

Abstract
Isosteres of cryptolepine (1) were synthesized and evaluated for their antiinfective activities. Overall, the sulfur isostere, 5-methyl benzothieno[3,2-b]quinoline (5b) was equipotent to 1 and has shown no cytotoxicity at 23.8 μg/ml. Compound 5b was also found to have a broad spectrum of activity. Both the carbon and oxygen isosteres were less potent than cryptolepine. A limited library of 2-substituted analogs of 5b has been synthesized and evaluated in antifungal screens but did not show increase in potency compared to the unsubstituted 5b. Similarly, evaluation of tricyclic benzothieno[3,2-b]pyridines while showing promise in individual screens did not produce an overall increase in potency. Overall, the evaluation of the activities of 5b compared with standard antifungal/anti-protozoal agents suggests that the benzothienoquinoline scaffold could serve as a lead for optimization.
Opportunistic infections are caused by pathogens that take advantage of a suppressed immune system. Such conditions as HIV AIDS disease, organ transplantation and long-term use of corticosteroids for example, cause either immune suppression or some disruption in the immune system.1 With an estimated 40.3 million people living with AIDS around the globe2 and the increasing development of resistance to current therapies, there is a continuing need for new antiinfective agents against opportunistic infections.
Previous studies in our laboratories3,4 and those of others5 have indicated that cryptolepine and other alkyl-substituted indolo[3,2-b]quinolines possess interesting biological activities including antiinfective activity against several opportunistic infectious organisms (OIs). These include Candida albicans (Ca), Cryptoccocus neoformans (Cn), and Aspergillus fumigatus (Af). These actions of the indoloquinolines appear to operate through intercalation into DNA, binding preferentially at GC-rich sequences and stimulating DNA cleavage by human topoisomerase II. On the basis of these initial observations, we embarked on Structure-Activity Relationship (SAR) studies to probe and identify structural features that enhance the potency of the indoloquinoline scaffold.
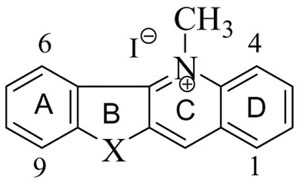
Compd 1; 5-Methyl indolo[3,2-b]quinolinium iodide; X = NH
Compd 5b; 5-Methyl benzothieno[3,2-b]quinolinium iodide; X = S
Several publications from our laboratories have indicated that alkylation of the nitrogen at the 5 position is required for the antiinfective activities associated with the quindoline scaffold.6,7 In particular, an ω-cycloalkyl pentyl group was associated with increased potency as antifungal, antibacterial and anti-protozoal agents. It was also observed that the absence of ring D appears to have little effect on antimicrobial activity8 but may alter the overall mode of action as both toxicity and spectrum of activity appeared to have increased with the tricyclic analogs. Overall however, increasing potency appears to be associated with increasing cytotoxicity.9 This observation has spurred our desire to modify the indoloquinoline scaffold so as to separate potency from cytotoxicity. In this paper, we report on the effect of isosteric replacements at the N-10 position and the results of further SAR investigation associated with these modifications.
Chemistry
The syntheses of cryptolepine (1) and other indoloquinolines were previously reported.10 5-Methyl 11H-indeno[3,2-b]quinolinium iodide (2), the carbon isostere of cryptolepine, was obtained by a series of reactions beginning with the reaction of anthranilic acid with 1-indanone to yield indenoquinolone (Scheme 1).11 The resulting indenoquinolone was treated with phosphorus oxychloride (POCl3) followed by hydrogenation with palladium on carbon to produce 11H-indeno[3,2-b]quinoline, which was methylated (MeI) to produce the desired product, 2. Attempts to alkylate with 5-phenylpentyl iodide were unsuccessful and often led to decomposition.
Scheme I.
