surprisingly, only 273 (22%) of the initial 1,234 binding regions overlap with such conserved elements, but partitioning the binding sites using this criterion showed that these 22% carry most of the conservation signal (Figure 8). Using size-matched random samples of genomic location we estimate the enrichment of conserved sites to be only approximately 13% (unpublished data).
Figure 8. Most of the Sequences Flanking the 1,234 ER Binding Sites Are Not Conserved through Evolution.

Measure of species conservation at all 1,234 ER binding sites from the center of the ChIP-PET cluster is depicted in the blue line. The green line measures species conservation of randomly selected fragments. The red line depicts the degree of conservation in 22% (273/1,234) of the binding sites bearing conserved elements, and the yellow line shows the degree of species conservation of the remaining 78% (961/1,234) of the binding sites that harbor no conserved elements. These results show that most of the conservation signal is driven by a minority of the binding sites. Conservation was measured by base-by-base comparisons.
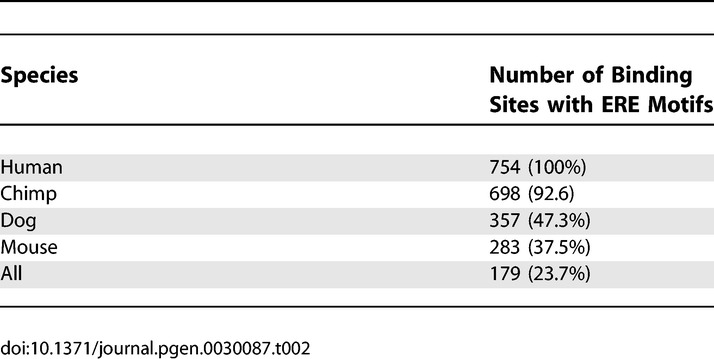
Since TFBS motifs are short, typically 10–20 bp, they may not necessarily be located in a conserved region as detected by standard algorithms. We sought, therefore, to identify ER binding sites with ERE-like sequences and determine whether the specific ERE-like motifs are conserved in homologous regions in chimpanzee, mouse, and dog regardless of conservation of surrounding sequences. We extracted the sequences associated with the 1,234 binding regions in human (hg17) and identified the corresponding homologous regions in chimpanzee (panTro1), mouse (mm5), and dog (canFam2) using the tool liftOver (UCSC Genome Browser utility tool, http://genome.ucsc.edu/cgi-bin/hgLiftOver). We then scanned for the presence of consensus EREs with up to two mutations. Using optimized 500-bp windows in human and chimpanzee and 1-kb windows in mouse and dog (see Table 2), 754 of the 1,234 human binding regions contained a full ERE. As expected, the vast majority (698 or 93%) of the homologous binding windows in chimpanzee also contain ERE-like sequences. The ERE motif is also found in 283 (38%) of the mouse homologous regions and in 357 (47%) of the dog windows. Because the ERE-like sequences are common in any genome, we considered a site bearing a conserved motif only if the ERE is retained in chimp, mouse, and dog. Using this more stringent criterion, we found 179 (24%) of the sites bearing motif conservation in all four species with a background of ~7%. This suggests that ~17% of the sites are under positive selection. Taken together, the sequence and motif conservation results indicate that the majority of binding sites identified in this study are poorly conserved between primates and other mammalian species, and the conservation of binding sites reported previously [15,16] likely resulted from a minority (22%–24%) of highly conserved sites when assessed by multiple conservation metrics. We should note that the actual conservation of binding sites may be higher than observed due to alignment errors [25]. Even with adjustment for this potential error, however, there will likely be a large number of nonconserved ER binding sites.
Table 2.
ERE Motif Conservation in Mammalian Species
ER Binding Regions Are Enriched for Other TFBS Motifs
Though the ERE appears to be the dominant recognition sequence for ER on DNA, other transcription factors and their binding sites are also involved in directing ER to their specific target sites. Indeed, Carroll and colleagues discovered an enrichment of forkhead binding site motifs within ER binding regions of human Chromosomes 21 and 22 and demonstrated a role for the FOXA1 transcription factor in facilitating ER's ability to bind EREs and regulate target gene expression. In other nongenomic based studies, ER is also known to bind DNA indirectly through interactions with Sp1 and AP-1 transcription factors [9,10]. To determine the presence of additional TFBS motifs in the 1,234 ChIP-PET binding sites across the genome, we analyzed the 1,234 cluster sequences for putative TFBS based on TRANSFAC (professional version 9.1) using the accompanying MATCH program [26] with the “minimize False Positive” setup. To compute the statistical significance, we generated a background sequence set, matching the total length of 1,234 clusters, using a third order Markov Chain sequence model trained on the whole human genome (hg17) and scanned them similarly for putative TFBS. This was done 1,000 times. For each TFBS matrix, the average number of sites found per nucleotide represents the background probability of finding its putative sites. The p-values were computed under the binomial distribution and were adjusted for multiple hypotheses testing using the conservative Bonferroni correction. Table 3 lists the top matrices (see Supporting Information for additional details of the analysis).
Table 3.
