

Abstract
The common ALDH2*2 polymorphism is associated with impaired ethanol metabolism and decreased efficacy of nitroglycerin treatment. These physiological effects are due to the substitution of Lys for Glu 487 that reduces the kcat for these processes and increases the KM for NAD+, as compared to ALDH2. In this study, we sought to understand the nature of the interactions that give rise to the loss of structural integrity and low activity in ALDH2*2 even when complexed with coenzyme. Consequently, we have solved the crystal structure of ALDH2*2 complexed with coenzyme to 2.5 Å. We have also solved the structures of a mutated form of ALDH2 where Arg 475 is replaced by Gln (475Q). The structural and functional properties of the 475Q enzyme are intermediate between those of wild type and the ALDH2*2 enzymes. In both cases, the binding of coenzyme restores most of the structural deficits observed in the apoenzyme structures. The binding of coenzyme to the 475Q enzyme restores its structure and catalytic properties to near wild-type levels. In contrast, the disordered helix within the coenzyme binding pocket of ALDH2*2 is reordered, but the active site is only partially reordered. Consistent with the structural data, ALDH2*2 showed a concentration-dependent increase in esterase activity and nitroglycerin reductase activity upon addition of coenzyme, but the levels of activity do not approach those of the wild-type enzyme or that of the 475Q enzyme. The data presented shows that Glu 487 maintains a critical function in linking the structure of the coenzyme-binding site to that of the active site through its interactions with Arg 264 and Arg 475, and in doing so, creates the stable structural scaffold conducive to catalysis.
INTRODUCTION
Mitochondrial aldehyde dehydrogenase (ALDH2)2 is most commonly associated with its role in ethanol metabolism, catalyzing the oxidation of ethanolderived acetaldehyde to acetate (Table 1) (1). Approximately 40% of the East Asian population has a semi-dominant polymorphism of ALDH2, ALDH2*2, that is essentially inactive in vivo (2). Those individuals who carry the gene for ALDH2*2 experience a disulfiram-like reaction to ethanol consumption, with a more severe reaction observed in individuals who are homozygous for ALDH2*2 (3). This reaction, also referred to as the flushing response, is associated with a low occurrence of alcoholism (4). In recent years, another role for ALDH2 has been identified as a bioactivator of nitroglycerin (GTN) employed in the treatment of angina pectoris (5). Individuals who are hetero- or homozygous for ALDH2*2 also experience reduced efficacy of nitroglycerin in the treatment of angina (6). These studies reveal a correlation between reduced ALDH2 activity and the effectiveness of nitroglycerin.
Table 1.
Reactions Catalyzed by ALDH2.
| Enzyme Function | Example Reaction Equation |
|---|---|
| dehydrogenase | acetaldehyde + NAD+ + Ered → acetate + NADH + Ered |
| esterase | p-nitrophenyl acetate + Ered → p-nitrophenol + acetate + Ered |
| reductase | GTN + Ered → 1,2-GDN + NO2- + Eox |
The low activity of ALDH2*2 is the result of a substitution of lysine for glutamate at position 487 of the 500-amino-acid mature enzyme or position 504 of the 517 amino acid precursor protein (7). This substitution yields an enzyme that exhibits a 200-fold increase in the KM for NAD+ as well as a 10-fold reduced kcat when compared to the wild-type enzyme (8). The KMNAD+ exceeds the in vivo concentration of the coenzyme by 15-fold, which, combined with the 10-fold lower catalytic rate, is responsible for its severely reduced activity in vivo. In addition to these kinetic changes, the ALDH2*2 enzyme also does not exhibit the same pre-steady-state burst of NADH production observed for the wild-type enzyme, which is evidence for a change in rate-limiting step from acyl-enzyme hydrolysis to some step preceding hydride transfer in ALDH2*2 (8).
The crystal structures of the wild-type ALDH2, both as apoenzyme and coenzyme-bound forms, and the ALDH2*2 enzyme in the apoenzyme state have been solved (9-11). In every case the enzymes are tetramers of four identical subunits. The arrangement of the subunits displays 222 symmetry where one of the two-fold axes conceptually divides the enzyme into dimer pairs, and the subunit interface within a dimer pair is distinct from that between dimers. Each subunit in the tetramer is comprised of three domains—the catalytic domain, the coenzyme-binding domain, and the oligomerization domain. The latter of which is important for both dimer and tetramer formation. Like all other known structures of ALDH family members, the binding of coenzyme to the enzyme does not elicit any large conformational changes in the protein structure. However, when bound to ALDH2 the coenzyme is observed to adopt one of two conformations, termed the ‘hydride transfer’ and ‘hydrolysis’ conformations (12-14). The ‘hydride transfer’ conformation is characterized by the nicotinamide mononucleotide (NMN) portion being extended toward the catalytic nucleophile, Cys 302. Whereas, the ‘hydrolysis’ conformation is distinguished by a more contracted NMN, which allows room for the hydrolysis of the acyl-enzyme intermediate (12-14). Magnesium appears to selectively stabilize both conformations through interactions across the pyrophosphate oxygen atoms, and its presence in enzyme assays increases the Vmax of ALDH2 (13,15).
When bound to ALDH, the adenosine portion of NAD+ is nestled between the αF and αG helices within the NAD+-binding pocket. The αG helix is situated at the dimer interface and interacts with the same helix across the interface (Figure 1). In wild-type ALDH2, Glu 487 forms hydrogen bonds with Arg 264 and Arg 475. Arg 264 lies C-terminal to the αG helix and is the first residue in the last β-strand (β11) of the coenzymedomain. Arg 475 is located within a loop in the opposing subunit. These interactions help to stabilize the dimer interface and maintain the integrity of both the NAD+-binding cleft and the floor of the active site (9).
Figure 1.
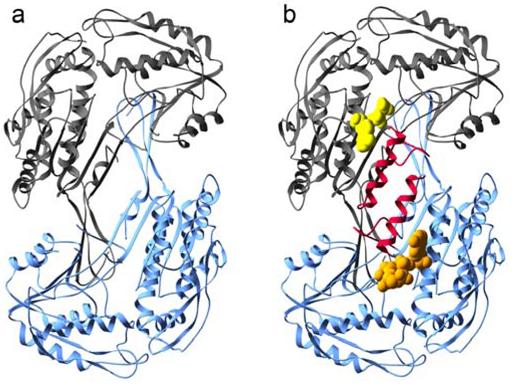
Structures of ALDH2*2. a) Subunits C (dark gray) and D (blue) of the apoenzyme ALDH2*2 structure (PDB ID 1ZUM) which lack ordered αG helices. b) Subunits A and B of the coenzyme-bound ALDH2*2 structure with the αG helices colored red. The bound coenzyme molecules are represented using space-filling atoms. The ordered portion of the NAD+ molecule in subunit A, modeled as ADP, is shown in yellow, and the bound NAD+ molecule in subunit B is shown in gold.
In the ALDH2*2 apoenzyme, the presence of a lysine at residue 487 disturbs these stabilizing hydrogen bonds and leads to disorder in the αG helix and the loop that includes Arg 475. The loss of these interactions severely compromises the structure of the NAD+-binding site and repositions or disorders several catalytically important residues (11). With the loss of the interactions that stabilize the αG helices at the dimer interface, the coenzyme-binding and catalytic domains rotate away from the interface by 2.5° along an axis that runs between the oligomerization and catalytic domains (11). The structural perturbations are propagated into the active site region of the opposing subunit and lead to a disordering of the general base Glu 268 as well as Glu 399, a residue that helps to properly position the NMN portion of the coenzyme.