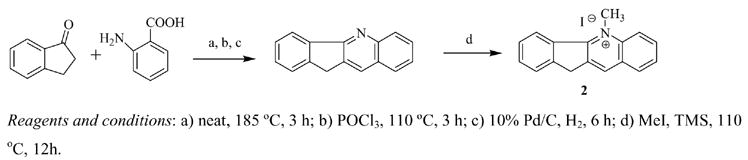
Preparation of 5-Methyl-11H-indeno[3,2-b]quinolinium Iodide, 2
The preparation of benzothienoquinoline and benzofuroquinoline was achieved using modifications of a previously published procedure10,12 outlined in Scheme 2. Briefly, substituted anthranilic acids, available from commercial sources, were acylated with acyl chlorides obtained by alkylating phenol or thiophenol. In a double intramolecular cyclization reaction with polyphosphoric acid (PPA), the acylated intermediates were converted to substituted-11-quinolones (3a–3f), which were subsequently converted to benzofuroquinoline (4a) and benzothienoquinolines (4b–4h) by chlorination with phosphorous oxychloride (POCl3) and hydrogenation on Pd/C to remove the chlorine. The targeted final products (5a–5h) were then obtained by methylation with methyl iodide. Compound 4b was also alkylated with methyl triflate and 5-phenylpentyl iodide to form 5-methylbenzothieno[3,2-b]quinolinium triflate (6) and 5-(5-phenylpentyl)benzothieno[3,2-b]quinolinium iodide (7) respectively.
Scheme 2.
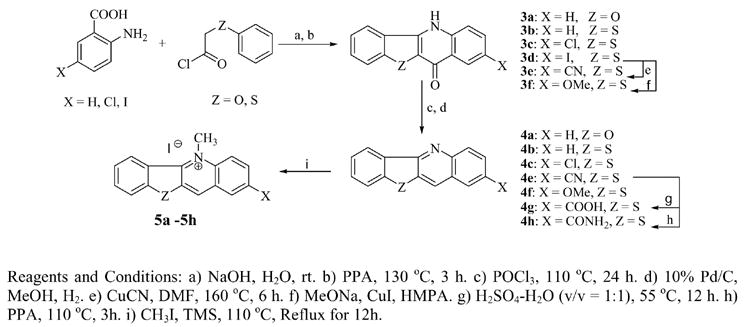
Synthesis of Benzothienoquinolinium Iodides and Benzofuroquinolinium Iodide
Construction of the tricyclic benzothieno[3,2-b]pyridine ring (10a–c) proved to be more challenging than we first thought. Eventually, 3-bromopyridine N-oxide13 was coupled with 4-nitrobenzene diazonium tetrafluoroborate14 to obtain 3-bromo-1-(4-nitrophenoxy)pyridinium tetrafluoroborate (8) in good yield (68%) and then converted to 3-bromo-2-(2-hydroxy-5-nitrophenyl) pyridine (9a). Carbamoylation with dimethylthiocarbamoyl chloride produced 9b, which when heated at 190 °C underwent rearrangement and subsequent cyclization in ethylene glycol to form 8-nitro-benzo[4,5]thieno[3,2-b]pyridine (10a). Attempts to obtain the unsubstituted benzothieno[3,2-b]pyridine using the same approach were unsuccessful. However, the unsubstituted compound (11c) was obtained using the amino derivative, 10b as the starting material. Diazotization of 10b and treatment of the resulting diazonium ion with hypophosphorous acid (H2PO3) yielded 10c. Compound 10a was methylated to 11a but was resistant to alkylation with 5-phenylpentyl iodide, apparently as a result of decreased pKa of the N atom. Compound 10c was similarly alkylated to produce the target compound, 11c. The amine substituent 11b, was prepared by reducing compound 10a with SnCl2 to yield the primary amine (10b) which was subjected to alkylation with methyl iodide to form 11b. Compound 11d was obtained by alkylation of amine 10c with 5-phenylpentyl iodide. The overall yields for these reactions were rather low although no attempts were made to optimize yields.
