

Abstract
Autophagy is the regulated process by which cytoplasmic organelles and long-lived proteins are delivered for lysosomal degradation. Increased numbers of autophagosomes and autolysosomes often represent prominent ultrastructural features of degenerating or dying neurons. This morphology is characteristic not only of neurons undergoing pathologic degeneration, but also during developmental programmed cell death of some neuronal populations. In recent years, a growing number of reports highlight potentially important roles for autophagy-related processes in relation to protein aggregation, regulated cell death pathways, and neurodegeneration. While starvation-induced autophagy involves nonselective bulk degradation of cytoplasm, mechanisms that regulate selective targeting of damaged organelles form an emerging area. As the study of autophagy evolves from physiologic homeostasis to pathologic situations, consideration of terminology and definitions becomes important. Increased autophagic vacuoles do not necessarily correlate with increased autophagic activity or flux. Instead, the striking accumulation of autophagic vacuoles in dying or degenerating neurons likely reflects an imbalance between the rates of autophagic sequestration and completion of the degradative process. In other words, these cells can be thought of as undergoing “autophagic stress.” The concept of autophagic stress may reconcile apparently conflicting roles of autophagy-related processes in adaptive, homeostatic responses and in pathways of neurodegeneration and cell death.
Keywords: Alzheimer disease, Autophagy, Huntington disease, Neurodegeneration, Mitochondria, Lysosomal storage disease, Parkinson disease, Programmed cell death
INTRODUCTION
It has been observed for more than 30 years that the predominant ultrastructural feature of degenerating or dying neurons often involves prominent accumulation of autophagosomes and autolysosomes (1). This morphology is characteristic of certain populations of neurons undergoing programmed developmental cell death, including the isthmo-optic nucleus of chicks, tail motoneurons of larval frogs, and moth motoneurons (1, 2). In addition, increased autophagic vacuoles (AVs) have been described in Parkinson disease (PD) (3, 4), Alzheimer disease (AD) (5), Lewy body dementia (4), Huntington disease (6), prion diseases (7), lysosomal storage diseases (8), and X-linked or toxic myopathies (9, 10).
While the appearance of AVs in degenerating neurons has been historically dismissed as a nonselective stress response, advances in recent years demonstrate that autophagy is a highly regulated process. Scientific interest in autophagy is rapidly expanding, due in part to identification of genes involved in the autophagic molecular machinery (reviewed in (11, 12), in part to recent studies suggesting important roles for autophagy in clearing protein aggregates associated with neurodegenerative diseases (reviewed in (13)), and in part to a growing number of studies implicating autophagy-related processes in neurodegeneration and certain forms of regulated cell death (reviewed in (13–15)). These trends further highlight the importance of studying autophagy regulation under pathologic as well as physiologic conditions.
AUTOPHAGY INVOLVES MATURATION OF AVs
There are 3 major forms of "autophagy," a term derived from Greek roots meaning "self eating." These forms of autophagy share in common the delivery of intracellular cargo for degradation within lysosomes. Chaperone mediated autophagy involves recognition of protein motifs by trans-membrane lysosomal proteins, and their direct import across the lysosomal membrane (reviewed in (16)). Microautophagy involves rearrangement of the lysosomal membrane to engulf portions of adjacent cytoplasm or nucleus (reviewed in (16, 17)). Macroautophagy, hereafter referred to as autophagy, involves the sequestration of cytoplasmic proteins and organelles into a double-membrane vesicle, followed by stepwise maturation involving dissolution of the inner membrane, acidification, delivery to and fusion with lysosomes (18–20) (Fig. 1). The term AV is used to encompass the entire spectrum of evolution from early double-membrane autophagosomes to secondary autolysosomes (21). The AV-rich morphology observed in neuropathologic situations most likely reflects dysregulation of macroautophagic processes and comprises the subject of this review.
FIGURE 1.
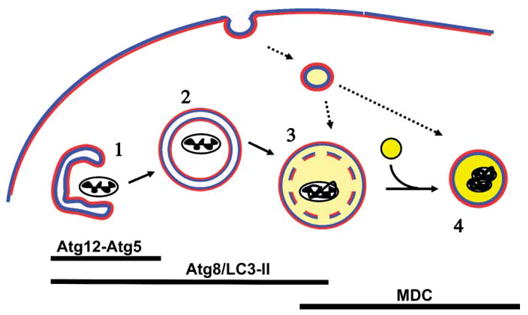
Schematic of AV maturation. Pre-autophagosomal membranes may be derived from smooth endoplasmic reticulum. Conjugation of Atg12-Atg5 complex and of Atg8/LC3 to the membrane is required for extension of the sequestering membrane (1). In contrast to other organelles such as endoplasmic reticulum, Golgi, or endosomes in which the lumen contacts ‘‘extracellular’’-type membrane (blue), the autophagosome (2) is a double membrane structure with cytoplasmic leaflets both inside and out (red). Upon completion of the nascent autophagosome, Atg12-Atg5 dissociates, while a portion of LC3-II remains associated with the AV until degraded. Early acidification (light yellow) and dissolution of the inner membrane (3) occurs, most likely through fusion with the endocytic pathway, and appears to be necessary for fusion with lysosomes (bright yellow) to form fully degradative autolysosomes (4). Acidification and membrane compaction results in staining of autolysosomes by MDC. Markers of late AVs are useful for measuring autophagic stress, but are less specific as convergence with endosomes and other vesicular pathways represent a normal consequence of AV maturation.
In a topographical sense, autophagosomes are unique, forming not from invagination of a unit membrane, but from extension of a double membrane or phagophore (22), around the cytoplasmic structures being enveloped (Fig. 1). Acidification and loss of the inner membrane occurs within several hours (18), probably through interaction with the endosomal pathway (20). Thus, ultrastructural observation of a double or single membrane enclosing recognizable cytoplasmic structures represents a gold standard for identifying an autophagosome (21). Evidence of organelle disorganization, compaction, or AV maturation should be sought, however, as invagination of a healthy neighboring cell process could theoretically result in a double membrane profile, and small neurites in cross section may appear similar to AVs containing mitochondria. Immunogold labeling for autophagy markers such as Atg8/LC3 (see below) can be useful (23, 24), although most immunolabeling protocols result in reduced ultrastructural resolution of membranous structures. Fusion of the AV with lysosomes, a process that appears to require both acidification by vacuolar ATPases and intact microtubule function (20, 25, 26), results in degradation of the contents within autolysosomes (19).