Transcription Factors with Binding Sites Enriched in the 1,234 ChIP-PET Clusters
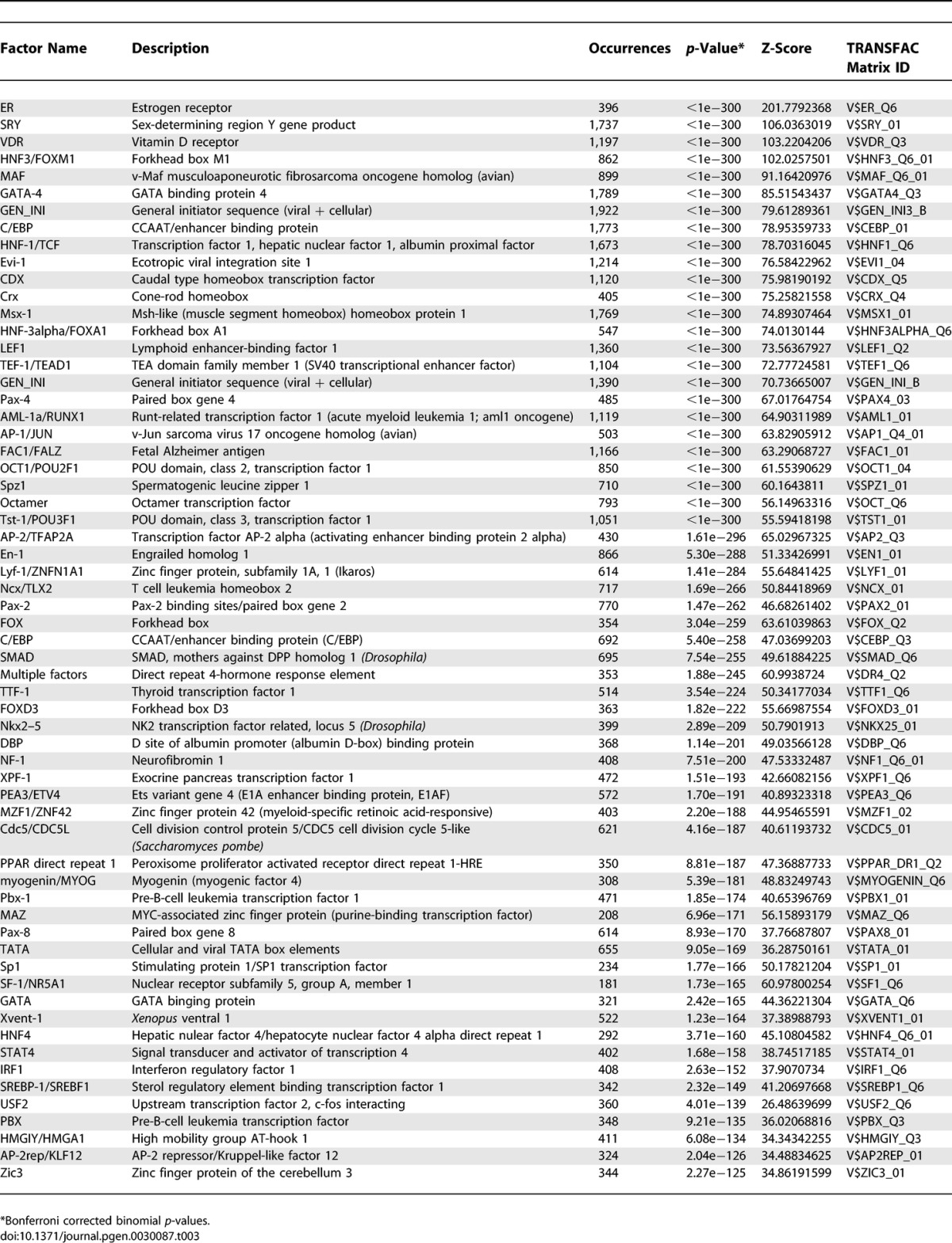
As expected, the predominant sequence motif enriched in ER binding sites is the ERE, Interestingly, however, a large number of transcription-factor binding site motifs are statistically enriched in these ER binding regions even when corrected for multiple sampling suggesting that ER may interact extensively with other transcription factors at the DNA sites. Previous investigations have shown that FOXA1 bound to forkhead binding site motifs adjacent to EREs and interacted with ER, as do AP-1 and Sp1. We also found FOXA1, AP-1, and Sp1 binding motifs significantly associated with the GIS-PET clusters. To further assess the functional significance of detected binding sites, we performed RNA interference experiments using small interfering RNA (siRNA) to knock-down Sp1 and then examined the expression of ten estrogen-responsive genes, which from our data were found to have adjacent ChIP-PET ER binding sites and predicted Sp1 binding sequences (Figure 9). Transfections with Sp1 siRNA constructs reduced Sp1 protein levels by 85% (see immunoblot, Figure 9A) as compared to the luciferase siRNA controls. Sp1 knock-down reduced basal expression levels of all the genes examined and had significant impact on estrogen-responsive induction of MN1, JMJD2B, TBX2, SL9A3R1, CDC6, and KIBRA and more moderate effects on GREB1, IGFBP4, RARA, and MYC as compared to the luciferase and vehicle-treated controls (Figure 9A). In ChIP experiments where we examined four estrogen responsive genes (Figure 9B and 9C), we observed that recruitment of ER to the ChIP-PET site was greatly increased by the presence of estrogen (E2) and that this ER recruitment was reduced after knock-down of Sp1 in three of the four genes (Figure 9B). By contrast to ER, Sp1 was present at a lower level at the ChIP-PET site (note the lower fold recruitment), and the recruitment level of Sp1 was not affected by treatment with E2 (Figure 9C). We also investigated these parameters in three estrogen-responsive genes in which their ChIP-PET region contained an ERE but was not enriched for Sp1 sites. For the three genes assessed (FOS, BCL2, and PGR), we found that E2 treatment increased their gene expression (mRNA level) and that Sp1 knock-down also reduced their gene expression after E2. In these genes, we saw a very low level of Sp1 at the ChIP-PET region that was not altered by E2 treatment (unpublished data). We believe that our observations are in keeping with the fact that these genes all contain Sp1 binding sites close to the promoter, shown previously to be important in their gene regulations [27–30], so that some Sp1 presence and impact of Sp1 knock-down would be expected. Looping of the distal enhancer to a proximal region that binds Sp1 would result in the presence of both ER and Sp1 in our ChIP assays. This might be similar to the direct interaction of a distal signal-specific enhancer binding factor (NF-κB) region with the proximal transcription factor Sp1 binding region, as reported recently for the tumor necrosis factor α-inducible regulation of the monocyte chemoattractant protein-1 gene, MCP-1 [31]. Our findings suggest that the predicted binding sites found in ER ChIP-PET clusters and their associated transcription factors are likely involved in ER-mediated transcriptional regulation, although the extent and impact of their involvement may differ due to gene-specific cis-regulatory architecture and transcription factor complex recruitment, and, in the case of Sp1, interactions between ER-associated effects and promoter proximal regulatory functions.
Figure 9. Impact of Sp1 Knock-Down in MCF-7 Cells on E2 Stimulation of Target Genes Adjacent to ChIP-PET Clusters with Predicted Sp1 Binding Sites.

(A) Cells were transfected with GL3 luciferase siRNA control or Sp1 siRNA constructs 72 h prior to treatment with 0.1% ethanol vehicle or 1nM E2 for 4 h. Expression of target genes was analyzed by quantitative real-time PCR. Values and error bars are based on the mean of three determinations.
(B) Knock-down of Sp1 impacts ERα recruitment to E2-regulated genes. ChIP assays using ERα antibody were performed after transfection of MCF-7 cells with GL3 luciferase siRNA control or Sp1 siRNA for 72 h followed by 45 min treatment with 0.1% ethanol vehicle or 1nM E2.
(C) Sp1 is present at ChIP-PET regions of E2-target genes, but its presence is not affected by E2 treatment. ChIP assays were performed using Sp1 antibodies after 45 min of 0.1% ethanol vehicle or 1nM E2 treatment. Enrichment of ERα or Sp1 at ChIP-PET regions was evaluated by quantitative real-time PCR and normalized to IgG control antibody. Results average two to four independent determinations.
Using these specific validated associations as reference points to assess the relative importance of other associated binding site motifs, we found that 46 other transcription factors are as significantly associated with the 1,234 GIS-PET binding sites as these three validated cis-partners of ER. This suggests that a wider range of transcription factors may partner with ER at the site of DNA binding than previously thought. Given these findings, we next asked whether there was discernable structure within the ER binding sites relative to the transcription factor response elements that are significantly over-represented.