Results and Discussion
Previous studies on N-alkylated δ-carbolines and their carbon isostere, azafluorenes,8 suggest that antifungal activity is modulated by isosteric replacement. While cryptolepine may be a useful lead for drug design, it has shown considerable cytotoxicity in Vero cell lines, prompting us to look for ways to attenuate its cytotoxicity. As part of that desire, we have embarked on various SAR approaches including isosteric replacement of the N-10 atom. Three isosteres were synthesized and evaluated first against two common opportunistic pathogens, C. albicans and C. neoformans and subsequently against other microorganisms. The microbes were selected for evaluation because of their known pathogenicity.15 Compounds which show inhibition (IC50) of the microorganism at 20 μg/ml or better were so evaluated and the results are recorded in Tables 1 and 2. The results indicate that cryptolepine is more active than its carbon (2) and oxygen (3) isosteres against C. albicans and C. neoformans. The sulfur isostere or 5-methyl benzothieno[3,2-b]quinolinium iodide (5b) was found to have similar potency as cryptolepine. All compounds except 7 were further evaluated against other pathogens including A. fumigatus and M. intracellulare. As shown in Table 2, most of the compounds have no activity against A. fumigatus or M. intracellulare (activity below 20 μg/ml). The compounds were also evaluated against leishmania, an infectious species causing infection in about 1.5 million people a year.16 All the compounds show significant activities against leishmania as compared to the standard pentamidine. In fact, 5a and 5b are twice as potent as pentamidine. In addition, because previous studies17 indicated compound 1 had activity against malaria parasites, the isosteres were also evaluated for activity against P. falciparum. Moderate activities were associated with the isosteres as shown in Table 2.
Table 1.
Physicochemical Data and Antifungal Activities of Synthetic Compounds
| IC50 (μg/mL)
|
|||||
|---|---|---|---|---|---|
| Compd | yield % | MP (°C) | formula | Cn | Ca |
| 1 | 2–15.6b | 2.- >20 | |||
| 2 | 68% | 251–252 | C17H14IN·0.3H2O | >20 | >20 |
| 5a | 81% | 204–205 | C16H12INO | >20 | >20 |
| 5b | 68% | 215–216 | C14H15IN2S | 2.5* | 1.0* |
| 5c | 15% | 185–186 | C16H11ClINS·0.2H2O | 15 | 2.0 |
| 5e | 74% | 204–205 | C17H11IN2S·2.5H2O | >20 | >20 |
| 5f | 51% | 216–217 | C17H14INOS·0.5H2O | 8.0–15>20 | |
| 5g | 64% | 203–204 | C17H12INO2S | >20 | >20 |
| 5h | 53% | 234–235 | C17H13IN2OS·1.25H2O | >20 | >20 |
| 6 | 84% | 237–238 | C17H12F3NO3S2·0.2H2O | >20 | 10.0 |
| 7 | 30% | 182–184 | C26H30INS·2.5H2O | 0.5* | 1.0* |
| 11a | 78% | 254–255 | C13H9IN2O2S | >20 | >20 |
| 11b | 65% | 207–208 | C14H15IN2S·3.8H2O | >20 | >20 |
| 11c | 71% | 194–196 | C12H10INS | >20 | >20 |
| 11d | 82% | 158–160 | C22H22INS·1.1H2O | 20 | >20 |
| Amphotericin B | 0.95 | 0.25 | |||
Table 2.
Antiinfective Activities of Synthetic Compounds
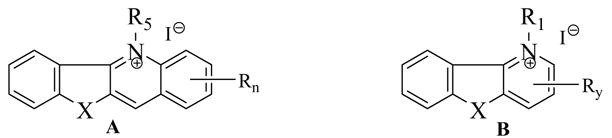
| STRUCTURE | IC50 (μ g/ml) |
P. falciparum (IC50 μg/ml) |
Cytotox (vero cells) |
||||
|---|---|---|---|---|---|---|---|
| Compd. | A/B X, R5/R1, Rn/Ry | Af | Mi | Leish | D6 Clone | W2 Clone | IC50 |
| 1 | A, NH, CH3, H | >20a | 15 | 0.23 | 0.18 | 0.35 | 3.2 |
| 2 | A, CH2, CH3, H | >20 a | >20 | NT | NT | NT | NT |
| 5a | A, O CH3, H | 15 | >20 | 2.2 | 0.28 | 0.28 | >23.8 |
| 5b | A, S CH3, H | >20 a | >20 | 0.35 | 1.6 | 2.0 | >23.8 |
| 5c | A, S CH3, 2-Cl | 4.0 a | >20 | 0.42 | 4.0 | 2.8 | >23.8 |