Typically, membranes from engulfed organelles become compacted, and can form heterogeneous electron dense contents and multilamellar whorls, particularly at the autolysosomal stages. This forms the basis for accumulation of the autofluorescent chemical monodansylcadaverine (MDC) within autolysosomes. MDC has an affinity for acidic, membrane-rich compartments. A combination of ultrastructural studies and density gradient subcellular fractionation indicate that MDC labels degradative AVs (amphisomes and secondary lysosomes), but not endosomes or endoplasmic reticulum (27). MDC labeling is not specific for autophagosomal processes, however, as several vesicular trafficking pathways converge at the level of amphisomes and lysosomes. MDC is useful for following accumulation of these later stages of AV maturation, but should be used in concert with more specific early AV markers (see below). An advantage of MDC labeling is a sharp vacuolar signal with low cytosolic noise, due to the increased fluorescence quantum yield that MDC exhibits in polar, membrane-rich environments (28), which facilitates quantitative image analysis (J.H. Zhu and C.T. Chu, unpublished data). Lysosomal markers have also been used to track the end stage of autophagy-related processes in non-phagocytic cells (21), although the possibility of heterophagic uptake of adjacent apoptotic cells must be considered in cell death studies. As increased AVs could result from both increased formation and impaired lysosomal fusion/degradation, parallel studies using techniques that track both earlier and later phases of AV evolution are desirable.
MOLECULAR REGULATION OF THE AUTOPHAGIC MACHINERY
While the morphology of autophagy-related structures was first characterized in mammalian cells, autophagy regulatory genes were identified from advances in yeast species (reviewed in (11, 12)). Following the first biannual Gordon Research Conference on Autophagy in Stress, Development, and Diseasein 2003, a uniform nomenclature for autophagy-related genes (ATG) was introduced (29). Two ubiquitin-like conjugating systems regulate autophagic sequestration (11). Deposition of the Atg12-Atg5 complex and of microtubule-associated protein 1 light chain 3 (LC3), the mammalian Atg8 homolog, initiates elongation of the isolation membrane or phagophore (Fig. 1) (reviewed in (30, 31)). Atg12-Atg5 is lost upon completion of the double membrane autophagosome, but LC3 remains within the AV until degraded (23). Thus, membrane associated LC3 is a highly useful AV marker that spans early autophagosome to early degradative stages (30) (Fig. 1). LC3 is modified by conjugation with phosphatidylethanolamine upon recruitment to the autophagosome membrane. This LC3-II form can be distinguished from cytosolic LC3-I by virtue of its faster migration during electrophoresis (23). However, as with morphologic evaluation of AV numbers, the degree of LC3-II in a cell reflects the balance between autophagy formation and degradation, rather than flux through the autophagic machinery (32). Although each technique has advantages and limitations, a combination of approaches including electron microscopy, LC3, MDC, and degradative assays would complement each other in comprehensive experimental studies of autophagic responses.
SIGNALING REGULATION OF AUTOPHAGY
The signaling regulation of autophagy has been the subject of several excellent reviews (for example, (33)). Two major upstream signaling pathways that regulate starvation-induced autophagy in mammals involve the mammalian target of rapamycin (mTOR) and type III phosphoinositide 3-kinase (PI3K), the mammalian VPS34 homolog. Activation of the TOR-p70 S6 kinase pathway by insulin receptors or amino acids results in tonic repression of autophagy under nutrient rich conditions. Rapamycin, which inhibits mTOR, promotes autophagy by releasing this inhibition. Class III PI3K lacks much of the N-terminal regulatory domains present in the more commonly studied class I PI3Ks that function downstream of growth factor receptors (34). Class III PI3K exists in a complex with beclin 1, the mammalian Atg6 homolog (35, 36), and produces only one product, phosphatidylinositol-3-phosphate (PtdIns(3)P). Administration of PtdIns (3) P stimulates autophagy, while inhibition of class III PI3K inhibits autophagy (37).
Many proteins identified in yeast serve similar roles in the mammalian autophagic machinery (30, 31), but important differences in signaling regulation, functional divergence of Atg homologues, and functional convergence (recruitment of unrelated gene products) are likely to exist (12). In yeast, the Atg1 kinase appears to link TOR inactivation to the pre-autophagosomal machinery, but structural or functional homologues remain to be discovered in mammals. Different members of the mitogen activated protein kinases appear to regulate autophagy in a cell type and stimulus dependent manner (33). The extracellular signal regulated protein kinases (ERK) can activate the inhibitory mTOR/p70S6K signaling pathway in neutrophils (38), although inhibition of autophagy by ERK signaling has not been demonstrated. In contrast, ERK promotes autophagy in colon cancer cells (39) and in neurotoxin treated cells (J.H. Zhu and C.T. Chu, unpublished data). Likewise, glucagon and cAMP stimulate liver autophagy (40), while high doses of cAMP inhibit autophagy in yeast (41). The effects of kinase signaling networks on autophagy are cell type- and context-specific, yet relatively little is known about the regulation of neuronal autophagy under either physiologic or pathologic conditions.
A neuron differs from most other cell types in that it depends almost exclusively on glucose (or ketones after induction of the appropriate enzymes) to provide both energy and carbon chains for protein synthesis. Thus, its ability to internalize and utilize glucose is independent of insulin. In most cell types, insulin-related signaling is a major regulator of cellular responses to nutrient status (42–44), and starvation-induced liver autophagy functions in part to supply the brain with fuel via amino acid deamination and conversion to glucose. Thus, autophagic responses in neurons may be more related to organelle damage or neuritic remodeling than in generating amino acids, and mechanisms of regulation may differ from other cells. Delineating the role of specific signaling pathways in autophagic responses to pathologic neuronal injury merit further study, and may yield therapeutically relevant insights to neurodegenerative disease mechanisms.
MOLECULAR DETERMINANTS OF CARGO SELECTION
While starvation-induced autophagy involves nonselective bulk degradation, processes that regulate more selective targeting of injured organelles form a significant emerging area. The most studied example of selective autophagic degradation is pexophagy, the removal of excess peroxisomes (17). In yeast, Atg1 regulates cargo selectivity or retrieval of Atg components in certain forms of yeast vacuolar targeting (45). In mammalian cells, peroxisomes and mitochondria are selectively degraded after toxic stimuli (46, 47) and mitochondria bearing mtDNA deletions are preferentially degraded during serum deprivation in an IGF-1 sensitive manner (44). It has been suggested that microtubule-dependent delivery of polyglutamine cargo, components of the Atg machinery and lysosomes into close proximity could represent a mechanism to confer increased selectivity (26). Additionally, relocation of the autophagy linked FYVE domain protein Alfy and interactions between LC3 and the polyubiquitin-binding protein p62/SQSTM1 may be involved in targeting cytosolic protein aggregates for autophagic degradation (48, 49). These studies provide evidence for the existence of mechanisms mediating specificity of cargo selection.