We attempted to rescue the ALDH2*2 enzyme by constructing a series of ALDH2 double mutants in which Arg 264 and Arg 475 were separately converted to a glutamine or glutamate in the context of the lysine at 487 (16). The corresponding triple mutant was also created. However, none of the additional mutations restored activity to wild-type levels. An unexpected outcome of this study was that substitution of Arg 475 by Glu or Gln in ALDH2*2 resulted in enzymes that exhibited cooperative binding of NAD+. This was also true when the R475Q mutation was introduced in the wild-type enzyme: this 475Q enzyme exhibited a Hill coefficient of 1.8, a KSNAD+ more than 20-fold higher than wild type, and a 2-fold reduction in kcat. Although, the 475Q mutation does not completely restore wild-type activity levels in ALDH2*2, the mutation-induced cooperativity may shed light on intersubunit communication that gives rise to the observed dominance of Lys 487 in ALDH2*2/ALDH2 heterotetramers (16,17).
Thermal stability assays of ALDH2 475Q have demonstrated that this enzyme is less thermally stable than wild type by approximately 15°C (16). Furthermore, the binding of NAD+ was found to increase the thermal stability of the 475Q enzyme, but had little effect on the stability of wild-type enzyme (16).
In order to study the role of residues 475 and 487 in the maintenance of structural integrity of the coenzyme-binding and active sites of ALDH2, we have solved the crystal structure of ALDH2*2 with bound coenzyme to 2.5 Å resolution and both the apo and coenzyme-bound structures of the 475Q mutant enzyme to 2.15 Å and 2.0 Å, respectively. Overall, these structures more closely resemble wild-type ALDH2 rather than the apoenzyme ALDH2*2. In particular, the main-chain atoms of the αG helix and the loop that includes position 475 are visible in the electron density maps. The structural deficits introduced by the Gln 475 mutation are similar to those produced by the Lys 487 mutation, but the extent of the structural perturbation is less severe. While binding of NAD+ does reconstitute much of the coenzyme binding site in the ALDH2*2 enzyme, the presence of coenzyme alone cannot completely overcome the effects of lysine at residue 487, which leads to the disordering of Glu 268 and Glu 399, residues essential for catalysis and effective coenzyme positioning, respectively. In contrast, the less severe structural perturbations observed in the 475Q mutant can be corrected through coenzyme binding in one subunit, which induces a reordering of the neighboring subunit and thereby confers the observed cooperativity of coenzyme binding. Thus, the Gln 475 mutant appears to be a structural and functional intermediate between that of the wild-type and ALDH2*2 enzymes. These data show that residues 264, 475, and 487 comprise a key structural cornerstone in ALDH2 that can transduce structural and kinetic information between neighboring subunits and underlies the mechanisms that give rise to both allelic dominance in ALDH2*2 and cooperativity in the 475Q mutant.
MATERIALS AND METHODS
Materials
DTT, DEAE-sepharose, p-hydroxyacetophenone, guanidine-HCL, and ethylene glycol were each purchased from Sigma Chemical Co. (St. Louis, MO). N-(2-acetamido)-2-aminoethanesulfonic acid (ACES), MgCl2, and propionaldehyde were purchased from Aldrich Chemical Co., Inc. (Milwaukee, WI). Bio-Gel P-6 DG and Bio-Rad Protein Assay reagent were each obtained from Bio-Rad Laboratories (Hercules, CA). Grade I NAD+ was purchased from Roche Diagnostics Corp. (Indianapolis, IN). PEG 6000 was from Hampton Research (Laguna Niguel, CA), and the 24-well sitting drop trays were from Charles Supper Co., Inc (Natick, MA).
Protein Preparation and Crystallization
Human ALDH2 and ALDH2*2 cDNA was expressed in E. coli BL21 (DE3) cells using the pT-7-7 expression system previously described (8,18). The R475Q mutation was introduced into the pT-7-7 expression system as previously published (16). E268Q and C302S mutant expression vectors were prepared as published (13). All enzymes were purified according to the established procedure (8,11,13), concentrated in an Amicon stirred cell, and either flash frozen in liquid nitrogen for storage at -80°C (ALDH2*2) or stored in 50% glycerol at -20°C (all others). Purification was monitored at 340 nm using a Beckman DU 640 spectrophotometer by following the aldehyde oxidation activity reaction at 25°C in 100 mM sodium pyrophosphate, pH 9.5, containing 10 mM NAD+ and 200 μM propionaldehyde.
To prepare ALDH2*2 for crystallization, the protein was removed from -80°C, thawed, and diluted from 8.4 mg/mL to 7.5 mg/mL with 10mM ACES buffer, pH 6.6. To prepare ALDH2 475Q for crystallization, protein was buffer exchanged on a P-6 DG column into 10mM ACES buffer, pH 6.6. Protein was then concentrated in a YM-30 Amicon Centricon filter device to 8.0 mg/mL and 7.8 mg/mL for the low and high resolution apoenzyme structures, respectively. For the NAD+-bound ALDH2 475Q enzyme, protein was utilized directly from purification, not stored on glycerol or frozen, and concentrated to 7.9 mg/mL. Protein concentrations were determined using the Bio-Rad Protein Assay and bovine serum albumin as the standard.
Sitting drop trays for crystallization were prepared as described (11) and contained the following components: 100 mM ACES, pH 6.6-6.8, 5-10 mM MgCl2, 100 mM guanidine HCl, 15% w/v PEG 6000, and 4-8 mM DTT. The equilibrium drop volume was 3 μL. Trays were stored at 20°C and crystals were harvested at 14-22 days.
Coenzyme was introduced into apoenzyme ALDH2*2 crystals through a two-step soaking procedure. Crystals were soaked for 10 minutes in mother liquor containing 35 mM NAD+ followed by a second 10 minute soak in mother liquor containing 70 mM NAD+. Coenzyme was introduced into apoenzyme ALDH2 475Q crystals through a 2 hour soak in mother liquor containing 10 mM NAD+. Cryoprotection of the crystals was accomplished using a rapid two-step procedure to introduce 19-22% ethylene glycol cryoprotectant. The crystals were immediately flash frozen at 100K in a N2 cryostream.
Data Collection, Processing, and Model Refinement for the NAD+-bound ALDH2*2 Structure
Data were collected at the Advanced Light Source (ALS) at Berkeley National Laboratory (Berkeley, CA) on the Molecular Biology Consortium beamline 4.2.2. The beamline was equipped with a NOIR-1 CCD detector, and the X-ray beam wavelength for data collection was 1.072Å (11,564 KeV). Data were indexed, integrated, and scaled with the D*TREK program suite (19).
The human wild-type apoenzyme ALDH2 structure served as the starting model (20) following its placement into the P1 cell through alignment of the wild-type coordinates with the three ALDH2*2 tetramers in the asymmetric unit (21,22). These coordinates were then used directly for refinement. This model was refined using the Crystallography and NMR System (CNS) suite (23). Model rebuilding was performed using σA-weighted electron density maps viewed through the program O (24). To verify placement of cofactor and reordered sections of the protein, simulated annealing omit maps were generated by excluding all cofactor molecules as well as all residues not included in the coordinates for apoenzyme ALDH2*2 (11). Rfree was evaluated using a randomly chosen 5% of the crystallographic data.
Data Collection, Processing, and Model Refinement for ALDH2 475Q Structures
The apoenzyme 2.75Å data and apoenzyme 2.15Å data were collected on a Rigaku Raxis IIC instrument and at the Argonne National Laboratory Advanced Photon Source BioCars beamline 14-BM-C, respectively. NAD+-bound enzyme data constitute a merged data set where lower resolution (30-2.5Å) data were collected on an Raxis IIC instrument, and higher resolution data (25-2.0Å) were collected at beamline X26C at the National Synchrotron Light Source at Brookhaven National Laboratories. Data collection wavelengths, detectors, and data reduction software are summarized in Table 1. Data were reduced using the HKL program suite (25).
Phases were solved using the molecular replacement method and wild-type apoenzyme as the starting model for apoenzyme. The 2.75 Å apoenzyme 475Q structure served as the starting model for the NAD+-bound enzyme. Molecular replacement was executed using the program AMoRe as implemented in the CCP4 program package (21,26). The resulting models were refined using the Crystallography and NMR System (CNS) suite (23). The 2.15 Å apoenzyme structure proved to be pseudo-merohedrally twinned, therefore, the program Shelx was utilized for refinement of this structure (27). Rfree was evaluated using a randomly chosen 5% of the crystallographic data (low resolution apoenzyme and NAD+-bound enzyme). For the twinned structure, 5% of the data was chosen by thin shells for inclusion in the test reflection set. Model rebuilding was performed using σA-weighted electron density maps viewed through the program O (24).