| 5e | A, S CH3, 2-CN | 20 a | >20 | 2.6 | NA | NA | >23.8 |
| 5f | A, S CH3, 2-OMe | 20a | >20 | 0.6 | 0.74 | 0.74 | >23.8 |
| 5g | A, S CH3, 2-CO2H | 10a | 20 | 0.39 | NA | 4.5 | 7.0 |
| 5h | A, S CH3, 2-CONH2 | >20a | >20 | 40 | NA | NA | >23.8 |
| 6 | 5a with OTf co-anion | 6.5 | 15 | 0.27 | 0.34 | 0.30 | >23.8 |
| 11a | B, S CH3, 8-NO2 | >20a | >20 | >40 | 0.4 | 0.46 | >23.8 |
| 11b | B, S CH3, 8-N(Me)2 | >20a | 15 | 3.0 | 0.18 | 0.20 | >23.8 |
| 11c | B, S CH3, H | >20a | >20 | NT | 1.5 | 1.2 | >23.8 |
| 11d | B, S, (CH2)5Ph, H | 20a | 1.5 | 1.2 | 0.27 | 0.22 | 20.0 |
| Amphotericin B | 0.85 | NT | NT | NT | NT | 6.5 | |
| Ciprofloxacin | 0.35 | NT | |||||
| Pentamidine | 0.7 | NT | |||||
| Chloroquine/Artemisinin | 0.02/0.02 | 0.14/0.02 | >4.7/>4.7 | ||||
= MIC values; Abbreviations: NA = Not active up to 4.76 μg/ml; NT = Not tested. Af = Aspergillus fumigatus; MI = Mycobacterium intracellulare; Leish = Leishmaniasis
The potential for selective toxicity of the compounds was also evaluated in Vero cell lines. All the compounds except for cryptolepine and 5g showed no cytotoxicity in Vero cell lines up to a concentration of 23.8 μg/ml. Compound 5g was less cytotoxic than 1 (IC50 of 7.0 and 3.2 μg/ml respectively). Finally, compounds 1 and 5b were further evaluated for cytotoxicity against several cell lines and, the results recorded in Table 3 showed compound 5b to be significantly less toxic than compound 1 in all cell lines.
Table 3.
Toxicity of Key Compounds
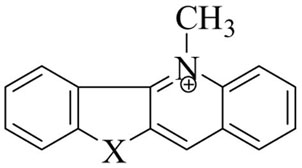
| STRUCTURE | Toxicity expressed as IC50 (μ g/ml) | ||||||
|---|---|---|---|---|---|---|---|
| Compd | X | Murine M109a | Human Hep 2a | Human HuVeca | Vero Cells | L6 Cytotoxb | BrdU Cytotoxb |
| Compd 1 | NH | 0.44 | <0.18 | 1.38 | 3.2 | 39.3 | <0.78 |
| Compd 5b | S | 1486 | 355 | 662 | >4.7 | >100 | 4.6 |
| Amph B | NT | NT | NT | 6.5 | NT | NT | |
= Data supplied by Bristol Myers Squibb.
= Data supplied by Eli Lilly & Co.
The broad spectrum of activity shown by compound 5b and its lower cytotoxicity spurred us on to evaluate a limited library of substituted analogs for their antiinfective properties. Position 2 which is para to the quaternary N-5 atom was of interest because of the effect substituents would have on the positive charge on the N-atom at position 5. The results of the evaluation are reported in Tables 1 and 2. Five such compounds (5c–5i) were synthesized to test the effect of electron withdrawing and electron donating substituents at the 2-position. These effects could be inductive or mesomeric. The results show that activities of the compounds (5c–5i) did not change substantially when the substituted 5-methyl benzothienoquinolines were compared with the unsubstituted compound (5b). Similarly, removal of ring D from 5b to form benzothieno[3,2-b]pyridines (11a–11c) did not result in much improvement in potency. However, as observed with the indoloquinolines, activity increased substantially when an ω-phenyl pentyl or an ω-cyclohexyl pentyl moiety was introduced onto the pyridine nitrogen in both rings (7 and 11d). There was also a general observation that by replacing nitrogen with a sulfur atom in the indole ring, cytotoxicity to Vero cells was attenuated. Compounds 5b and 6 enabled a comparison of the effect of different co-anions on activity. In terms of potency, there was little difference in the use of the triflate as compared to iodide. However, the use of different anions might affect solubility.