The number of mitochondria undergoing permeability transition has been proposed to determine whether a cell undergoes mitochondrial autophagy, apoptosis, or necrosis (50), and indeed a protein that induces permeability transition stimulates autophagy in malignant glioma cells (51). Live cell imaging studies suggest that mitochondria are surrounded by LC3 immediately before loss of membrane potential (Personal communication, Insil Kim and John J. Lemasters, University of North Carolina at Chapel Hill, 2006), suggesting additional upstream signals in regulating autophagic sequestration of damaged mitochondria. Cardiolipin, a component of the mitochondrial inner membrane that is sensitive to oxidation, plays an important role in apoptosis (52), and early loss of cardiolipin occurs in NGF-deprived sympathetic neurons prior to permeability transition (53). Recently, loss of the outer mitochondrial membrane protein Uth1p was shown to inhibit rapamycin-induced mitochondrial turnover without affecting degradation of other cytosolic proteins (54). This interesting study demonstrates that mitochondria are not passively and non-selectively degraded during starvation. It is unknown whether alterations in Uth1p might also be involved in sensing mitochondrial damage during injury-induced mitophagy. As mitochondrial turnover was not completely abolished in Δuth1 yeast, other mechanisms are likely to exist.
Recently, there has been growing interest in the role of mitochondrially targeted kinases and phosphatases in regulating neuronal injury responses (55, 56). In particular, degenerating human substantia nigra neurons in PD and Lewy body dementia show punctate cytoplasmic accumulations of phosphorylated ERK and other signaling proteins (57, 115). Immunogold labeling studies demonstrate intense labeling of abnormal mitochondria, some of which are present in autophagosomes (4). It is not possible to determine from postmortem studies whether phosphoproteins passively accumulate as a result of impaired degradation or whether they play an active role in regulating mitophagy. Nevertheless, alterations in subcellular localization of ERK is also observed in culture models of PD (58), and inhibition of ERK phosphorylation results in decreased AV content in toxin treated cells (Zhu and Chu, unpublished observations). Other MAP kinases such as c-Jun N-terminal kinases are also known to show mitochondrial localization (55), and may function to regulate autophagy (59).
AUTOPHAGIC STRESS: A PATHOLOGIC CONCEPT DISTINCT FROM AUTOPHAGIC FLUX
There is growing evidence that autophagy-related processes play important roles in the degeneration of neuronal processes and certain regulated forms of cell death, particularly those involving large neurons such as Purkinje cells, sympathetic neurons, motor neurons, and substantia nigra neurons (see below). It is unclear, however, whether increases in AV content within injured or degenerating neurons necessarily reflect increases in autophagic flux (as measured by catabolism of organelles or long-lived proteins). Indeed, it is possible that impaired completion of lysosomal degradation contributes to an imbalance between rates of AV formation and degradation. In other words, these cells can be thought of as undergoing “autophagic stress.”
Autophagic stress develops due to dysregulation of autophagic responses or when excessive autophagic demand cannot be balanced by cellular reserves. Autophagic stress is defined as a relatively sustained imbalance in which rates of AV formation exceed rates of AV degradation (Fig. 2). As these systems are in dynamic flux, a transient increase of AVs, as observed during the early phase of nitrogen deprivation would not stress the cell, and the degradative systems are able to keep pace. Homeostasis may be achieved either because the stimulus for AV formation was transient and/or because compensatory upregulation of degradative systems allows the cell to achieve a new level of equilibrium, successfully recycling the sequestered material. If equilibrium is not achieved in the face of sustained high demand and/or impaired clearance of AVs, a condition of autophagic stress develops. Although excessive or unchecked autophagy could hypothetically lead to cell death through energy consumption or by altering the beclin 1/bcl-2 balance (60), autophagic stress can also arise from pathologic impairment of lysosomal function in the context of normal basal rates of autophagy. As with oxidative stress, different cell and tissue types are expected to possess different levels of tolerance to increased autophagic demand. Moreover, some cell types may be able to tolerate a greater level of AV accumulation for longer periods before physiologic processes become interrupted.
FIGURE 2.
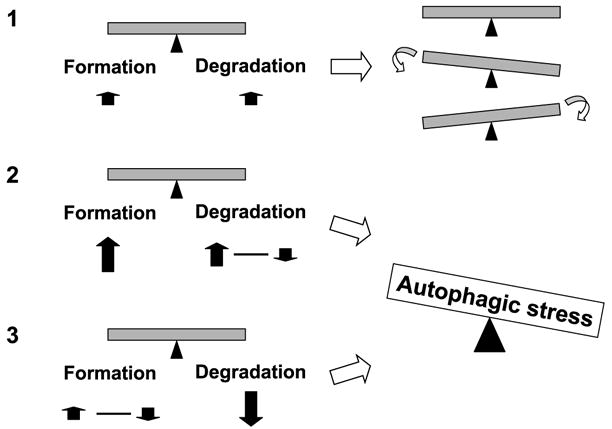
Autophagic stress: A balancing act. Increased catabolic turnover is induced by increases in both AV formation and degradation (1). Depending upon the cell type, there may be transient accumulation of AVs, or even a transient decrease in basal AV/LC3-II levels in cells with a large degradative potential. Increased autophagy induction in cells that exceeds the degradative reserve of the cell (2), or basal levels of autophagy induction in cells with marked impairment of lysosomal delivery/degradation (3), leads to autophagic stress, accompanied by accumulation of AVs. Hypothetically, unchecked increases in autophagic proteolysis could also result in a form of autophagic stress (2). The potential consequences of autophagic stress include impaired recycling of essential cellular components, expenditure and depletion of energy/biosynthetic resources, or expansion of enzymatic compartments involved in amyloidogenic processing.
In analogy to the concept of oxidative stress, which denotes an imbalance between formation and scavenging of reactive oxygen species, the concept of autophagic stress may serve to reconcile the established role of autophagy in normal cellular homeostasis with the rapidly increasing numbers of studies demonstrating a role for excessive AV accumulation in promoting pathologic disease mechanisms. There is a growing recognition that reactive oxygen species are essential for normal cellular communication, yet perturbations involving increased formation that exceeds the scavenging capacity of the system actively contributes to tissue injury and cell death. Likewise, accumulation of AVs in injured cells would occur when excessive autophagosomes are formed in relation to the degradative reserve of the system. Moreover, as with oxidative modifications, the degree of AV accumulation that can be tolerated may vary depending on the cell type and context (e.g. the other stimuli the cell has received in the recent past). In susceptible cell types, autophagic stress may kill the cell, while other cell types may merely become more susceptible to a “second hit.”