To this end, we assessed whether some motifs were nonrandomly distributed within a 500-bp window encompassing the center of the PET defined binding site or centered on the main ERE or half ERE (Tables S3, S4, and S5). Our results show that factors such as CDX, PAX, AP-1, SF1, and MAF are distributed within these sites in a nonrandom fashion. To dissect the anatomy of these adjacent binding motifs, we examined the specific position of these motifs vis-à-vis the central ERE (Figure 10). We plotted the position of the second transcription factor binding motif against the frequency of such an occurrence and found that SF1, PAX2, PAX3, MAF, and AP-1 co-exist with the central ERE in an ordered manner. Surprisingly, all these factors appear to have significant overlap specifically at the ERE site itself, the most striking being SF1 and PAX3. It did not escape our attention that the observed overlap could be arising from the inherent similarity of the other comotif with ERE or half-site ERE. However, if we use a validated cofactor AP-1 as a model, upon alignment of all the sequences of the AP-1 binding sites and their associated ERE (see Table 4), we discovered an inordinate number of AP-1 sites positioned in the place of a cognate ERE half site. These associated AP-1 sites could represent either truly functional AP1 sites, or degenerate ERE recognition sequences but with sufficient similarity to potentially be recognized by ER, or both. This half-site mimicry by the ERE of other transcription factor binding motifs was seen with MAF/BACH, and most intense with SF1 and PAX3 where a large proportion of those binding sites replace an ERE half site at ER binding regions. This was not seen in other adjacency candidates such as AML-1 where no internal ordering was noted. For CDX where nonrandom order was detected, the structure appeared to be an under-representation within 50 bp to around the EREs. Examination of the consensus binding motifs of these transcription factors reveal that SF-1, BACH/MAF, and PAX3 contain sequences usually just one base different from the ERE half site and could by chance generate an acceptable ERE half site (Figure 11). Moreover, as in the case with AP-1, the 5′ flanking sequences of these sites all contain the AP-1 consensus dinucleotide TG, which renders the ERE half site into a good AP-1 consensus. That these 8- to 13-mer recognition consensus sequences can be so frequently found as part of an ERE suggest that these factors may interact with ER in binding cis-regulatory sequences of target genes.
Figure 10. Nonrandom Positional Distribution of AP-1, SF1, MAF, PAX3, PAX2, CDX, and AML1 (as Negative Control) Binding Sites in the 500-bp Window Centered on the Main ERE.

The y-axis represents the cumulative frequency of the specific transcription factor motif, and the x-axis represents the position of that motif relative to the ERE centered at position 250. Motif hits are marked in red “+” and green “X” indicating forward and reverse strand hits respectively. Multiple hits on the same sequence are depicted as multiple marks on the same y-value sequence.
Table 4.
Alignment of ERE and AP-1 Motifs in ChIP-PET Clusters
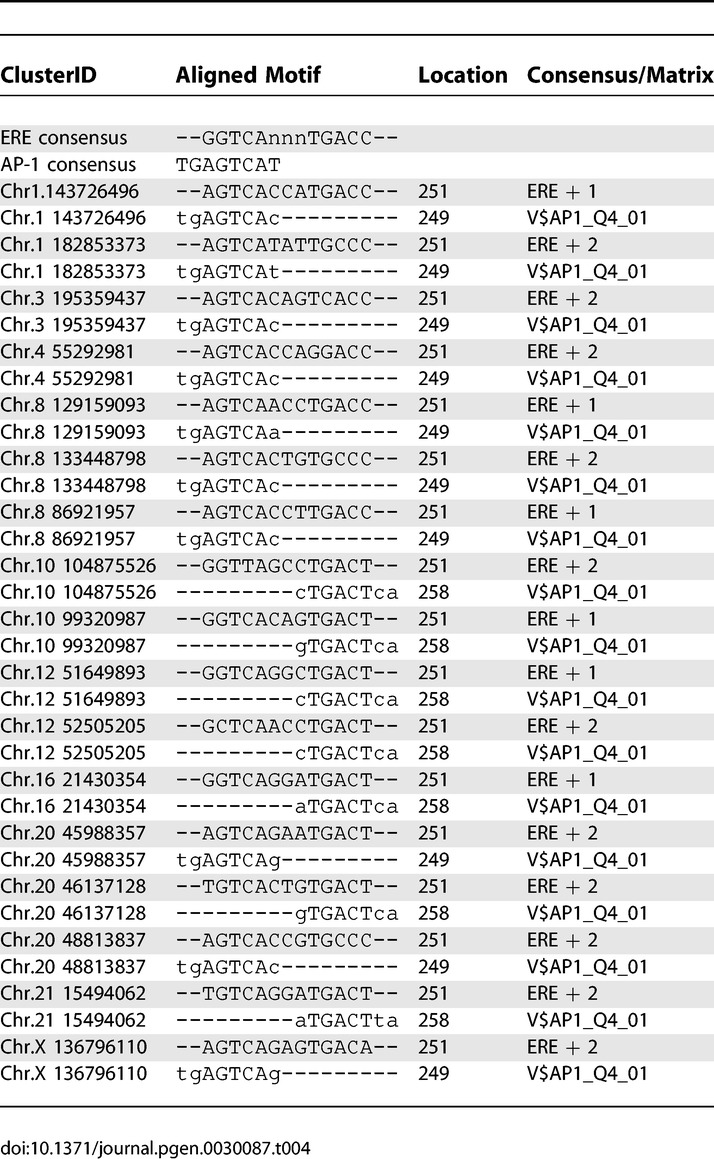
Figure 11. Alignment of TFBSs Enriched in ChIP-PET Clusters with Overlapping Sequence Motifs with the ERE.

The consensus string is a representation of the matrix based on the following rules [48]: A single nucleotide is shown if its frequency is greater than 50% and at least twice as high as the second most frequent nucleotide; a double-degenerate code indicates that the corresponding two nucleotides occur in more than 75% of the underlying sequences, but each of them is present in less than 50%; all other frequency distributions are represented by the letter “n;” the letters in red indicate bases identical to the ERE consensus.
It has been determined that ER can interact with AP-1 and Sp1 factors to regulate gene expression through a tethering mechanism where the DNA binding moiety is AP-1/Sp1. In our genome-wide analysis, we asked whether our ER binding sites without discernable EREs had a predominant transcription factor binding motif. Our results show that predominantly forkhead transcription factors, followed by SRY recognition sequences are significantly enriched in these regions (Table 5). AP-1 sites, though not on the enriched list is however very similar to the MAF recognition sequences, which appear as borderline significant after SRY. Since AP-1 can bind to MAF sites, AP-1 involvement in these purely tethered sites is projected. Thus, surprisingly, ER binding sites without EREs appear highly enriched for recognition motifs for the forkhead family of transcription factors and above that of the known AP-1 interacting factors.
Table 5.