In a search for an explanation as to why isosteres might have different activities, we evaluated the charge density on the N-5 atom of each isostere. The rationale for focusing on the N+ atom is 2-fold. First, we have shown that N-alkylation which leads to a quaternary nitrogen atom formation is required for activity.4,6 & 7 Secondly, Aymani18 and others have reported a role for the N+ charge in its binding to DNA fragments. The 1H and 13C NMR chemical shift (δ) of the methyl group on each quaternary N atom was used in this evaluation since the density of the positive charge on the N-5 atom should have a deshielding effect on the methyl group. In addition, the proton attached to C-11 may reflect the overall electron density in the C ring as a result of isosteric replacement of the N-10 atom. The results of this investigation are recorded in Table 4. Evaluation of the results shows that despite its electron donating capacity, which was thought to reduce the positive charge density on N+-5, the N-Me in 5-methyl benzo[3,2-b]thienoquinolinium iodide had the highest chemical shift. It is possible that the presence of sulfur may lead to increased aromaticity and a dispersal of the electron density in 5b than in other isosteres, i.e., N, O or carbon and thus, explain the higher chemical shift value. The trend of increasing proton chemical shift values appears to roughly correlate with the observed activities against C. albicans and C. neoformans and may thus reflect the positive nature of the N atom. On the other hand, the 13C-NMR chemical shift values of the N-methyl group do not correlate well with the activities. Overall though, the role of the positive charge in modulating activity may be confounded by other factors including lipophilicity and topology of the tetracyclic ring.
Table 4.
1H and 13C NMR chemical shift (δ) of the methyl groups on the N-5 atom of Isosteres.
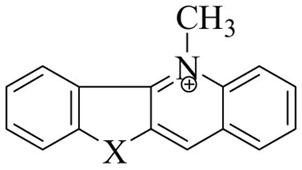
| STRUCTURE | 1H Chemical Shift (δ) | 13C NMR Chemical Shift (δ) | ||
|---|---|---|---|---|
| Compound | X | N+-CH3 | -C11H | N+-CH3 |
| Compd 1 | NH | 5.05 | 9.25 | 40.14 |
| Compd 2 | CH2 | 4.88 | 9.24 | 41.42 |
| Compd 5a | O | 5.01 | 9.65 | 41.28 |
| Compd 5b | S | 5.09 | 10.00 | 43.28 |
Conclusion
The results of the current investigations have shown that carbon (indenoquinoline) and oxygen (benzofuroquinoline) isosteres are significantly less potent as antiinfectives when compared with the parent nitrogen (indoloquinolines) isostere. The sulfur isostere (benzothienoquinoline) on the other hand, appears is as potent as the indoloquinolines. In addition to potency, the benzothienoquinolines have a broad spectrum of activity against not only fungi but also bacteria and protozoa. More importantly, they have significantly lower cytotoxicity than the indoloquinolines in several cell lines. In general, all isosteres have significant activity against leishmania, and hence further development of these compounds against this microorganism is warranted. At the current time, it is unclear to us why there are such significant differences in the activity profiles of the isosteres. The possibility that the positive charge density on the nitrogen might correlate with potency is not currently completely supported by the data. Further investigation to understand the effect of isosteric replacement in the indoloquinoline scaffold is also warranted.