Experimental tools that can be employed for determining the cause of autophagic stress in injured cells are still under development. While degradation of pulse-labeled long-lived proteins or organelles remains a gold standard for measuring autophagic flux through the entire system, these techniques depend upon normal AV maturation, and may not reflect increases in AV formation in pathologic situations. AV formation may be followed by assessing LC3-II levels in the presence of bafilomycin, microtubule destabilizers, or cysteine protease inhibitors to arrest fusion or degradation (25, 32, 61). Inhibition of protein synthesis has also been reported to inhibit AV maturation (62). AV maturation can be followed by pH, acquisition of MDC staining, or acquisition of amphisome and lysosome markers. While fluorescence techniques may provide insight into impaired AV trafficking and cargo delivery to lysosomes (59), quantifying lysosomal degradative activity in isolation of sequestration and fusion rates is more complex. Current assays for lysosomal enzyme activity are more closely related to expression levels and lysosomal capacity. It may be difficult to precisely dissect the relative contributions of specific processes to development of autophagic stress, particularly as these processes are likely to exist in a dynamic flux. While time course studies using a panel of inhibitors may offer useful information, the potential effects of these inhibitors themselves in altering neuronal function and health must also be considered. Regardless of the underlying cause, strategies to restore the balance by inhibiting excessive formation of AVs and enhancing lysosomal delivery/degradation may prove beneficial.
AUTOPHAGIC STRESS IN NEURONAL INJURY AND DEGENERATION
There are several excellent reviews concerning autophagy in neuronal cells and tissues (13, 21). In addition to potential differences in signaling regulation and autophagic responses to nutrient and insulin status (discussed above and in (42)), neurons are highly polarized cells, critically dependent on axonal transport over long distances. Given the importance of microtubule function for trafficking of AVs and their clearance (25, 26), it is likely that neurons are particularly susceptible to developing autophagic stress (Fig. 3).
FIGURE 3.
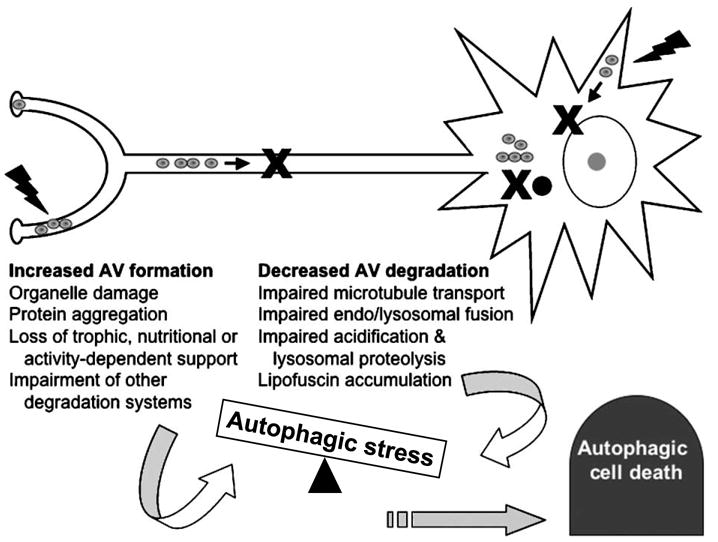
Autophagic stress, neurodegeneration, and autophagic cell death. Autophagy-related pathways may play important roles in degeneration of neuronal processes and in certain regulated forms of cell death. Increases in AV content within injured neurons can be caused by increased induction of autophagy or by impaired completion of lysosomal degradation. Most likely, both factors are involved in tipping the balance. While transient increases in AV content may reflect beneficial clearance of damaged or no longer useful cytoplasmic constituents, sustained alterations in susceptible cells result in autophagic stress. Autophagic cell death denotes an autophagosome-rich morphology of dying cells, without implying a particular role for autophagy. In some cases, this may reflect failure of autophagy to prevent cell death. In other cases, autophagy could be stimulated by signaling crosstalk involving a common upstream factor that also initiates cell death. Dysregulated autophagic processes could prove harmful to the cell through metabolic effects. Autophagy may also be directly harnessed by death programs to assist in the dismantling of large regions of cytoplasm.
Increased or continued demand for autophagy in the face of impaired transport, fusion, or degradative efficiency contributes to autophagic stress. Mitochondrially targeted injury, as observed in PD and its models, triggers degradation and loss of mitochondria prior to cell death (J.H. Zhu and C.T. Chu, unpublished data). In addition, protein aggregation that is characteristic of neurodegenerative diseases, and deficits in proteasomal or chaperone-mediated degradation pathways act to trigger compensatory increases in autophagic sequestration (63, 64). Accumulation of the aging pigment lipofuscin, which reflects indigestible lysosomal contents, not only is an indicator of impaired clearance of damaged constituents (65), but also may contribute to further impairments in handling effete or damaged mitochondria (63, 66). Furthermore, oxidative crosslinking contributes to formation of lipofuscin and neuromelanin, and iron associated with both these pigments may contribute to damage to sequestering membranes (66, 67). All of these factors may synergize in promoting autophagic stress in age-related neurodegenerative diseases.
Alzheimer disease (AD) is characterized by abnormalities in processing of β-amyloid precursor protein and tangled deposition of intracellular aggregates containing the micro-tubule associated protein tau. Abnormal accumulation of lysosomal structures has been observed for many years in susceptible populations of neurons in the form of granulovacuolar degeneration, or by staining for lysosomal cathepsins (68). More recently, it has been shown that AVs are abundant in neuritic processes in AD brains, suggesting a deficit of AV transport and maturation (5). This is interesting in light of other observations that an early axonopathy develops far in advance of other disease-related pathology in a mouse model of AD, implicating deficits in microtubule dependent transport as a key pathogenic factor (69). The accumulation of AVs may also contribute directly to neurodegenerative mechanisms by contributing to overproduction of pathogenic β-amyloid species (70). This represents an elegant example of how expansion of an organellar compartment to handle one type of stress may increase disease susceptibility due to other factors (which has been more fully described in relation to smooth ER/P450 enzyme induction and production of toxic drug metabolites in the liver).