Structure Analysis
Tetramer, subunit, and domain alignments were performed using the program LSQKAB as implemented in the CCP4 package (21,22). Root-mean-square deviations (RMSDs) were calculated for the Cα atoms present in each alignment. Residues 244-271, 424-425, and 463-478 were omitted for all pairings with apoenzyme ALDH2*2 (PDB ID 1ZUM). In alignments with wild-type ALDH2, NADH-bound ALDH2 (PDB ID 1O02) and apoenzyme ALDH2 (PDB ID 1O05) were used (13,20). To determine the extent of domain reorientation relative to apoenzyme ALDH2*2 and wild-type enzyme, the point of reference was established as the cofactor-binding domain. This domain was aligned to either apoenzyme wild type, NAD+-bound wild type, or apoenzyme ALDH2*2. The rotated ALDH2*2 subunit was then realigned to the compared molecule using either the catalytic or oligomerization domain. The extent of domain rotation is therefore expressed as the amount of rotation that is necessary to align the domain of interest relative to the common positions of the coenzyme-binding domains.
Esterase Activity Assays
The esterase activities of ALDH2 and ALDH2*2 were determined by monitoring the rate of p-nitrophenol formation at 400 nm in 25 mM BES (pH 7.5) with 800 μM p-nitrophenyl acetate as the substrate at room temperature in the absence or presence of added NAD+(15). A pH-dependent molar extinction coefficient of 16 mM-1cm-1 at 400 nm for nitrophenol was used. The concentration of NAD+ was determined by using a molar extinction coefficient of 18 M-1cm-1 at 260 nm.
Nitroglycerin Activity Assays
Purified ALDH2 enzyme (1-2 μg) and ALDH2 mutants (50-100 μg) were incubated for 5-10 minutes at 37°C with 1 μM GTN in 100 mM potassium phosphate buffer, pH 7.5, containing 0.5 mM DTT, and 0-20 mM NAD+. After incubation, the reactions were stopped. Extraction and analysis of GTN and 1,2-GDN were performed as previously described (5).
RESULTS
Structure Solution and Refinement
NAD+ was introduced into the ALDH2*2 crystals through a two-step soaking procedure, after which the crystal diffracted to 2.5 Å. The resulting data has been refined to an Rfree of 0.27 and Rwork of 0.23. Like apo-ALDH2*2, and wild-type ALDH2, NAD+-bound ALDH2*2 is a tetramer of four identical subunits, and three independent copies of the tetramer comprise the asymmetric unit. The lower resolution of the coenzyme-bound structure as compared to the apoenzyme structure (2 Å) is due to the fact that only smaller crystals of apo-ALDH2*2 survived the soaking process, and the data were collected at beamline 4.2.2 located at ALS which has a slightly lower flux than beamline 19-ID located at APS, where the apoenzyme data were collected. Two independent structures of the apoenzyme form of the 475Q mutant were determined from differing space groups. Nearly all forms of ALDH2 crystallize in two related, but distinct space groups: P21 and P212121. Both crystal forms have similar morphology and generally diffract to similar limits of resolution. The P21 crystals exhibit pseudo-C2221 symmetry in which the cell dimensions are similar to the primitive orthorhombic setting. However, approximately 50% of all P21 crystals are pseudo-merohedrally twinned, necessitating special treatment of the data during refinement. In the case of the apoenzyme form of the 475Q mutant, we were unable to obtain data of comparable resolution to the coenzyme-bound structure (2 Å, Rfree and Rwork of 0.24 and 0.20, respectively) except from pseudo-merohedrally twinned crystals. Therefore, we have included both the 2.15 Å pseudo-merohedrally twinned structure refined to an Rfree and Rwork of 0.31 and 0.25, respectively, and the 2.75 Å orthorhombic structure refined to an Rfree and Rwork of 0.27 and 0.23, respectively, since the structures provide supportive and complementary information. All data collection and refinement statistics are provided in Table 2.
Table 2.
Data Collection Statistics for ALDH2*2 and ALDH2 R475Q.
| Enzyme | NAD+ ALDH2*2 | apo R475Q | apo R475Q | NAD+ R475Q |
|---|---|---|---|---|
| Resolution (Å) | 49 - 2.50 | 38 - 2.75 | 25 - 2.15 | 30 - 2.0 |
| X-ray Source | ALS 4.2.2 | Rigaku Raxis IIC | APS 14-BM-C | RaxisIIC / NSLS X26C |
| Total Observations | 405,858 | 276,968 | 664,669 | 1,151,308 |
| Unique Observations | 208,258 | 95,356 | 186,854 | 251,665 |
| Completeness (%) | 97.6 (97.0) | 96.2 (82.0) | 94.6 (77.6) | 97.3 (87.5) |
| < I / σ(I) > | 9.1 (2.0) | 11.9 (2.8) | 14.1 (3.6) | 20.3 (3.5) |
| Rmerge (%) | 5.8 (32.5) | 7.6 (29.8) | 5.2 (18.7) | 6.1 (26.4) |
| Space Group | P1 | P212121 | P21 | P212121 |
| Cell Dimensions | ||||
| a, b, c (Å) | 96, 105, 162 | 141, 151, 177 | 101, 176, 101 | 143, 151, 177 |
| a, ß,γ ( ° ) | 79, 82, 89 | 90, 90, 90 | 90, 95, 90 | 90, 90, 90 |
| Asymmetric Unit | 3 tetramers | 2 tetramers | 2 tetramers | 2 tetramers |
| Structure Refinement | CNS | Xplor / CNS | Shelx (huge) | CNS |
| Rwork (%) | 23.0 | 22.6 | 24.9 | 20.4 |
| Rfree (%) | 27.1 | 26.8 | 31.4 | 23.9 |
| RMSD bonds (Å) | 0.008 | 0.008 | 0.005 | 0.010 |
| RMSD angles ( ° ) | 1.4 | 1.3 | 1.2 | 1.5 |
| Luzzati Coord. Error (Å) | 0.34 | 0.34 | 0.33 | 0.24 |
| NCS RMSD (Å) | 0.065 | 0.086 | 0.343 | 0.091 |
| overall B-factor (Å2) | 58 | 31 | 21 | 31 |
| # of solvent molecules | 1931 | 737 | 1019 | 2646 |
| PDB ID code | 2ONM | 2ONN | 2ONO | 2ONP |
ALDH2*2 with bound coenzyme
Coenzyme Binding
Cofactor is present in all twelve subunits in the asymmetric unit. The cofactor molecule is modeled in the hydrolysis conformation (13) in five of these subunits (subunits B, C, F, G, and H, Figure 2a), while at one standard deviation of the electron density map, the remaining seven subunits lack sufficient density for confident placement of the NMN portion of NAD+. Atoms were considered ordered and were included in the final coordinates if the electron density maps contoured at one standard deviation of the map supported their placement. Therefore, only ADP molecules are modeled in seven subunits (A, D, E, I, J, K, and L) with the position of the diphosphate modeled using the hydrolysis conformation (Figure 2b). In subunits A and D, the electron density for the diphosphates provides evidence for more than a single conformation, with the second conformation consistent with that of the hydride transfer conformation. Even though magnesium was present in the crystallization and crystal soaking conditions, no clearly interpretable density was observed for these cations at or near their wild-type positions where they are bound to the diphosphate group of the coenzyme.
Figure 2.