TFBS Enrichment in ER Binding Clusters Lacking ERE Motifs

Discussion
Whole genome analysis of transcription factors provides an unbiased view of their regulatory dynamics. Here we present a genome-wide analysis of the DNA binding sites of ERα as present in the MCF-7 breast cancer cell line and map these sites to transcripts regulated by estrogen. We used a cloning and sequencing based technology and identified 1,234 high probability binding sites using an algorithm that minimizes false positives from amplified regions of the genome. That 94% of a sample of these sites could be validated by standard ChIP suggests that the majority of the 1,234 sites identified by ChIP-PET represent bona fide binding regions for ERα. Of note is that 96% of the validated binding sites harbored either full ERE-like (71%) or solely half-ERE motifs (25%). Only 4% had no ERE-like sequences detectable using a two-position degeneracy cut-off, and therefore a pure tethered mechanism of ER transcriptional regulation must occur infrequently.
This dispersed nature of these 1,234 sites vis-à-vis the TSSs makes the direct molecular assessment of whether these adjacent genes can be regulated by ER highly impractical. We sought to resolve this problem by examining the clinical behavior of these genes adjacent to ER binding sites. We posited that if these adjacent genes were under ER regulation, then their expression in breast cancers should readily determine ER status of primary breast cancers. Our results using a cohort of 251 breast cancers showed that these putative ER regulated genes can significantly separate ER status in breast tumors and therefore represent a transcriptional regulatory cassette that appears to affect ER response. We further examined this question by studying the behavior of these genes in MCF-7 cells as assessed using expression arrays. Though only 23% of the genes proximal to an adjacent ER binding site are responsive following estrogen treatment, this represents a significant enrichment of bona fide ER binding sites adjacent to estrogen responsive versus unresponsive genes (p = 3.018433e − 51) (unpublished data). Therefore, our in vitro and in vivo probabilistic analysis all point to the biological significance of the ER binding sites identified by our ChIP-PET analysis in the regulation of gene expression by ER.
It is important to note the ability of ChIP-PET to identify, in an unbiased manner, bona fide ER binding sites among nearby EREs predicted only by computational methods. For example, for the carbonic anhydrase XII (CA12) gene, matrix-based computational approaches used to identify potential cis-regulatory elements directing ER-regulation of CA12 indicate that five putative EREs reside in the proximal 5′ 5 kb with an additional ERE found in the first intron of the gene. However, we have identified a moPET 5′ binding site approximately 6 kb 5′ to the gene, which was found to be the major regulatory site directing ER-mediated transactivation as a distal enhancer (D.H. Barnett and B.S. Katzenellenbogen, unpublished data). CA12 mRNA is up-regulated by estradiol in MCF-7 breast cancer cells [3,32] and in other ER-positive cells [33] and is positively associated with ERα status in primary breast tumors [34]. Hence, our findings highlight the ability of ChIP-PET to identify previously undiscovered enhancers of biologically relevant target genes.
Much of the research of ER transcriptional regulation has focused on a few EREs located within the proximal promoter. We have shown with our global binding site data that, in fact, the vast majority of sites are located in distal or intragenic regions relative to the nearest regulated transcripts. Our genome wide analysis confirmed the more limited observations previously seen in Chromosomes 21 and 22 that only a small portion (5%) of the binding sites are within 5 kb of the TSS and consistent with our previous predictions [6,35]. Intriguingly, however, detailed analyses revealed that the statistical preponderance of genes responsive in MCF-7 cells to E2 adjacent to ChIP-PET identified ER binding sites were up-regulated rather than down-regulated. Moreover, the location of these sites next to E2 induced genes showed an obvious enrichment around the TSS both in prestart locations and in 5′ introns and within 50 kb from the TSS (Figure 6B). The number of these sites is small when the entirety of genes regulated by ER is considered and therefore would have been missed by a less specific analysis. This distribution of the ER binding sites relative to the induced transcripts indicates diversity in both proximal and distal mechanisms in regulating RNA polymerase activity and suggests that the proposed looping mechanisms [15] may play a more prominent role in ER-mediated transcriptional regulation than previously thought. We have further mapped the entire transcriptome of the MCF-7 cell line using a full-length cDNA library sequencing approach [17]. In sequencing pair-end tags of over 500,000 full-length cDNA equivalents, we found that 13% of the 22,115 individual transcripts identified were novel. When novel transcripts from MCF-7 are accounted for, 90% of the 1,234 high quality ER binding sites are within 100 kb of transcript boundaries (G. Bourque, C.L. Wei, and E.T. Liu, unpublished data). This apparent distance restriction may reflect structural and spatial constraints on the distal effects of the bound ER on promoters.
Equally intriguing is the possibility that ER-mediated gene repression may use mechanisms very different than gene induction, and that genomic topography (i.e., binding site location and affinity) may have a significant role. Consistent with this is our quantification study using ChIP-qPCR on ChIP-PET identified ER binding sites where genes repressed by ER uniformly had ER binding sites that had the lowest fold induction after E2 exposure (~1–25), as compared to those binding sites adjacent to induced genes (~25–473), and were less likely to harbor a full ERE-like motif. When all sites are taken into account and measured by the number of overlapping PETs (moPETs) up-regulated genes have significantly higher moPET counts than down-regulated genes (p = 4.575e − 4, unpublished data). Moreover, in reporter assays performed with the 11 candidate ER binding sites, the only three that did not induce transcription off a TATA promoter were sites associated with repressed genes. These observations are consistent with previous findings that deviations from the full ERE motif reduces ER binding affinity and that the binding site dynamics may differ in genes that are induced by ER than those repressed by ER [11]. The large number of bona fide and nonproximal ER binding sites reported here represents ideal candidates for further characterization of these distinct mechanisms.
It is known that ER can regulate gene expression not by direct DNA binding but through association with an intermediary transcription factor such as AP-1. Theoretically, this mechanism of ER transcriptional regulation does not require ER binding to an ERE. Our motif searches in these non-ERE sites revealed that the predominant motifs in the pure tethered bin are those for the forkhead transcription factors, SRY, with MAF reaching borderline statistical significance (p = 0.056). MAF recognizes sequences related to the AP-1 target site and are considered as part of the larger AP-1 family of transcription factors and, therefore, our results suggest that AP-1 and MAF can bind to these sites [36]. The interesting observation is that in the absence of a minimum of an ERE half site, the fold enrichment of ER binding in these sites is lower (a median of 51-fold enrichment of binding as compared to 81-fold for ERE; Student's t-test p = 0.027). Moreover, our analysis of AP-1 sites within EREs show perfect orientation with one half site with similarities to AP-1 recognition sequences, and the second (cognate) half site primarily an ERE recognition sequence. These “hybrid” sites show higher levels of ER binding. This suggests that AP-1–associated tethering may favor sites with ERE half-site “anchors.” Indeed, previous analysis of the ERE half site associated with the AP-1 site found in the progesterone receptor promoter showed that the integrity of the ERE half site is required for ER and AP-1 binding and estrogen responsive promoter activity [37].