GFP-LC3 transgenic mice (24) are particularly promising for studying autophagy in vivo and in primary cultures, revealing movement of AVs along neuritic processes (personal communication, Zsolt Talloczy and David Sulzer, Columbia University, New York; Zhenyu Yue, Mount Sinai School of Medicine, New York, 2006). LC3-I is particularly abundant in the brain as a microtubule associated protein. Although AVs are not readily observed in the brain of either fed or starved mice (71), it is possible that the high LC3-I/ LC3-II ratio reflects high constitutive autophagic flux in the normal brain with rapid degradation of AVs. A high demand for successful autophagic recycling would render this organ particularly susceptible to developing autophagic stress with minor impairments in AV degradative efficiency, explaining its susceptibility to lysosomal storage diseases. However, it is also possible that LC3 may play additional roles in the nervous system. A particular advantage of the GFP-LC3 mice is the potential for studying AV trafficking with respect to degeneration of neuronal circuits.
The midbrain dopaminergic neurons of the nigrostriatal projection, which degenerate in PD, are prone to developing autophagic stress during injury to either soma or terminals. Increased AVs are observed in genetic models of familial PD and in toxin models relevant to PD. PC12 cells overexpressing the A53T mutant form of α-synuclein exhibit a non-apoptotic form of cell death characterized by increased AVs and decreased capacity for both proteasomal and lysosomal degradation (72). As α-synuclein may play a role in regulating dopamine transport within the cell (73), and autophagy may be involved in formation of neuromelanin to sequester oxidized dopamine metabolites (67), it is interesting to note that dopamine treatment of SH-SY5Y cells also results in an autophagosome-rich form of cell death (74). Methamphetamine, which injures the striatal projection of dopamine neuron terminals, elicits multilamellar whorls in both primary rat midbrain cultures (75) and in mice (76). 1-Methyl-4-phenylpyridinium (MPP+), the active metabolite of the parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydroxypyridine (77, 78), produces mitochondrial injury and autophagic stress in SH-SY5Y cells and primary midbrain dopaminergic neurons (Chu et al, unpublished observation). Moreover, autophagosomes are increased in substantia nigra neurons in PD (3) and related Lewy body diseases (4).
Autophagy appears to play an important role in clearing protein aggregates in several models of neurodegenerative diseases, such as trinucleotide-repeat diseases (79, 80) and peripheral neuropathy in Schwann cells (81). Moreover, rapamycin treatment reduces toxicity of polyglutamine expansion in vivo (80). It has been shown that amorphous collections of aggregated proteins (aggresomes) accumulate when misfolded proteins overwhelm the proteasomal system (82). α-Synuclein, which forms Lewy body and neurite aggregates in PD, is also degraded by autophagic mechanisms (83, 84), particularly in the presence of pathogenic mutations that interfere with normal chaperone-mediated degradation of this protein (64, 85). Not only are aggresomes degraded by autophagy (81), but also lactacystin, a commonly used proteasome inhibitor, can inhibit lysosomal hydrolases. Thus, viability effects attributed to proteasome inhibitors may result from a combination of proteasomal inhibition and impairment of backup autophagolysosomal clearance. As induction of autophagy may be promising from a therapeutic standpoint, a better understanding of factors that influence the balance of formation and degradation in different neurodegenerative contexts becomes critical. Stimulation of AV formation may not be desirable unless potential blockages in trafficking/maturation are first removed, and the degradative potential of the neuron is restored or augmented.
AUTOPHAGIC CELL DEATH: CONTROVERSIES
In the periphery, autophagy plays a homeostatic role for the organism, sacrificing dispensable replicative cells to maintain blood nutrient levels. Autophagy may also play an important degradative role for protein aggregates. Indeed, mice conditionally lacking Atg7, the E1-like activating enzyme for Atg12 and Atg8, accumulate ubiquitinated aggregates (86). Likewise, autophagocytosis of mitochondria may represent a protective response making cells more tolerant to anaerobic stress or limiting release of apoptogenic mediators (50, 87). How can this information be reconciled with growing numbers of studies implicating autophagy-related processes in disease?
As discussed above, mechanisms to promote autophagic clearance of harmful organelles or protein aggregates are likely to be effective only if completion of autophagic degradation can be accomplished. Under situations where successful recycling of cellular components is impaired, robust autophagic responses could become harmful. Indeed, it is well established that mutations in lysosomal surface proteins (88) and a variety of deficits in lysosomal enzymes result in prominent neurodegeneration accompanied by accumulation of late AVs and lysosomes (8). Direct inhibition of lysosomes also results in neuronal cell death (89), although accumulation of AVs do not necessarily trigger cell death (90). Studies in fibroblasts derived from patients with mucolipidoses indicate that impaired lysosomal function sensitizes cells to subsequent injury by affecting mitochondrial calcium buffering capability (Kiselyor and Chu, unpublished observation). Moreover, while inhibiting AV formation reduces temozolomide chemotherapeutic efficacy, using bafilomycin to inhibit AV maturation to autolysosomes sensitizes glioma cells to chemotherapeutic cell death (reviewed in [91]). Thus, situations that promote development of autophagic stress appear to predispose neuronal and glial cells to death.
In recent years there has been increased recognition that autophagy may play a more active role in programmed or regulated cell death (15, 92). The controversies concerning the concept of autophagic cell death have been the subject of several excellent recent reviews (14, 93, 94), and only selected studies are included in this discussion. While earlier studies often utilized less specific methods of inhibiting autophagy to infer a role for autophagy in cell death, several recent studies illustrate that specific Atg genes are necessary for execution in certain contexts (95–97). Although Atg genes might be involved in non-autophagic processes, the possibility of cell death assisted or mediated by autophagic degradation deserves serious consideration.
From a broader pathologic viewpoint, the apparently paradoxical role of autophagy-related processes in neuro-protection and neuronal cell death is not unexpected. Many important, essential biologic processes represent double-edged swords. Moreover, reparative responses frequently include mechanisms to actively send a cell into a regulated death pathway, as death of an individual cell can be advantageous for survival of multicellular organisms. An AV-rich morphology of cell death would be predicted to result from any process that creates autophagic stress (Fig. 3). Autophagic cell death, if defined to denote cell death accompanied by autophagic stress, could result from a combination of factors that impair AV degradation or overactivate AV formation relative to the degradative reserve of the cell.