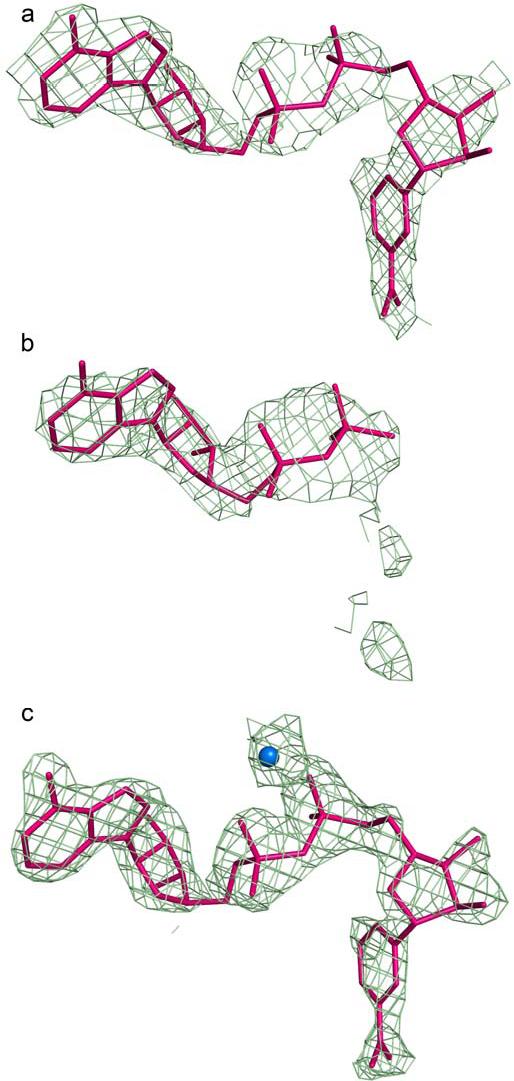
Electron density for coenzyme molecules in ALDH2*2 and ALDH2 475Q. a) A 2Fo-Fc omit map in which residues 240-260, 460-478, and coenzyme were omitted from the structure factor calculations, contoured at one standard deviation, for NAD+ in subunit F of the ALDH2*2 structure. b) A 2Fo-Fc omit map in which residues 240-260, 460-478, and coenzyme were omitted from the structure factor calculations, contoured at one standard deviation, for ADP modeled in subunit I of the ALDH2*2 structure. c) A 2Fo-Fc omit map in which residues 240-260, 460-478, and coenzyme were omitted from the structure factor calculations, contoured at one standard deviation, for NAD+ and magnesium in subunit D of the ALDH2 475Q structure.
Order is Restored to the Dimeric Interface
The most remarkable attribute of the holoenzyme structure is the reordering of both the αG helix at the dimer interface and the loop comprised of residues 463-478. Electron density for nearly all main-chain atoms of these secondary structures is observed, while some side chains are still disordered (Figure 3). Temperature factors for these structural features are elevated 1.5 fold as compared to the remainder of the molecule. While the electron density maps clearly indicate the chain path for these secondary structures, elevated temperature factors for these residues, in addition to missing side-chain density, suggest that the helix and loop possess more flexibility than those same structures within wild-type enzyme. The αG helix is slightly shifted into the cofactor binding site with an average 0.4 Å shift of alpha carbon atoms, as compared to wild-type ALDH2; whereas in apoenzyme ALDH2*2, the same helix is disordered in eleven of twelve subunits, and in the one subunit where the helix is observed, it is shifted as much as 3 Å into the cofactor binding site (11). In concert with the reordering of the αG helix, the overall structure of the holoenzyme tetramer is more similar to the wild-type enzyme, such that the domain rotations observed in the apoenzyme structure are no longer present in the coenzyme-bound structure.
Figure 3.
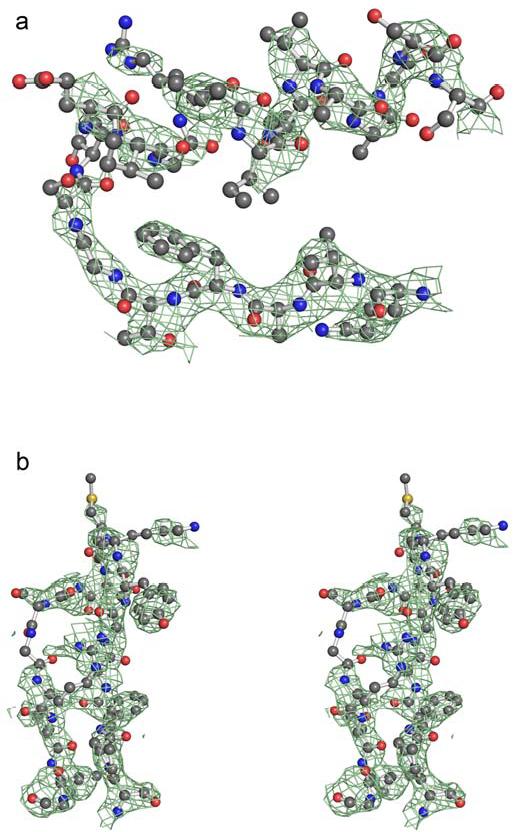
Electron density for the αG helix and loop at the dimer interface of ALDH2*2 complexed with coenzyme. a) A 2Fo-Fc omit map in which residues 240-260, 460-478, and coenzyme were omitted from the structure factor calculations, contoured at one standard deviation, for the αG helix. b) A 2Fo-Fc omit map, contoured at one standard deviation, for the loop comprised of residues 463-478.
Catalytic and Structural Residues
The conformation and observed electron density for Lys 487 is identical to that observed in the apoenzyme structure, with all but the Nζ atom well ordered in most subunits. The side chain of Arg 264 is disordered in most subunits with little electron density for definite placement of its side chain. Arg 475 is also disordered in most subunits, however, it is observed in subunits D and F where the side chain occupies the same conformation as in wild-type ALDH2. In wild-type enzyme, the two αG helices interact through a hydrogen bond between the side chain of Arg 251 in one subunit and the main chain oxygen atoms of residues 259 and 260 in the opposite subunit. In this structure, the side chain of Arg 251 is observed in only two subunits, E and F, where it forms interactions similar to that in the wild-type structure. Within the active site, the catalytic nucleophile, Cys 302, is well ordered and occupies a wild-type-like conformation, yet the side chain for the general base, Glu 268, lacks any interpretable electron density in all but one subunit (B), where the side-chain is pointing toward Cys 302 and its side chain carboxylate is 5.5 Å away from the sulfhydryl group.
In wild-type ALDH2, Glu 399 and Phe 401 support and help stabilize the position of the NMN portion of the coenzyme. In NAD+-bound ALDH2*2, Phe 401 occupies wild-type-like conformations in eleven of the twelve subunits and interacts with the nicotinamide ring in those subunits where it has well-defined electron density. In the remaining subunits (D and L), the side chains are disordered, as sparse electron density does not clearly support either conformation. Glu 399 adopts a wild-type-like conformation in four subunits (C, F, G, and H), each of which demonstrates density for the entire coenzyme molecule. The remaining eight subunits of the NAD+-bound ALDH2*2 structure lack sufficient electron density for definitive placement of Glu 399. Seven of these same subunits also lack clear placement of the NMN of the coenzyme. In two subunits (I and J), Glu 399 is best modeled in the ALDH2*2 apoenzyme-like conformation. Despite the lack of interpretable electron density for Glu 399 in some subunits, the main-chain atoms of residues 269-272 are properly positioned to interact with a wild-type conformation of Glu 399 in all the subunits (Figure 4). In one subunit (L), there is evidence for the rotated conformation of main chain atoms of residues 269-272, although the major conformation is wild-type-like.
Figure 4.