These genome wide approaches to nuclear hormone receptor binding sites are revealing in that the large number of validated binding sites provide statistical power in assessing underlying motif structure in the binding sites. The results of our motif search analysis also point to the potential involvement of a number of other transcription factors participating in ER transcriptional regulatory activity. Included in the list of putative transcriptional coregulators is FOXA1, which has been previously shown to be required for ER functions [15]. However, the fact that 46 other factors are enriched in the ER binding sites with the same probability as the proven interactors of FOXA1, AP-1, and Sp1 suggests the potential for highly complex interactions. Of course not all cis-partner transcription factors will be expressed in every cell type. But though it would be highly improbably that each co-occurrence will predict binding by both factors, our analysis of Sp1 action on ten estrogen-responsive genes with adjacent ChIP-PET ER binding sites and predicted Sp1 binding sequences showed down-regulation of all ten. Moreover, we have validated the effect of adjacent GATA3 and BACH interactions in ER binding to EREs (J. Thomsen and E.T. Liu, unpublished data). This suggests that our algorithms to predict adjacent transcription-factor binding are potentially highly accurate.
Perhaps even more interesting is the systematic order of these potential partner transcription factors relative to the position of the central ERE in bona fide ER binding sites. Consistent with the model where AP-1 binding appears “anchored” by an adjacent ERE is that AP-1 is distributed in a nonrandom manner within a 500-bp window of an ER binding site. In this distribution, the sequence of a number of full EREs are actually composite binding elements with an AP-1 site posing as an ERE half site. These composite EREs are seen with SF-1, MAF/BACH, AP-1, and PAX2 and PAX3. All these factors have recognition sequences that overlap with (but are distinct from) the ERE half site. Unexpectedly, highly skewed positioning was found with the SF-1 and PAX3 recognition sequences (Figure 10), where a large proportion of these response elements are positioned as the second ERE half site within bona fide ER binding sites. Although such overlap may be cues for inherent similarity of the computational model between ER binding sites and other factor binding sites, in the case with SF-1, it has been previously observed that SF-1 response elements can also bind ERα, but not ERβ [38]. Interestingly, SF-1 knock-out mice exhibited ovarian abnormalities and sterility resembling tissues from ER and aromatase knock-out animals, further suggesting an interaction between SF-1 and the ER-estrogen axis [39]. Thus, such composite sites are potential points of exchange for transcription factors possibly switching to and from homodimer and heterodimer states of occupancy and represent a potential mechanism to augment heterogeneous response to estrogen exposure.
We have previously reported very little conservation of ERE motifs within promoter regions of human and mouse genes even though conserved and nonconserved sites both bind ER [6]. In the promoter regions of putative direct target genes, approximately 6% of predicted EREs were conserved in the mouse. In contrast, Carroll and colleagues reported conservation in sequences flanking ER binding sites they experimentally mapped to human Chromosomes 21 and 22 and in their whole-genome study [15,16]. To reconcile these apparent differences, we examined the 1,234 ChIP-PET ER binding sites and determined conservation in both flanking sequences and detected ERE motifs. Using similar analytical approaches as those used by Carroll et al., we also find evidence of conservation within the 500-bp windows around the discovered binding sites. However, a more in-depth analysis showed that the conservation signal observed was driven by only 22% of all sites tested. There was limited conservation regardless of whether local sequence similarity or presence of an ERE motifs were used as the metric for conservation (Table 2). Thus, the conservation also observed by Carroll et al. is likely due to a small number of highly conserved sequences and does not represent global conservation of binding sites [15,16]. We have noted that the conservation may be underestimated due to alignment errors in the comparative analysis of whole-genome sequence data [25], but these errors will not fully explain the large number of nonconserved sites. The list of genes with conserved ER binding sites does not appear to have functional coherence (unpublished data). Genes classically thought of as prototypes of ER responsiveness, such as pS2/TFF1, and the progesterone receptor have bona fide ER binding sites in the human MCF-7 cell line that are not conserved by sequence or motif presence across mammalian species. Moreover, both conserved and nonconserved sites are associated with ER-regulated genes. A total of 287 of all 1,234 binding sites (23.3%) are associated with ER-regulated genes, while 63 of the 273 conserved binding sites (23.1%) are associated with regulated genes (not significantly different).
The limited conservation of ER binding sites does not imply that the genes that are important in ER function are not regulated by ER, but that the precise DNA targets may differ. Given the distance of 100 kb, in which an occupied ERE can potentially regulate its associated promoter, there is much flexibility in the placement of ER regulatory elements. Nevertheless, these observations indicate that there are likely species-specific differences in the components and the dynamics of estrogen action and that results from animal studies need to be interpreted with this caveat in mind.
In summary, our work provides a new cartography of ER binding on a genome-wide scale. The collective configuration of these binding sites has revealed fundamental rules that describe the characteristics of a bona fide ER recognition motif. The dominance of the ERE, the distributed nature of the binding sites distant to their associated genes, the separate nature of up- versus down-regulated genes, the importance of adjacent binding motifs of other transcription factors, and the frequency of composite ER response elements are all findings that would have been difficult to assess on a gene-by-gene basis. Data from this work will provide the experimental targets that will further dissect the intricacies of ER transcriptional regulation.
Materials and Methods
Cell culture and treatments.
MCF-7 cells were grown to 80% confluence in D-MEM/F-12 (Invitrogen/Gibco, http://www.invitrogen.com) supplemented with 10% FBS (Hyclone, http://www.hyclone.com). Cells were washed with PBS and incubated in phenol red-free D-MEM/F-12 medium (Invitrogen/Gibco) supplemented with 0.5% charcoal-dextran stripped FBS (Hyclone) for 24 h in preparation for 17β-estradiol (E2; Sigma, http://www.sigmaaldrich.com) treatment.
ChIP.
Estrogen-deprived MCF-7 cells were treated with 10 nM E2 for 45 min prior to the ChIP procedures. ChIP was carried out as described previously [6] using the HC-20 anti-ERα antibody (Santa Cruz Biotechnology). Following ChIP, DNA fragments were either pooled for PET library generation or analyzed for ER binding at specific sites by real-time PCR. Proper DNA fragment length and ER binding to the known pS2/TFF1 ERE were confirmed by gel electrophoresis and real-time PCR, respectively. ChIP assays using antibodies to Sp1 were performed as previously described [40]. The antibodies used were from Santa Cruz Biotechnology (Sp1 PEP-2, rabbit IgG) and Upstate Biotechnology (Sp1) (http://www.upstate.com). DNA obtained from ChIP was analyzed by quantitative real-time PCR using specific primers for the ER binding sites closest to selected ER-regulated genes.
Real-time PCR.
PCR quantification was performed on the ABI7500 Real-time PCR System (Applied Biosystems, http://www.appliedbiosystems.com) with 20 μl reaction volume consisting of 20 ng of ChIP samples or 20 ng of input DNA as templates, 0.2 μM primer pairs, and 10 μl of 2× SYBR Green PCR Master Mix (Applied Biosystems). For each PCR run, the samples underwent 40 amplification cycles. Fluorescence was acquired at the conclusion of each cycle at 60 °C during the amplification step.