It is interesting to note that the neuronal cell types in which autophagic stress has been implicated in a degenerative or pro-death role, the sympathetic ganglion cell (98), cerebellar Purkinje cell (99–101), retinal tectal projection neurons (1), motor neurons (2), pyramidal neurons of the hippocampus (102, 103), and substantia nigra dopaminergic neurons (75), tend to be projection neurons. In the lurcher mouse, Purkinje cells degenerate and die as a result of altered interactions involving a mutant glutamate receptor and beclin 1 (99). This degeneration occurs in a retrograde fashion, and GFP-LC3 puncta are observed in the target nuclei of Purkinje cells (Personal communication, Zhenyu Yue, Mount Sinai School of Medicine, NY, 2006). Neurotrophin receptors and death domain receptors may also regulate autophagic cell death in Purkinje neurons (100). In contrast, the small granular neurons in the same culture preparations undergo classic apoptotic execution. It is possible that degeneration of these larger neurons require autophagic dismantling during regulated cell death. Alternatively, larger projection neurons may be more susceptible to processes that interfere with completion of autophagic degradation, such as microtubule dependent axonal transport.
It has been observed that studies showing that Atg gene products are necessary for cell death typically involve apoptosis-deficient cells (94). Interestingly, several lines of evidence suggest that mature neurons may be relatively apoptosis-deficient compared to developing neurons. While neonatal brain ischemia in humans frequently results in abundant pontosubicular apoptosis (104), apoptotic morphology is not commonly observed in adult brain ischemia or in neuro-degenerative diseases (105). There is also age-related development of a non-apoptotic form of 6-hydroxydopamine induced neuron death in rats (106). It has been observed that neurons that have never formed synapses die by classic apoptosis, while neurons that have interacted with the periphery exhibit a vacuolated form of programmed cell death (107), similar to that observed in primary striatal neurons (108). This may relate in part to expression of inhibitors of apoptosis proteins in mature neurons (109). While anti-apoptotic mechanisms function to inhibit loss of these crucial post-mitotic cells, they may also promote diversity in neuronal death pathways once death becomes inevitable.
While autophagy can be prominent in non-apoptotic forms of regulated cell death, changes of apoptosis, autophagy and necrosis often occur together (50), particularly in vivo (110). Additionally, there are several points of potential intersection between autophagic and apoptotic pathways (12, 33, 97). As either too little or too much autophagy may promote cell death, it has been suggested that the Bcl-2/beclin 1 interaction serves a rheostat function to prevent over-activation of autophagy (60). Autophagy is elicited by classic apoptotic stimuli in neurons, and has been suggested to represent an alternative execution pathway in sympathetic neurons (98, 111). Although caspase inhibitors delay cell death, mitochondrial autophagy persists and the neurons eventually die (47). In other cell types, autophagy appears to precede apoptosis (97, 112). While this may be consistent with autophagy in either anti- or pro-apoptotic roles, Atg5 appears necessary for both autophagy and apoptotic death (97). During serum/potassium deprivation, ultrastructural studies show that the cytoplasm of dying Purkinje neurons becomes filled with enlarged autophagosomes, and knockdown of p75ntr or transfection with dominant negative death domains reduced autophagic stress (100), implicating signaling crosstalk between death receptors and autophagy (97). Other studies indicate that autophagy opposes or delays apoptosis (90, 113). Divergent roles of autophagy in injury and death may reflect cell type and signaling context, the nature and intensity of the insult, and downstream lysosomal degradative capacity, or in other words, the level of autophagic stress developed in the cell.
AUTOPHAGIC STRESS: A BALANCING ACT
To summarize, autophagy plays an important role in physiological homeostasis: mediating turnover of damaged organelles, mobilizing amino acids during periods of nutrient deprivation, and executing tissue regression during development or in response to hormonal cycles (12, 30). Yet changes in autophagy are implicated in aging (63, 114), and autophagy-related processes may contribute to neuronal injury and neurodegenerative diseases. With age-related alterations in lysosomal function (63, 66), normal autophagic responses may become less effective in maintaining homeostasis, serving instead to create autophagic stress. Robust autophagic responses could be detrimental, particularly when successful recycling of cellular components is impaired, by acting as a sink for cellular resources (60, 66) or potentiating harmful side reactions as with pathogenic β-amyloid formation (70). Under other circumstances, autophagy may be recruited to promote cell death, as evidenced by studies showing a requirement for Atg genes in some model systems (95–97).
The concept of autophagic stress considers the dynamics of formation, maturation, and degradation, and may serve to reconcile apparently conflicting roles for autophagy-related processes in both adaptive/homeostatic and degenerative or death-associated pathways. Identifying mechanisms that may help restore balance to the system will be important for guiding potential future therapies for nervous system injuries and diseases.
Footnotes
Research in the author’s laboratory is supported by the National Institutes of Health (NS040817, NS053777), the Pittsburgh Foundation John F. and Nancy A. Emmerling Fund, and the Center for the Environmental Basis of Human Disease.