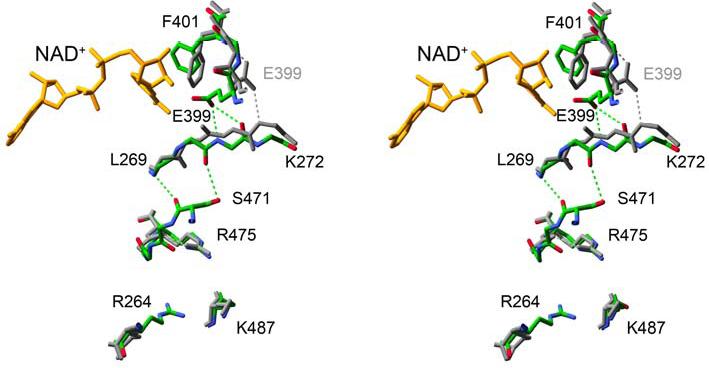
A stereo diagram of the aligned apo- and coenzyme-bound structures of ALDH2*2. The structure of the apoenzyme ALDH2*2 between residue 487 and the active site in the opposing subunit is colored grey with the same region of structure in the coenzyme-bound form of ALDH2*2 colored using atom-type colors. Hydrogen bonds are depicted using dashed lines.
Esterase and Nitroglycerin Reductase Activities in ALDH2*2
Because the dehydrogenase, esterase, and reductase reactions of ALDH2 utilize the same active site (5,28), the reordering of much of the ALDH2*2 structure as a consequence of coenzyme binding led us to examine whether high concentrations of coenzyme could activate the enzyme for the hydrolysis of nitrophenylacetate and the nitroglycerin reductase activity. Both reactions are known to be activated 6-10 fold in the wild-type enzyme by the presence of saturating coenzyme concentrations. In the case of ALDH2*2, the hydrolysis of nitrophenylacetate and the nitroglycerin reductase activity were at or just above the limits of detection for each assay in the absence of coenzyme. However, as observed for the wild-type enzyme (5), the presence of coenzyme activated both reactions in ALDH2*2 (Figure 5). In the absence of added coenzyme, the ALDH2*2 enzyme catalyzed the hydrolysis of nitrophenylacetate 70-fold lower than the wild-type enzyme (4.56 versus 320 nmol/min/mg, respectively). The esterase reaction is activated 36-fold by 20 mM NAD+ in the ALDH2*2 enzyme (165 nmol/min/mg), compared to a maximal activation of 7.2-fold for the wild-type enzyme at 5 mM NAD+ (2,310 nmol/min/mg). The concentration for half-maximal activation was about 150 μM for the wild-type enzyme and about 7,000 μM for ALDH2*2, reflecting the severe deficit in coenzyme binding associated with the ALDH2*2 enzyme. The maximal catalytic rates for ester hydrolysis differ between wild type and ALDH2*2 by about 14-fold, consistent with their 12-fold differences in dehydrogenase activity (9). The ability of ALDH2*2 to catalyze the nitroglycerin reductase activity at physiological concentrations of NAD+ (0.5 mM) is also reduced nearly two orders of magnitude compared to the wild-type enzyme (0.34±0.02 versus 28.1±3.3 nmol/min/mg, respectively). The level of nitroglycerin reductase activity catalyzed by ALDH2*2 at 0.5 mM NAD+ was similar to an ALDH2 mutant in which the general base, Glu 268, was mutated to glutamine (Q268-0.32±0.11 nmol/min/mg), but not as low as a mutant where the catalytic nucleophile was mutated to serine (S302-0.057±0.026 nmol/min/mg). Neither the S302 nor the Q268 mutants show deficits in coenzyme binding (29,30). Consequently, the nitroglycerin reductase rates determined for these enzymes are under conditions of maximal activation. Similar to that observed for the wild-type enzyme (5), the addition of near saturating amounts of coenzyme to ALDH2*2 activated the nitroglycerin reaction (0.22±0.012 nmol/min/mg at 0 mM NAD+ versus 0.93±0.01 nmol/min/mg at 20 mM NAD+). However, the extent of activation (4-fold) was less than that for wild-type enzyme (10-fold) (5).
Figure 5.
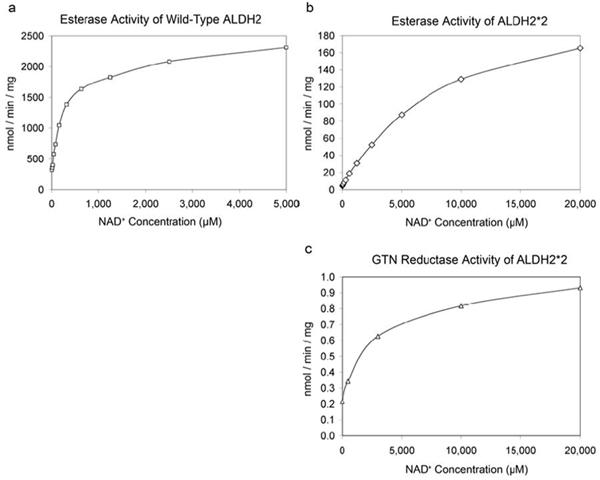
Esterase and nitroglycerin reductase activity of ALDH2*2 as a function of NAD+ concentration. a) The wild-type ALDH2 esterase activity. b) The ALDH2*2 esterase activity. c) The ALDH2*2 nitroglycerin reductase activity.
475Q Mutant Structures
475Q Apoenzyme Structure
The 475Q mutant exhibits weak or discontinuous density for the αG helices and the loop of residues in which position 475 resides, albeit not to the same extent as in ALDH2*2 (Figure 6).
Figure 6.
Electron density for the αG helix in the 475Q mutant. a) A 2Fo-Fc omit map in which residues 240-260 and 460-478 were omitted from the structure factor calculations, contoured at one standard deviation, for the αG helix of the apoenzyme ALDH2 475Q structure (orthorhombic space group). b) A 2Fo-Fc omit map in which residues 240-260, 460-478, and coenzyme were omitted from the structure factor calculations, contoured at one standard deviation, for the αG helix in the coenzyme-bound ALDH2 475Q structure.
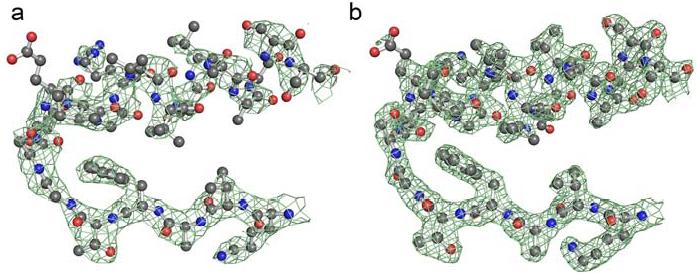
Only the structure from the pseudo-merohedrally twinned data shows interpretable electron density for the Gln 475 side chain where it is oriented towards the side chain of Gln 483 about 4.5 Å away (Figure 7). Both crystal forms exhibit sparse density for the side chain of Arg 264, and the available density is variable (Figure 8b). In contrast, Glu 487 is well-ordered, and occupies a wild-type-like conformation. The side chain of Arg 251, which caps the C-terminus of the αG helix in the opposing subunit, is disordered in most subunits with only discontinuous electron density present for the guanidinium group. Similarly, the general base, Glu 268, displays little or no electron density in most subunits: only one subunit displays complete electron density for 268 where the side-chain carboxyl group is oriented toward and 4.8 Å away from the catalytic nucleophile, Cys 302 (subunit E in the P21 structure). As was observed in the ALDH2*2 apoenzyme, Cys 302 retains a wild-type like position in all subunits. In contrast to the ALDH2*2 apoenzyme structure, the side chains of Glu 399 and Phe 401 are generally well-ordered and the main chain atoms of residues 269-272 retain their wild-type orientations.
Figure 7.
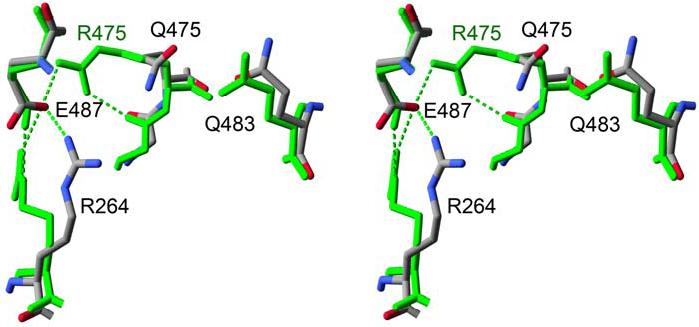
Glutamine 475 in the 475Q mutant. Stereodiagram of the apoenzyme wild-type ALDH2 structure (green) aligned to the ALDH2 475Q apoenzyme structure (atom-type coloring). Hydrogen bonds are shown using dashed lines.