ChIP-PET library construction and sequencing.
Around 140 ng of ChIP DNA were used for construction of the ChIP-PET library for mapping ER binding sites in the human genome, following a procedure described previously [18]. Briefly, End-It DNA End-Repair Kit (Epicentre, http://www.epibio.com) was used to repair the ends of the ChIP DNA. DNA fragments larger than 500 bps were selected by using cDNA size fractionation columns (Invitrogen) and cloned into pGIS-3a vector [18], which contains the Mme I cassettes flanking the cloning site (XhoI). The ligation mixture was transformed into the One Shot Top10 Electrocomp Cells (Invitrogen). A total of 2.3 million clones were obtained. Around 90% of the clones contained inserts. We plated out 1.2 million clones on LB-agar (ampicillin 50 ng/ml) and scraped off the cells for plasmid DNA isolation. Around 10 μg of purified plasmid DNA mixture was digested with MmeI and end-polished with T4 DNA polymerase to remove the 3′-dinucleotide overhangs. The resulting plasmids containing a signature tag from each terminal of the original ChIP DNA insert were self-ligated to form single-PET plasmids. These were then transformed into One Shot Top10 Electrocomp Cells (Invitrogen) to form a “single-PET library.” We plated out 1.2 million clones from this library on LB-agar (ampicillin 50 ng/ml) and extracted plasmid DNA from the cells. Around 250 μg of plasmid DNA were digested with BamHI to release the 50 bp PETs. About 600 ng of single-PETs were PAGE-purified, then concatenated and separated on 4%–20% gradient TBE-PAGE. Appropriate size fraction (600–1,100 bps) of the concatenated DNA was excised, extracted, and cloned into EcoRV-cut pZErO-1 (Invitrogen) to form the final ChIP PET library. The clones were grown on LB-Agar (Zeocin 25 μg/ml). The plasmids were prepared and sequenced using ABI3730 DNA analyzer.
Microarray experiments and analysis.
All microarray experiments were carried out on Affymetrix U133 A and B GeneChips. MCF-7 cells were treated with 10 nM E2 for 12, 24, and 48 h and RNA extraction, labeling, and hybridizations were performed according to manufacturer protocols. Affymetrix analysis software was used to perform the preliminary probe-level quantitation of the microarray data. These data were further normalized using the RMA [41] normalization method. The default option of RMA (with background correction, quantile normalization, and log transformation) was used to generate the normalized intensity for each probeset.
Differentially expressed genes were identified at each time point separately using the three untreated at the time point as controls against the three treated samples. The SAM [42] statistical method was used to select differentially expressed genes. Genes were selected based on the q-value less than 2%. Experiments using patient samples were performed as described in a previous publication [23], and the data used in this study were obtained from the Uppsala cohort from the previous study.
Construction of plasmids and luciferase reporter assay.
ER ChIP-PET binding sites were amplified from MCF-7 genomic DNA by PCR and cloned into the pGL4-TATA vector (a minimal TATA box upstream of pGL4-Basic) by homologous recombination using the In-Fusion CF Dry-Down PCR Cloning kit (Clontech, http://www.clontech.com). Putative EREs were mutated using the QuickChange Site Directed Mutagenesis kit according to the manufacturers instructions (Stratagene, http://www.stratagene.com). MCF-7 cells, grown in hormone-depleted medium for at least 3 d, were cotransfected with the ChIP-PET constructs and HSV-TK renilla with Fugene (Roche, http://www.roche.com). After the cells were treated with 10 nM estradiol or ethanol for 18–24 h, cell lysates were harvested and assessed for firefly and renilla luciferase activity using the Dual Luciferase Reporter Assay system (Promega, http://www.promega.com).
Sp1 RNA interference.
Estrogen-deprived MCF-7 cells were transfected with Sp1 SMARTpool or GL3 luciferase control siRNA (Dharmacon, http://www.dharmacon.com), according to the manufacturer's instructions. After 72 h, cells were treated with 1nM E2 for 4 h. Total RNA was harvested and prepared using Trizol reagent (Invitrogen). Quantitative real-time PCR was performed as previously described [33]. The fold change in expression was calculated using the ribosomal protein 36B4 as an internal control as previously described [3,35]. Primer sequences are available upon request. Proteins were extracted from MCF-7 cells using RIPA buffer, separated on SDS-PAGE, transferred to nitrocellulose membrane, and immunoblotted using anti-Sp1 antibodies (Upstate Biotechnology).
ChIP-PET mapping and primary annotations.
PET sequence extraction and mapping were done as described previously [18,19], using the PET-Tool [43]. The mapped PET sequences were further processed, annotated, and visualized using the T2G genome browser, our in-house genome browser developed based on the UCSC genome browser.
Library saturation analysis.
To assess the saturation of the library, we fitted a Hill Function:
where x is the number of PETs sequenced and f(x) is the number of distinct PETs mapped into the genome among x PETs sequenced. The parameters were chosen to ascertain the completeness of the library and to gain insight on the sequencing effort required for attaining higher saturation level. Using the nonlinear least-square Marquardt-Levenberg algorithm [44] and the historical sequencing data, we obtained a fit with a = 185,915 (±4.362), b = 1.04144 (±2.704e−5), and c = 239,414 (±12.07).
Identification of high quality binding regions.
The underlying aberrant genome of MCF-7 presented an additional challenge in determining which of the ChIP-PET clusters were truly bound by ER. Presence of amplified regions [45], with high and varying copy numbers, increased the probability of those regions being sampled during the ChIP assays, which translated into unusual overall ChIP-PET enrichments in multiple genomic pockets. Relying solely on the raw count of overlapping PETs would introduce undesirable false positives. To address this issue, we have developed a binding region identification algorithm (unpublished data) that produces lower false positives when predicting binding clusters in amplified regions, compared to using raw counts. When assessing the likelihood of a given ChIP-PET cluster being bound by ER, the two-stage algorithm first estimates the amount of noisy PETs surrounding the cluster of interest within its 25-kb flanking regions. Based on the estimated noise level and the neighborhood size (i.e., 50 kb), a moPET cut-off value can be calculated, such that the false positive probability is less than 1e−2. If the given ChIP-PET cluster has a stronger overlapping region (i.e., higher moPET value) than the calculated cut-off, we consider the cluster to be truly bound by ER.
Comotif analysis.
The rich presence of putative EREs points to the canonical and dominant theme of direct ER-DNA interaction. Nevertheless, ER interplays with other transcription factors have previously been reported and are expected, for it to exert wider and more diverse regulatory roles. These high quality binding regions present an unprecedented opportunity for the study of regulatory partners of ER. We employed a three-pronged approach to mine the binding regions for potential enrichment of binding motifs of other transcription factors, where the first assessed the enrichment of certain motifs in a given set of sequences, the second tested whether putative motifs of other transcription factors exhibited certain spatial correlation with respect to the main ERE or half ERE motif, and lastly the Genomatix suite was used for a low-throughput high quality semi-automatic assessment and visualization of potential comotifs.