References
- 1.Clarke PG. Developmental cell death: Morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 2.Kinch G, Hoffman KL, Rodrigues EM, Zee MC, Weeks JC. Steroid-triggered programmed cell death of a motoneuron is autophagic and involves structural changes in mitochondria. J Comp Neurol. 2003;457:384–403. doi: 10.1002/cne.10563. [DOI] [PubMed] [Google Scholar]
- 3.Anglade P, Vyas S, Javoy-Agid F, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 4.Zhu J-H, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13:473–81. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 6.Sapp E, Schwarz C, Chase K, et al. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann Neurol. 1997;42:604–12. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- 7.Sikorska B, Liberski PP, Giraud P, Kopp N, Brown P. Autophagy is a part of ultrastructural synaptic pathology in Creutzfeldt-Jakob disease: A brain biopsy study. Int J Biochem Cell Biol. 2004;36:2563–73. doi: 10.1016/j.biocel.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Koike M, Shibata M, Waguri S, et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–28. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishino I. Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep. 2003;3:64–9. doi: 10.1007/s11910-003-0040-y. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki T, Nakagawa M, Yoshikawa A, et al. The first molecular evidence that autophagy relates rimmed vacuole formation in chloroquine myopathy. J Biochem (Tokyo) 2002;131:647–51. doi: 10.1093/oxfordjournals.jbchem.a003147. [DOI] [PubMed] [Google Scholar]
- 11.Ohsumi Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–16. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 12.Levine B, Klionsky DJ. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 13.Rubinsztein DC, DiFiglia M, Heintz N, et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- 14.Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1–2:66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]
- 15.Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40:401–13. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]
- 16.Cuervo AM. Autophagy: Many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 17.Farre JC, Subramani S. Peroxisome turnover by micropexophagy: An autophagy-related process. Trends Cell Biol. 2004;14:515–23. doi: 10.1016/j.tcb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 18.Shelburne JD, Arstila AU, Trump BF. Studies on cellular autophagocytosis. The relationship of autophagocytosis to protein synthesis and to energy metabolism in rat liver and flounder kidney tubules in vitro. Am J Pathol. 1973;73:641–70. [PMC free article] [PubMed] [Google Scholar]
- 19.Dunn WA., Jr Studies on the mechanisms of autophagy: Maturation of the autophagic vacuole. J Cell Biol. 1990;110:1935–45. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eskelinen E-L. Maturation of autophagic vacuoles in mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 21.Larsen KE, Sulzer D. Autophagy in neurons: A review. Histol Histopathol. 2002;17:897–908. doi: 10.14670/HH-17.897. [DOI] [PubMed] [Google Scholar]
- 22.Stromhaug PE, Berg TO, Fengsrud M, Seglen PO. Purification and characterization of autophagosomes from rat hepatocytes. Biochem J. 1998;335(Pt 2):217–24. doi: 10.1042/bj3350217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–28. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webb JL, Ravikumar B, Rubinsztein DC. Microtubule disruption inhibits autophagosome-lysosome fusion: Implications for studying the roles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol. 2004;36:2541–50. doi: 10.1016/j.biocel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and Micro-tubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 27.Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14. [PubMed] [Google Scholar]
- 28.Niemann A, Takatsuki A, Elsasser HP. The lysosomotropic agent monodansylcadaverine also acts as a solvent polarity probe. J Histochem Cytochem. 2000;48:251–58. doi: 10.1177/002215540004800210. [DOI] [PubMed] [Google Scholar]
- 29.Klionsky DJ, Cregg JM, Dunn WA, Jr, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–45. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 30.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–29. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 31.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–18. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 33.Codogno P, Meijer AJ. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005;12 (Suppl 2):1509–18. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 34.Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22:267–72. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- 35.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidyli-nositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–35. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tassa A, Roux MP, Attaix D, Bechet DM. Class III phosphoinositide 3-kinase-Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J. 2003;376:577–86. doi: 10.1042/BJ20030826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–98. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 38.Lehman JA, Gomez-Cambronero J. Molecular crosstalk between p70S6k and MAPK cell signaling pathways. Biochem Biophys Res Commun. 2002;293:463–69. doi: 10.1016/S0006-291X(02)00238-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:39090–95. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- 40.Shelburne JD, Arstila AU, Trump BF. Studies on cellular autophagocytosis. cyclic AMP- and dibutyryl cyclic AMP-stimulated autophagy in rat liver. Am J Pathol. 1973;72:521–40. [PMC free article] [PubMed] [Google Scholar]
- 41.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–66. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 42.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: Cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–48. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 43.Kanazawa T, Taneike I, Akaishi R, et al. Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem. 2004;279:8452–59. doi: 10.1074/jbc.M306337200. [DOI] [PubMed] [Google Scholar]
- 44.Gu Y, Wang C, Cohen A. Effect of IGF-1 on the balance between autophagy of dysfunctional mitochondria and apoptosis. FEBS Lett. 2004;577:357–60. doi: 10.1016/j.febslet.2004.10.040. [DOI] [PubMed] [Google Scholar]
- 45.Nair U, Klionsky DJ. Molecular mechanisms and regulation of specific and nonspecific autophagy pathways in yeast. J Biol Chem. 2005;280:41785–88. doi: 10.1074/jbc.R500016200. [DOI] [PubMed] [Google Scholar]
- 46.Yokota S. Degradation of normal and proliferated peroxisomes in rat hepatocytes: Regulation of peroxisomes quantity in cells. Microsc Res Tech. 2003;61:151–60. doi: 10.1002/jemt.10324. [DOI] [PubMed] [Google Scholar]
- 47.Tolkovsky AM, Xue L, Fletcher GC, Borutaite V. Mitochondrial disappearance from cells: A clue to the role of autophagy in programmed cell death and disease? Biochimie. 2002;84:233–40. doi: 10.1016/s0300-9084(02)01371-8. [DOI] [PubMed] [Google Scholar]
- 48.Simonsen A, Birkeland HC, Gillooly DJ, et al. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci. 2004;117:4239–51. doi: 10.1242/jcs.01287. [DOI] [PubMed] [Google Scholar]
- 49.Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemasters JJ, Qian T, He L, et al. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–81. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- 51.Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–93. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 52.Kagan VE, Borisenko GG, Tyurina YY, et al. Oxidative lipidomics of apoptosis: Redox catalytic interactions of cytochrome c with cardio-lipin and phosphatidylserine. Free Radic Biol Med. 2004;37:1963–85. doi: 10.1016/j.freeradbiomed.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 53.Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. Loss of cardiolipin and mitochondria during programmed neuronal death: Evidence of a role for lipid peroxidation and autophagy. Neuroscience. 2002;115:587–602. doi: 10.1016/s0306-4522(02)00512-2. [DOI] [PubMed] [Google Scholar]
- 54.Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–74. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 55.Horbinski C, Chu CT. Kinase signaling cascades in the mitochondrion: A matter of life or death. Free Radic Biol Med. 2005;38:2–11. doi: 10.1016/j.freeradbiomed.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 56.Dagda RK, Zaucha JA, Wadzinski BE, Strack S. A developmentally regulated, neuron-specific splice variant of the variable subunit Bbeta targets protein phosphatase 2A to mitochondria and modulates apoptosis. J Biol Chem. 2003;278:24976–85. doi: 10.1074/jbc.M302832200. [DOI] [PubMed] [Google Scholar]