Figure 8.
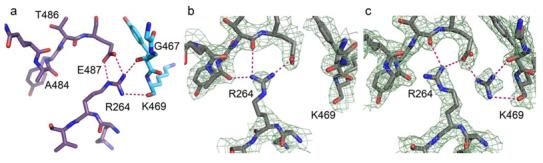
Arginine 264 in the 475Q mutant. a) In wild-type ALDH2, the interactions among Glu 487 and Arg 264 of one subunit (violet) with the main-chain carbonyl oxygen atoms of 469 and 467 in the neighboring subunit (blue). b) A 2Fo-Fc omit map in which residues 240-260 and 460-478 were omitted from the structure factor calculations, contoured at one standard deviation, showing the intrasubunit interactions of Arg 264 in the apoenzyme 475Q structure. c) A 2Fo-Fc omit map in which residues 240-260, 460-478, and coenzyme were omitted from the structure factor calculations, contoured at one standard deviation, showing the intrasubunit interactions of Arg 264 and the guanidine solvent molecule in the coenzyme-bound 475Q structure.
475Q with NAD(H) Bound
NAD(H) is well defined in the electron density maps of all eight subunits and occupies the hydrolysis conformation (Figure 2c) (13). The binding of coenzyme to the 475Q mutant enzyme is associated with the acquisition of a more ordered structure for the main-chain atoms of αG and the loop in which residue 475 resides (Figure 6). Gln 475 is well ordered and is found in two distinct conformations in different subunits. One conformation, as observed in the apoenzyme structure, has the glutamine amide pointing toward 483 (subunits B-H, Figure 7), and the other conformation is oriented toward Arg 264 (subunit A). In all subunits, Arg 264 is well ordered and occupies a conformation in which the side chain is oriented toward the carbonyl atoms of residues 483, 484, and 486 within the same subunit (Figure 8c). This conformation is such that the guanidinium group is close enough to residue 486 and the side chain of Glu 487 to form hydrogen bonds with both of them in nearly all subunits. In addition, another peak of electron density corresponding to the wild-type position for Arg 264 is also observed in each subunit, athough the density is not contiguous with the remainder of the side chain. We have modeled guanidine, which is present in our crystallization buffers, into each of these locations, but we cannot rule out the possibility that these peaks of electron density represent an alternate conformation for Arg 264. This guanidine molecule is unique to the NAD+-bound 475Q structure. Glu 487 and Arg 251 are both well ordered and occupy wild-type-like conformations. As in the apoenzyme structure, Glu 399 and Phe 401 are well-ordered in all subunits and display wild-type like conformations. However, coenzyme binding does not reorder the general base, Glu 268.
DISCUSSION
The structures of both the 475Q and 487K mutant forms of ALDH2 are more severely affected in their respective apoenzyme states, than in their coenzyme-bound states. In both situations, the binding of coenzyme to the apoenzyme form of each mutant rescues the majority of the structural deficits induced by the mutations, and this agrees well with functional measurements of both enzymes. In particular, the consequences of the mutations would suggest that the 475Q enzyme is a structural and functional intermediate between the wild-type enzyme and the ALDH2*2 enzyme. The 475Q enzyme exhibits only a 2-fold reduction in kcat relative to the wild-type enzyme, compared to 12-fold reduction for the ALDH2*2 enzyme, and its KSNAD+ is increased 23-fold over wild type, compared to a 200-fold increase for ALDH2*2. These intermediate functional parameters are consistent with the observed structural changes, as the 475Q enzyme exhibits many of the same disordered regions of protein structure, but to a lesser extent than observed in the ALDH2*2 enzyme.
It is interesting, though, that mutation of position 475 would have a greater effect on coenzyme binding than on kcat, especially when one considers that residue 475 is positioned much closer to the active site than to the coenzyme-binding site (Figure 9a). Mutation of Arg 264 to glutamine, which resides very close to the coenzyme-binding site, affects kcat and the KMNAD+ only 2-fold (16). Thus, the interactions between residues 264, 475 and 487 are not equivalent in their contributions to structural and enzymatic properties of ALDH2 and its mutated forms. This is likely due to the unique arrangement of residues 487 and 475 and the loop in which it resides within the structure of ALDH2. Residues 466-475 are positioned between residues 268 and 270 in one subunit and residues 260-264 in the opposing subunit and forms van der Waals contacts with residues 247 and 251 (Figure 9).
Figure 9.
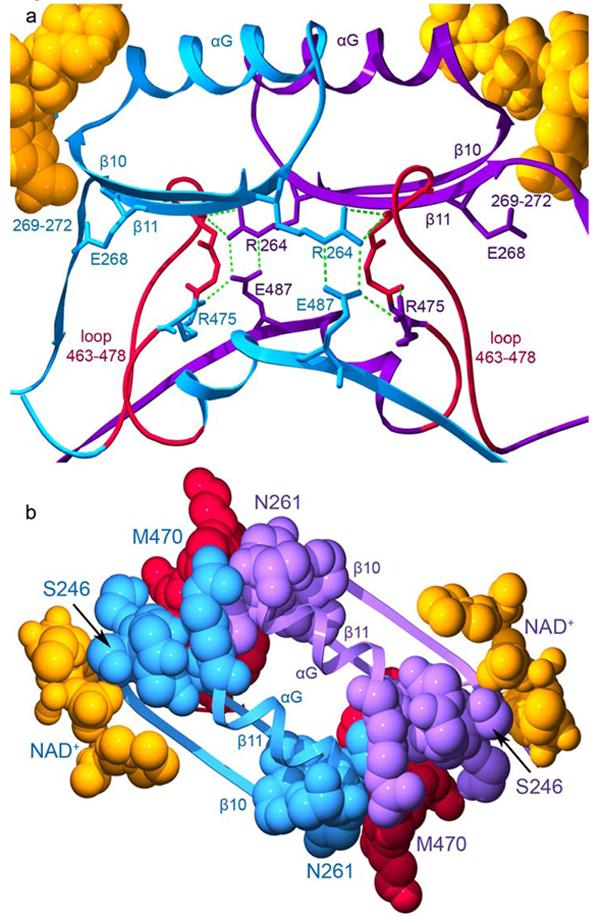
The interactions across the dimer interface contributed by residues 463-478. The structure of the wild-type ALDH2 enzyme with coenzyme-bound (PDB ID 1O02) is used for this representation. Subunits A (blue) and B (violet) are shown. a) The loop comprised of residues 463-478 is shown in red for both subunits with the side chain for residue 475 colored according to its subunit. Hydrogen bonds are represented by green dashed lines. b) Contacts among residues 463-478, the αG helices, and β-strands at the interface. The view in this panel is rotated 90° about a horizontal axis with respect to panel a. The elements of secondary structure are labeled. Residues 246 and 261, which mark the beginning and end of αG are labeled as is residue 470 within the loop which contacts these regions. The bound coenzyme molecules are shown using space-filling atoms and are colored gold.
Position 487 is central to this region of local structure as it serves to anchor and stabilize the contacts between residues 246-270 and residues 466-475. Substitution of this critical glutamate residue to lysine uncouples these regions and leads to the disorder and conformational changes observed in the apo-ALDH2*2 structure (Figure 4). Binding of coenzyme to ALDH2*2 provides sufficient stabilizing interactions to reorder residues 246-270 and through their contact with each other, residues 466-475. However, without Glu 487 to link Arg 264 and Arg 475 to a common, stable structural scaffold, the active site and the NMN-portion of the coenzyme do not achieve sufficient productive encounters to approach wild-type rates of catalysis.