For the first and second sets of analysis, putative binding sites were identified based on weight matrices available in TRANSFAC (professional version 9.1) and using the accompanying MATCH program [26] with the “minimize False Positive” configuration. To compute the statistical significance of motif enrichment in a given set of sequences, a background sequence set, with its total length matching that of the sequence set, was generated using a third-order Markov Chain sequence model (trained on the whole human genome [hg17]) and was similarly scanned for putative TFBS. This was done 1,000 times, and for each TFBS matrix, the average number of sites found per nucleotide represents the background probability of finding its putative sites. The p-values for motif enrichment were computed under the Binomial distribution and were adjusted for multiple hypotheses testing using the conservative Bonferroni Correction procedure. Evaluation of spatial correlation between main ERE or ERE half sites was carried out using Kolmogorov-Smirnov test. A 500-bp sequence window was defined for each binding region, centering on its main ERE or ERE half site. The putative binding sites of each transcription factor were tested whether they were uniformly distributed within the sequence window.
Conservation analysis.
PhastCons scores are base-by-base values between 0 and 1 that give a measure of evolutionary conservation in eight vertebrate genomes (human, chimp, mouse, rat, dog, chick, fugu, and zebrafish) based on a phylogenetic hidden Markov model, phastCons [24], and Multiz alignments [46]. PhastCons Conserved Elements identify regions of the genome with high conservation scores. These tracks were obtained through the UCSC Genome Browser [47]. A binding site is identified as sequence conserved if its overlapping region overlaps any PhastCons Conserved Elements. Motif conservation analysis was carried out as follows: (1) sequences centered on the middle of the overlapping region of the 1,234 binding regions in human (hg17) were identified; (2) corresponding homologous regions in chimpanzee (panTro1), mouse (mm5), and dog (canFam2) were identified using the tool liftOver (UCSC Genome Browser utility tool); (3) corresponding fasta sequences were extracted; and (4) all sequences were scanned for the consensus ERE motif allowing for two mismatches. Process was repeated for various window sizes in human varying from 250 bp to 5 kb (unpublished data).
Supporting Information
The number of distinct unique PETs of the library, representing nonredundant information, is plotted against the number of PET sequenced (in chronological order) to attain it. A Hill Function, shown adjacent to the curve, is then fitted to the empirical data to estimate the total number of distinct PETs attainable should the PETs continue to be sequenced indefinitely. From that estimate, we conclude that the ER ChIP PET library used in this study reaches a saturation level of 73.24%.
(1.2 MB AI).
(1.2 MB AI).
(A) Comparison of number of ChIP-PET binding sites (open bars) to chromosome size (closed bars) is presented.
(B) Binding site distribution (open bar) as compared to gene density (closed bars) on each chromosome is presented.
(C) Location of ER binding sites relative to the nearest genes in the UCSC KG database shows a large majority of sites distal to the genes (>5 kb) or within intragenic regions.
(1.3 MB AI).
(210 KB XLS)
(47 KB XLS)
The Kolmogorov-Smirnov Test was employed to test whether the observed putative binding sites locations follow uniform distribution.
(15 KB XLS)
(16 KB XLS)
(15 KB XLS)
Acknowledgments
We thank Senali Abayratna for generating the pGL4-TATA and pGL4-2ERE-TATA constructs used in the study.
Abbreviations
- ChIP
chromatin immunoprecipitation
- ER
estrogen receptor
- ERα
estrogen receptor α
- ERE
estrogen response element
- GIS
gene identification signature
- KG
Known Gene
- moPET
maximum overlap PET
- PET
paired end diTag
- siRNA
small interfering RNA
- TFBS
transcription factor binding sites
- TSS
transcriptional start site
Footnotes
¤ Current address: Department of Microbiology and Molecular Biology, Brigham Young University, Provo, Utah, United States of America
A previous version of this article appeared as an Early Online Release on April 17, 2007 (doi:10.1371/journal.pgen.0030087.eor).
Author contributions. CYL, VBV, BSK, YR, and ETL conceived and designed the experiments. CYL, JST, TZ, SLK, MX, DHB, FS, AY, YKL, NP, LDM, EC, YR, and CLW performed the experiments. CYL, VBV, JST, KPC, LL, FS, JG, VAK, THC, LDM, EC, BSK, GB, CLW, and ETL analyzed the data. VBV, JST, DHB, FS, VAK, NP, LDM, EC, YR, GB, and CLW contributed reagents/materials/analysis tools. CYL, VBV, JST, BSK, GB, and ETL wrote the paper.
Funding. This study was supported by the Singapore Agency for Science Technology and Research (A*STAR), the National Institutes of Health grant CA18119 (BSK), and a grant from The Breast Cancer Research Foundation (BSK).
Competing interests. The authors have declared that no competing interests exist.