- 57.Zhu J-H, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated kinase in Lewy body diseases. Am J Pathol. 2002;161:2087–98. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Bichem. 2004;271:2060–66. doi: 10.1111/j.1432-1033.2004.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 61.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 62.Lawrence BP, Brown WJ. Inhibition of protein synthesis separates autophagic sequestration from the delivery of lysosomal enzymes. J Cell Sci. 1993;105(Pt 2):473–80. doi: 10.1242/jcs.105.2.473. [DOI] [PubMed] [Google Scholar]
- 63.Keller JN, Dimayuga E, Chen Q, Thorpe J, Gee J, Ding Q. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol. 2004;36:2376–91. doi: 10.1016/j.biocel.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 64.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–95. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 65.Sullivan PG, Dragicevic NB, Deng JH, et al. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem. 2004;279:20699–707. doi: 10.1074/jbc.M313579200. [DOI] [PubMed] [Google Scholar]
- 66.Brunk UT, Terman A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med. 2002;33:611–19. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- 67.Sulzer D, Bogulavsky J, Larsen KE, et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci U S A. 2000;97:11869–74. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cataldo AM, Barnett JL, Berman SA, et al. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: Evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14:671–80. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 69.Stokin GB, Lillo C, Falzone TL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–88. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 70.Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy-a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–60. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. Faseb J. 2001;15:916–26. doi: 10.1096/fj.00-0334com. [DOI] [PubMed] [Google Scholar]
- 74.Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res. 2003;73:341–50. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- 75.Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Meth-amphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–60. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fornai F, Lenzi P, Gesi M, et al. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J Neurochem. 2004;88:114–23. doi: 10.1046/j.1471-4159.2003.02137.x. [DOI] [PubMed] [Google Scholar]
- 77.Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord. 1998;13(Suppl 1):35–38. [PubMed] [Google Scholar]
- 78.Beal MF. Experimental models of Parkinson’s disease. Nat Rev Neurosci. 2001;2:325–34. doi: 10.1038/35072550. [DOI] [PubMed] [Google Scholar]
- 79.Qin ZH, Wang Y, Kegel KB, et al. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet. 2003;12:3231–44. doi: 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- 80.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 81.Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23:10672–80. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Waelter S, Boeddrich A, Lurz R, et al. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 84.Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–62. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 85.Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem. 2005;280:23727–34. doi: 10.1074/jbc.M503326200. [DOI] [PubMed] [Google Scholar]
- 86.Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol. 2001;11:361–65. doi: 10.1016/s0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- 88.Eskelinen EL, Tanaka Y, Saftig P. At the acidic edge: Emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003;13:137–45. doi: 10.1016/s0962-8924(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 89.Shacka JJ, Klocke BJ, Shibata M, et al. Bafilomycin A1 Inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol. 2006 doi: 10.1124/mol.105.018408. in press. [DOI] [PubMed] [Google Scholar]
- 90.Boya P, Gonzalez-Polo RA, Casares N, et al. Inhibition of macro-autophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:72634. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 92.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–81. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 93.Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int J Biochem Cell Biol. 2004;36:2405–19. doi: 10.1016/j.biocel.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 94.Levine B, Yuan J. Autophagy in cell death: An innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yu L, Alva A, Su H, Dutt P, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 96.Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–28. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 97.Pyo JO, Jang MH, Kwon YK, et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–29. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 98.Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signalling in sympathetic neurons: An alternative mechanism of death execution. Mol Cell Neurosci. 1999;14:180–98. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- 99.Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: Implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–33. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- 100.Florez-McClure ML, Linseman DA, et al. The p75 neurotrophin receptor can induce autophagy and death of cerebellar Purkinje neurons. J Neurosci. 2004;24:4498–4509. doi: 10.1523/JNEUROSCI.5744-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Selimi F, Lohof AM, Heitz S, et al. Lurcher GRID2-induced death and depolarization can be dissociated in cerebellar Purkinje cells. Neuron. 2003;37:813–19. doi: 10.1016/s0896-6273(03)00093-x. [DOI] [PubMed] [Google Scholar]
- 102.Uchiyama Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch Histol Cytol. 2001;64:233–46. doi: 10.1679/aohc.64.233. [DOI] [PubMed] [Google Scholar]
- 103.Borsello T, Croquelois K, Hornung JP, Clarke PG. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci. 2003;18:473–85. doi: 10.1046/j.1460-9568.2003.02757.x. [DOI] [PubMed] [Google Scholar]
- 104.Rossiter JP, Anderson LL, Yang F, Cole GM. Caspase-3 activation and caspase-like proteolytic activity in human perinatal hypoxic-ischemic brain injury. Acta Neuropathol (Berl) 2002;103:66–73. doi: 10.1007/s004010100432. [DOI] [PubMed] [Google Scholar]
- 105.Roth KA. Caspases, apoptosis, and Alzheimer disease: Causation, correlation, and confusion. J Neuropathol Exp Neurol. 2001;60:829–38. doi: 10.1093/jnen/60.9.829. [DOI] [PubMed] [Google Scholar]
- 106.Marti MJ, Saura J, Burke RE, et al. Striatal 6-hydroxydopamine induces apoptosis of nigral neurons in the adult rat. Brain Res. 2002;958:185–91. doi: 10.1016/s0006-8993(02)03694-6. [DOI] [PubMed] [Google Scholar]
- 107.Pilar G, Landmesser L. Ultrastructural differences during embryonic cell death in normal and peripherally deprived ciliary ganglia. J Cell Biol. 1976;68:339–56. doi: 10.1083/jcb.68.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Castro-Obregon S, Rao RV, del Rio G, et al. Alternative, nonapoptotic programmed cell death: mediation by arrestin 2, ERK2, and Nur77. J Biol Chem. 2004;279:17543–53. doi: 10.1074/jbc.M312363200. [DOI] [PubMed] [Google Scholar]
- 109.Potts PR, Singh S, Knezek M, Thompson CB, Deshmukh M. Critical function of endogenous XIAP in regulating caspase activation during sympathetic neuronal apoptosis. J Cell Biol. 2003;163:789–99. doi: 10.1083/jcb.200307130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clarke PG, Posada A, Primi MP, Castagne V. Neuronal death in the central nervous system during development. Biomed Pharmacother. 1998;52:356–62. doi: 10.1016/s0753-3322(99)80002-x. [DOI] [PubMed] [Google Scholar]
- 111.Yu LY, Jokitalo E, Sun YF, et al. GDNF-deprived sympathetic neurons die via a novel nonmitochondrial pathway. J Cell Biol. 2003;163:987–97. doi: 10.1083/jcb.200305083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guimaraes CA, Benchimol M, Amarante-Mendes GP, Linden R. Alternative programs of cell death in developing retinal tissue. J Biol Chem. 2003;278:41938–46. doi: 10.1074/jbc.M306547200. [DOI] [PubMed] [Google Scholar]
- 113.Bauvy C, Gane P, Arico S, Codogno P, Ogier-Denis E. Autophagy delays sulindac sulfide-induced apoptosis in the human intestinal colon cancer cell line HT-29. Exp Cell Res. 2001;268:139–49. doi: 10.1006/excr.2001.5285. [DOI] [PubMed] [Google Scholar]
- 114.Bergamini E, Cavallini G, Donati A, Gori Z. The role of macroautophagy in the ageing process, anti-ageing intervention and age-associated diseases. Int J Biochem Cell Biol. 2004;36:2392–2404. doi: 10.1016/j.biocel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 115.Chalovich EM, Zhu ZH, Caltagarone J, et al. Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cell. J Biol Chem Cell. 2006 doi: 10.1074/jbc.M602632200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]