We suggest that the lower catalytic rates toward all substrates are due to either the inability to stably bind the coenzyme in position for hydride transfer or to activate Cys 302. The observed conformational changes then affect both the dehydrogenase reaction, where the bound coenzyme is integral to the chemistry being catalyzed, as well as the esterase and nitroglycerin bioactivation reactions (Figure 5), where the bound coenzyme augments the catalytic rate by increasing the nucleophilicity of Cys 302 (29), but does not participate chemically. These processes might be linked as others have shown that the binding of oxidized coenzyme to ALDH family members is associated with the lowering of the pKa of the active site cysteine from 8.5 to 6.1 (29). In the case of ALDH2*2, the rate-limiting step would not appear to be hydride transfer per se, but some process preceding hydride transfer as the oxidation of chloroacetaldehyde was increased 2-fold over that of acetaldehyde (8). For the esterase reaction, the formation of the initial enzyme-bound intermediate is considered rate-limiting. Consequently, any process that perturbs the reactivity of Cys 302 will diminish the observed catalytic rate.
We could surmise from the structures of the wild-type enzyme in the presence and absence of coenzyme that Arg 475 helps to stabilize the loop in which it resides and that this loop forms the floor of the active site (Figure 9). However, the manner in which its substitution to glutamine induced cooperativity in coenzyme binding could previously only be guessed. Here we see that the substitution of glutamine for arginine at 475 destabilizes the loop in which it resides as well as residues 246-260, which comprise the αG helix (Figure 6). Similar to what is observed for the ALDH2*2 enzyme, the apoenzyme form of the 475Q enzyme lacks appreciable esterase activity (16). Thus, in the absence of bound coenzyme, the reactivity of Cys 302 is also severely impaired. The addition of saturating amounts of NAD+ activates the esterase activity of the 475Q enzyme 75-fold to within a factor of two of the wild-type enzyme activity (16). Similarly, the presence of high concentrations of NAD+ activates the esterase activity of ALDH2*2 36-fold, but this is still an order of magnitude lower than the wild-type activity (Figure 5). Consequently, both the 475Q and ALDH2*2 enzymes achieve greater structural stability upon coenzyme binding, and this increased stability is translated into greater catalytic power. However, because Glu 487 in the 475Q enzyme can still couple Arg 264 to residues 466-475 and provide at least part of the structural scaffold required for catalysis, the maximal catalytic rate is only modestly affected, and the rate-limiting step remains unchanged (16).
The basis for cooperativity in the 475Q enzyme is a consequence of the ability of coenzyme to directly stabilize the position of the αG helix with which it interacts. This binding stabilizes the position of the αG helix from the neighboring subunit. The decreased mobility of these helices reinforces residues 466-475 through the van der Waals contacts at the ends of the αG helices and from the hydrogen-bonds that form between Arg 264 and residues 467-469 when the guanidinium group is correctly positioned by Glu 487 (Figure 9). These interactions increase the affinity for coenzyme in the neighboring subunit and give rise to the observed positive cooperativity.
From these results alone one would be tempted to speculate that it is the ability of Glu 487 to structurally couple Arg 264 to residues 466-475 following coenzyme binding that provides the sole basis for cooperativity. However, mutation of 475 to glutamine in the context of the ALDH2*2 enzyme also produces cooperative binding of coenzyme. Thus, the main effect of cooperativity must be due to the physical stabilization of the adjacent αG helices produced by the binding of the first coenzyme molecule. Yet, the extent to which residues 466-475 are stabilized by the binding of coenzyme influences the observed kcat value, and this is clearly linked to whether or not Lys 487 is present. When Arg 475 is mutated to Gln, either in the context of the wild-type enzyme or the ALDH2*2 enzyme, the resulting enzyme exhibits positive cooperativity toward NAD+ with KS values differing by only 2-fold (16), but the respective kcat value is more similar to the parent enzyme from which each 475Q mutant was derived (8). Thus, the van der Waals contacts provided by the ends of the αG helices are insufficient to completely reorder residues 466-475 without the added ability of Glu 487 to couple Arg 264 to this loop. Therefore, in the absence of Glu 487, residues 466-475 show additional mobility which impacts the stability of residues 268-271 and ultimately residues in and around the NMN binding pocket (Figures 2 & 4). The inability to properly position the NMN moiety and possibly the general base, Glu 268, is thus the physical basis for the reduced kcat values associated with the Lys 487 substitution.
The structural instability associated with residues 466-475, as well as their impact on the mobility of the NMN moiety of NAD+, occurs across the subunit interface from residue 487 (Figure 9). This is the likely explanation for the dominance of the ALDH2*2 allele. We have previously proposed a model where the ALDH2*2 subunit within a heterodimer of a tetramer would inactivate both itself and its neighboring wild-type subunit (17). However, no such inactivation between dimer pairs in a tetramer was observed. The structural explanation for this ‘within dimer dominance’ is likely that Lys 487 leads to structural disorganization of residues 466-475 in the opposing subunit, which negatively impacts the stability of both αG helices in the dimer, which in turn leads to increased mobility of residues 466-475 within the same subunit by virtue of their interactions with the opposing end of the αG helices (Figure 9). Apparently, the presence of a single Glu 487 per dimer is not sufficient to stabilize the structure of either subunit. The kinetic data supports this since the apparent kcat values for the heterotetrameric pairs is proportional to the number of Lys 487-containing subunits (reduced 2-fold, 8-fold, 20-fold and 25-fold for tetramers containing 1, 2, 3 and 4 copies of the E487K mutation, respectively) (17). Thus, the effect on kcat is dominant, while the increase in the KMNAD+ is recessive, since elevated KMNAD+ values were only observed after three or more of the subunits in the tetramer contained the E487K substitution (17). Further explanation for the structural underpinnings of the dominant effect will require structure determination of a heterotetrameric species.
As mentioned above, the ALDH2*2 enzyme has no apparent esterase activity in the absence of added coenzyme, and its ability to catalyze the bioactivation of nitroglycerin is much impaired (Figure 5). Considerably less is known about the actual chemistry of this reaction, but residue exchanges in the enzyme affect the nitroglycerin reductase reaction in a manner similar to the esterase and dehydrogenation reactions, though not to the same extent. Thus, the nitroglycerin reaction utilizes the same catalytic machinery in the ALDH2 active site, but the relative importance of the residues in the reaction chemistry differs. This is also true for the impact that the substitution of Glu 487 by lysine in the ALDH2*2 enzyme has on this reaction. In the absence of coenzyme, ALDH2*2 catalyzes the reductase reaction at a very low rate. The addition of high concentrations of coenzyme activates this reaction about 4-fold, but is still about 30-fold lower than the wild-type enzyme. The response to coenzyme activation is similar to wild type where coenzyme activates the reaction about 10-fold (5), but the instability of the active site and the inability to properly position the coenzyme and fully activate Cys 302 diminishes the catalytic efficiency for the reaction. This observation is supported by the reduced clinical efficacy of nitroglycerin administration associated with the ALDH2*1/2*2 and ALDH2*2/2*2 genotypes during treatment of angina as assessed by relief of angina and by improvements in blood flow (6,30). That the decrease in efficacy is not complete may indicate that the levels of nitric oxide produced by the mutant enzyme under physiological conditions may be close to the clinical threshold or that other enzymes can contribute to the bioactivation reaction (31-33).
CONCLUSIONS
The dimer interface of ALDH2 is intricately interlaced, with residues 264, 487, and 475 playing essential roles in interface stability and subsequent structural integrity of the active site. Disruption of this intersubunit scaffolding, through mutation of either residue 487 or 475, perturbs both the coenzyme-binding and catalytic sites of ALDH2. These residues together with Arg 264 link the two active sites of a dimer through their interactions at the subunit interface, utilizing the αG helices and residues 466-475 to relay structural communication. Both the dominance of ALDH2*2 and cooperativity of 475Q employ this intersubunit conduit, and the differing coenzyme-binding properties of these enzymes are due to the roles of the residues mutated.