References
- Charpentier AH, Bednarek AK, Daniel RL, Hawkins KA, Laflin KJ, et al. Effects of estrogen on global gene expression: Identification of novel targets of estrogen action. Cancer Res. 2000;60:5977–5983. [PubMed] [Google Scholar]
- Finlin BS, Gau CL, Murphy GA, Shao H, Kimel T, et al. RERG is a novel ras-related, estrogen-regulated and growth-inhibitory gene in breast cancer. J Biol Chem. 2001;276:42259–42267. doi: 10.1074/jbc.M105888200. [DOI] [PubMed] [Google Scholar]
- Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, et al. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- Gruvberger S, Ringner M, Chen Y, Panavally S, Saal LH, et al. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001;61:5979–5984. [PubMed] [Google Scholar]
- Inoue A, Yoshida N, Omoto Y, Oguchi S, Yamori T, et al. Development of cDNA microarray for expression profiling of estrogen-responsive genes. J Mol Endocrinol. 2002;29:175–192. doi: 10.1677/jme.0.0290175. [DOI] [PubMed] [Google Scholar]
- Lin CY, Strom A, Vega VB, Li Kong S, Li Yeo A, et al. Discovery of estrogen receptor alpha target genes and response elements in breast tumor cells. Genome Biol. 2004;5:R66. doi: 10.1186/gb-2004-5-9-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulez M, Parker MG. Identification of novel oestrogen receptor target genes in human ZR75–1 breast cancer cells by expression profiling. J Mol Endocrinol. 2001;27:259–274. doi: 10.1677/jme.0.0270259. [DOI] [PubMed] [Google Scholar]
- Ali S, Coombes RC. Estrogen receptor alpha in human breast cancer: Occurrence and significance. J Mammary Gland Biol Neoplasia. 2000;5:271–281. doi: 10.1023/a:1009594727358. [DOI] [PubMed] [Google Scholar]
- Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, et al. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- Porter W, Saville B, Hoivik D, Safe S. Functional synergy between the transcription factor Sp1 and the estrogen receptor. Mol Endocrinol. 1997;11:1569–1580. doi: 10.1210/mend.11.11.9916. [DOI] [PubMed] [Google Scholar]
- Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Ng P, Wei CL, Sung WK, Chiu KP, Lipovich L, et al. Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation. Nat Methods. 2005;2:105–111. doi: 10.1038/nmeth733. [DOI] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Kuznetsov VA. Mathematical analysis and modeling of SAGE. In: Wang SM, editor. SAGE: Current technologies and applications. Norwich, United Kingdom: Horizon BioScience; 2005. pp. 139–180. [Google Scholar]
- Stack G, Kumar V, Green S, Ponglikitmongkol M, Berry M, et al. Structure and function of the pS2 gene and estrogen receptor in human breast cancer cells. Cancer Treat Res. 1988;40:185–206. doi: 10.1007/978-1-4613-1733-3_8. [DOI] [PubMed] [Google Scholar]
- Bourdeau V, Deschenes J, Metivier R, Nagai Y, Nguyen D, et al. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol Endocrinol. 2004;18:1411–1427. doi: 10.1210/me.2003-0441. [DOI] [PubMed] [Google Scholar]
- Miller LD, Smeds J, George J, Vega VB, Vergara L, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard DA, Moses AM, Iyer VN, Eisen MB. Detecting the limits of regulatory element conservation and divergence estimation using pairwise and multiple alignments. BMC Bioinformatics. 2006;7:376. doi: 10.1186/1471-2105-7-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, et al. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan R, Porter W, Safe S. Estrogen-induced c-fos protooncogene expression in MCF-7 human breast cancer cells: Role of estrogen receptor Sp1 complex formation. Endocrinology. 1998;139:1981–1990. doi: 10.1210/endo.139.4.5870. [DOI] [PubMed] [Google Scholar]
- Dong L, Wang W, Wang F, Stoner M, Reed JC, et al. Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J Biol Chem. 1999;274:32099–32107. doi: 10.1074/jbc.274.45.32099. [DOI] [PubMed] [Google Scholar]
- Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, et al. Differential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol. 2004;88:113–122. doi: 10.1016/j.jsbmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Schultz JR, Petz LN, Nardulli AM. Estrogen receptor alpha and Sp1 regulate progesterone receptor gene expression. Mol Cell Endocrinol. 2003;201:165–175. doi: 10.1016/s0303-7207(02)00415-x. [DOI] [PubMed] [Google Scholar]
- Teferedegne B, Green MR, Guo Z, Boss JM. Mechanism of action of a distal NF-kappaB-dependent enhancer. Mol Cell Biol. 2006;26:5759–5770. doi: 10.1128/MCB.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, et al. Selective estrogen receptor modulators: Discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004;64:1522–1533. doi: 10.1158/0008-5472.can-03-3326. [DOI] [PubMed] [Google Scholar]
- Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, et al. Transcriptional profiling of estrogen-regulated gene expression via estrogen receptor (ER) alpha or ERbeta in human osteosarcoma cells: Distinct and common target genes for these receptors. Endocrinology. 2004;145:3473–3486. doi: 10.1210/en.2003-1682. [DOI] [PubMed] [Google Scholar]
- van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Vega VB, Lin CY, Lai KS, Kong SL, Xie M, et al. Multi-platform genome-wide identification and modeling of functional human estrogen receptor binding sites. Genome Biol. 2006;7:R82. doi: 10.1186/gb-2006-7-9-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Kyo M, Kamiya T, Tanaka T, Engel JD, et al. Predictive base substitution rules that determine the binding and transcriptional specificity of Maf recognition elements. Genes Cells. 2006;11:575–591. doi: 10.1111/j.1365-2443.2006.00965.x. [DOI] [PubMed] [Google Scholar]
- Petz LN, Ziegler YS, Loven MA, Nardulli AM. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology. 2002;143:4583–4591. doi: 10.1210/en.2002-220369. [DOI] [PubMed] [Google Scholar]
- Vanacker JM, Pettersson K, Gustafsson JA, Laudet V. Transcriptional targets shared by estrogen receptor- related receptors (ERRs) and estrogen receptor (ER) alpha, but not by ERbeta. Embo J. 1999;18:4270–4279. doi: 10.1093/emboj/18.15.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyasuria P, Ikeda Y, Jamin SP, Zhao L, De Rooij DG, et al. Cell-specific knockout of steroidogenic factor 1 reveals its essential roles in gonadal function. Mol Endocrinol. 2004;18:1610–1619. doi: 10.1210/me.2003-0404. [DOI] [PubMed] [Google Scholar]
- Stossi F, Likhite VS, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J Biol Chem. 2006;281:16272–16278. doi: 10.1074/jbc.M513405200. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu KP, Wong CH, Chen Q, Ariyaratne P, Ooi HS, et al. PET-Tool: A software suite for comprehensive processing and managing of Paired-End diTag (PET) sequence data. BMC Bioinformatics. 2006;7:390. doi: 10.1186/1471-2105-7-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerter KE, Munson PJ, McBride WO, Rodbard D. Computerized estimation of size of nucleic acid fragments using the four-parameter logistic model. Anal Biochem. 1990;189:235–243. doi: 10.1016/0003-2697(90)90114-o. [DOI] [PubMed] [Google Scholar]
- Naylor TL, Greshock J, Wang Y, Colligon T, Yu QC, et al. High resolution genomic analysis of sporadic breast cancer using array-based comparative genomic hybridization. Breast Cancer Res. 2005;7:R1186–R1198. doi: 10.1186/bcr1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, et al. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006;34:D590–D598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavener DR. Comparison of the consensus sequence flanking translational start sites in Drosophila and vertebrates. Nucleic Acids Res. 1987;15:1353–1361. doi: 10.1093/nar/15.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The number of distinct unique PETs of the library, representing nonredundant information, is plotted against the number of PET sequenced (in chronological order) to attain it. A Hill Function, shown adjacent to the curve, is then fitted to the empirical data to estimate the total number of distinct PETs attainable should the PETs continue to be sequenced indefinitely. From that estimate, we conclude that the ER ChIP PET library used in this study reaches a saturation level of 73.24%.
(1.2 MB AI).
(1.2 MB AI).
(A) Comparison of number of ChIP-PET binding sites (open bars) to chromosome size (closed bars) is presented.
(B) Binding site distribution (open bar) as compared to gene density (closed bars) on each chromosome is presented.
(C) Location of ER binding sites relative to the nearest genes in the UCSC KG database shows a large majority of sites distal to the genes (>5 kb) or within intragenic regions.
(1.3 MB AI).
(210 KB XLS)
(47 KB XLS)
The Kolmogorov-Smirnov Test was employed to test whether the observed putative binding sites locations follow uniform distribution.
(15 KB XLS)
(16 KB XLS)
(15 KB XLS)