The binding of NAD+ to either the ALDH2*2 (E487K) or 475Q enzyme ameliorates the structural disorder observed for the respective apoenzyme structures. However, the structure of ALDH2*2 demonstrates that increased disorder of the NMN moiety of NAD+ likely produces the decreased catalytic activity observed for the dehydrogenase, esterase, and reductase reactions. Although coenzyme binding certainly assists in reordering the ALDH2*2 enzyme, the fact that coenzyme only moderately stimulates nitroglycerin reductase activity is consistent with partial recapitulation of the wild-type structure in ALDH2*2.
ACKNOWLEDGEMENTS
The authors wish to thank Susan Carlson for assistance with the ALDH2 475Q protein purification and Elisabeth Lasater for assistance with the crystallization of ALDH2*2. This work was supported by the National Institutes of Health grant R01 AA 11982. We thank Robert Sweet and the staff of beamline X26C. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. We thank the staff of BioCARS beamline 14-BM-C. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. We thank Jay Nix and the staff of MBC beamline 4.2.2. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. All figures for publication were created using either the PyMol Molecular Graphics program (34) or Deep View Swiss PDB Viewer (35) and Pov-Ray (36).
Footnotes
- ALDH2
- mitochondrial aldehyde dehydrogenase
- ALDH2*2
- semidominant polymorphism of ALDH2
- R475Q and 475Q
- laboratory-created mutant of ALDH2 in which position 475 has been mutated from R to Q
- GTN
- glyceryl trinitrate (nitroglycerin)
- 1,2-GDN
- 1,2-glyceryl dinitrate
- NMN
- nicotinamide mononucleotide portion of NAD+
- ACES
- N-(2-Acetamido)-2-aminoethanesulfonic acid
REFERENCES
- 1.Hurley TD, Edenberg HJ, Li TK. Pharmacogenomics of Alcholism. In: Licinio J, Wong M, editors. Pharmacogenomics: The Search for Individualized Therapies. Culinary and Hospitality Industry Publications Services; 2002. [Google Scholar]
- 2.Seitz HK, Matsuzaki S, Yokoyama A, Homann N, Vakevainen S, Wang XD. Alcoholism: Clinical & Experimental Research. 2001;25(5 Suppl ISBRA):137S–143S. doi: 10.1097/00000374-200105051-00024. [DOI] [PubMed] [Google Scholar]
- 3.Peng GS, Wang MF, Chen CY, Luu SU, Chou HC, Li TK, Yin SJ. Pharmacogenetics. 1999;9(4):463–476. [PubMed] [Google Scholar]
- 4.Higuchi S, Muramatsu T, Shigemori K, Saito M, Kono H, Dufour MC, Harford TC. J Stud Alcohol. 1992;53(2):170–175. doi: 10.15288/jsa.1992.53.170. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Zhang J, Stamler JS. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(12):8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Zhang D, Jin W, Shao C, Yan P, Xu C, Sheng H, Liu Y, Yu J, Xie Y, Zhao Y, Lu D, Nebert DW, Harrison DC, Huang W, Jin L. Journal of Clinical Investigation. 2006;116(2):506–511. doi: 10.1172/JCI26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goedde HW, Harada S, Agarwal DP. Human Genetics. 1979;51:331–334. doi: 10.1007/BF00283404. [DOI] [PubMed] [Google Scholar]
- 8.Farres J, Wang X, Takahashi K, Cunningham SJ, Wang TT, Weiner H. Journal of Biological Chemistry. 1994;269(19):13854–13860. [PubMed] [Google Scholar]
- 9.Steinmetz CG, Xie P, Weiner H, Hurley TD. Structure. 1997;5(5):701–711. doi: 10.1016/s0969-2126(97)00224-4. [DOI] [PubMed] [Google Scholar]
- 10.Hurley TD, Steinmetz CG, Weiner H. Advances in Experimental Medicine & Biology. 1999;463:15–25. doi: 10.1007/978-1-4615-4735-8_3. [DOI] [PubMed] [Google Scholar]
- 11.Larson HN, Weiner H, Hurley TD. J Biol Chem. 2005;280(34):30550–30556. doi: 10.1074/jbc.M502345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore SA, Baker HM, Blythe TJ, Kitson KE, Kitson TM, Baker EN. Structure. 1998;6(12):1541–1551. doi: 10.1016/s0969-2126(98)00152-x. [DOI] [PubMed] [Google Scholar]
- 13.Perez-Miller S, Hurley TD. Biochemistry. 2003;42:7100–7109. doi: 10.1021/bi034182w. [DOI] [PubMed] [Google Scholar]
- 14.Hammen PK, Allali-Hassani A, Hallenga K, Hurley TD, Weiner H. Biochemistry. 2002;41(22):7156–7168. doi: 10.1021/bi012197t. [DOI] [PubMed] [Google Scholar]
- 15.Ho KK, Allali-Hassani A, Hurley TD, Weiner H. Biochemistry. 2005;44(22):8022–8029. doi: 10.1021/bi050038u. [DOI] [PubMed] [Google Scholar]
- 16.Wei B, Ni L, Hurley TD, Weiner H. Biochemistry. 2000;39(18):5295–5302. doi: 10.1021/bi000028n. [DOI] [PubMed] [Google Scholar]
- 17.Zhou J, Weiner H. Biochemistry. 2000;39(39):12019–12024. doi: 10.1021/bi001221k. [DOI] [PubMed] [Google Scholar]
- 18.Zheng CF, Wang TT, Weiner H. Alcoholism: Clinical & Experimental Research. 1993;17(4):828–831. doi: 10.1111/j.1530-0277.1993.tb00849.x. [DOI] [PubMed] [Google Scholar]
- 19.Pflugrath JW. Acta Crystallographica Section D-Biological Crystallography. 1999;55(Pt 10):1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 20.Hurley T, Perez-Miller S, Breen H. Enzymology and Molecular Biology of Carbonyl Metabolism. 2001;130-132:3–14. [Google Scholar]
- 21.Collaborative Computational Project, N. Acta Crystallographica Section D-Biological Crystallography. 1994;50:760–763. [Google Scholar]
- 22.Kabsch W. Acta Crystallographica Section A. 1976;32(5):922–923. [Google Scholar]
- 23.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallographica Section D-Biological Crystallography. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 24.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Cryst A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter J, Sweet RM, editors. Methods in Enzymology, Volume 276: Macromolecular Crystallography, part A. Academic Press; 1997. C.W. [DOI] [PubMed] [Google Scholar]
- 26.Navaza J. Acta Cryst A. 1994;A50:157–163. [Google Scholar]
- 27.Sheldrick G. SHELX: applications to macromolecules. In: Dordrecht FS, editor. Direct Methods for Solving Macromolecules. Kluwer Academic Publishers; 1998. [Google Scholar]
- 28.Feldman RI, Weiner H. Journal of Biological Chemistry. 1972;247(1):267–272. [PubMed] [Google Scholar]
- 29.Marchal S, Branlant G. Biochemistry. 1999;38:12950–12958. doi: 10.1021/bi990453k. [DOI] [PubMed] [Google Scholar]
- 30.Mackenzie IS, Maki-Petaja KM, McEniery CM, Bao YP, Wallace SM, Cheriyan J, Monteith S, Brown MJ, Wilkinson IB. Arteriosclerosis, Thrombosis & Vascular Biology. 2005;25(9):1891–1895. doi: 10.1161/01.ATV.0000179599.71086.89. [DOI] [PubMed] [Google Scholar]
- 31.Ignarro LJ. PNAS. 2002;99(12):7816–7817. doi: 10.1073/pnas.132271799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fung H-L, Bauer J. Cardiovasc Drugs Ther. 1994;8:489–499. doi: 10.1007/BF00877927. [DOI] [PubMed] [Google Scholar]
- 33.Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, Kitagawa K, Nakayama KI, Hess DT, Stamler JS. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(34):12159–12164. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 35.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 36.Persistence of Vision Raytracer. Persistence of Vision Pty. Ltd.; 2004. (Version 3.6) [Google Scholar]