Experimental
Melting points were determined on a Gallenkamp (UK) apparatus and are uncorrected. NMR spectra were obtained on a Varian 300 MHz Mercury or a Bruker 270 MHz NMR Spectrometer. Elemental analyses were carried out by Atlantic Microlab, Inc., Norcross, GA and are within 0.4% of theory unless otherwise noted. Flash chromatography was performed with Davisil grade 634 silica gel. N,N-Dimethylformamide was distilled from CaSO4 and stored over 4 Å molecular sieves. Sulfolane was dried over 4 Å molecular sieves. 5-Cyclohexylpentyl bromide and 5-phenylpentyl bromide were prepared by treatment of the corresponding alcohols with PBr3. The remaining alkyl halides were obtained from either Sigma–Aldrich Chemicals or Fisher Scientific and were used without further purification. Quindoline (10H-indolo[3,2-b]quinoline), the starting material in several of the syntheses below was obtained as previously reported.4
5.1. General method for the synthesis of N-10 alkylated quindolines
5-Methyl-11H-indeno[1,2-b]quinolin-5-ium iodide, 2: Method A
A mixture of anthranilic acid (5.0 g, 36.5 mmol), 1-indanone (7.5 g, 56.8 mmol) was heated with stirring for 3 h at 185 °C. The resulting mixture was filtered to obtain a solid which was washed with pyridine (25 mL), Et2O (2× 25 mL) to give yellow solid, 5.65 g, 66%. The yellow solid (1.0 g, 4.3 mmol) was dissolved in POCl3 (15 mL) and was refluxed for 3 h. The excess POCl3 was removed in vacuo. The residue was neutralized with saturated NaHCO3 and the aqueous solution was extracted with CH2Cl2 (300 mL). The organic layer was washed with brine, dried (Na2SO4), and filtered. The filtrate was concentrated in vacuo and the residue was purified on silica gel with 20% EtOAc-hexane to provide a white solid, 0.88 g, yield 82%. The white solid (340 mg) was dissolved in THF (50 mL), the solution was charged with 10% Pd/C (100 mg) and was hydrogenated in H2 atmosphere (50 psi) for 6 h to yield the desired product, 11H-Indeno[1,2-b]quinoline, 251 mg, 85%. 1H NMR (300 MHz, CDCl3): δ8.31 (1H, brs), 8.20 (1H, m), 8.14 (1H, s), 7.75 (1H, d, J = 8.1 Hz), 7.64 (1H, t, J = 8.7 Hz), 7.54 (1H, m), 7.43 (3H, m), 3.96 (2H, s). Calcd for: C16H11N: C 88.45, H 5.10, N 6.45; Found: C 88.55, H 5.12, N 6.43.
Method B
A mixture of 11H-Indeno[1,2-b]quinoline (100 mg, 0.46 mmol) and MeI (0.2 mL) in tetramethylene sulfone (TMS) (0.5 mL) was heated in a sealed tube for 12h at 110 °C. After cooling to room temperature, EtOAc was added to induce precipitation. The precipitate was collected by filtration followed by recrystalization from MeOH-Et2O to afford 5-Methyl-11H-indeno[1,2-b]quinolin-5-ium iodide 2, 112 mg, 68%. Mp 251-252 °C. 1H NMR (300 MHz, DMSO-d6: δ 9.24 (1H, s), 8.71 (2H, m), 8.42 (1H, d, J = 8.4 Hz), 8.21 (1H, t, J = 8.7 Hz), 7.98 (2H, m), 7.89 (1H, t, J = 7.5 Hz), 7.76 (1H, t, J = 7.5 Hz), 4.88 (3H, s), 4.43 (2H, s). Calcd for C17H14IN·0.3H2O: C 56.00, H 3.87, N 3.84; Found: C 55.75, H 3.64, N 3.98.
Synthesis of Benzofuro[3,2-b]quinolinium Iodide 5a and Benzo[b]thieno[3,2-b]quinolinium Iodides, 5b–5h
5-Methyl-benzo[b]thieno[3,2-b]quinolin-5-ium iodide 5b: General Procedure Method C
A mixture of anthranilic acid (7.4 g, 54 mmol), sodium hydroxide (4.32 g, 108 mmol), and phenylthioacetyl chloride (10.0 g, 54 mmol) in H2O (300 mL) was stirred at 0 °C for 1 h, and then at room temperature for 3 h. The mixture was acidified (pH = 4–5) with 2% aq. HCl and then extracted with EtOAc (2×500 mL). The combined organic layer was dried (Na2SO4), filtered and solvent was evaporated in vacuo. The residue was crystallized in EtOAc to give a white solid, 13.5 g, 86%. The white solid (7.7 g, 26.6 mmol) was mixed with polyphosphoric acid (PPA, 120 g) and heated at 130–135 °C for 3 h. After cooling to rt, the mixture was poured onto ice/water (200 mL), neutralized with saturated solution of Na2CO3 and the solid was collected. The solid was washed with H2O, EtOAc, and Et2O to give quinolone 3b, 5.75 g, 86%. A suspension of 3b (5.75 g, 26.6 mmol) in POCl3 (15 ml) was refluxed with stirring at 120 °C for 3 h. After evaporating the POCl3, the reaction mixture was poured onto ice/water (50 mL) and then neutralized with 10 % NaOH. The aqueous solution was extracted with CH3Cl (3× 200 mL) and the combined organic layer was washed with brine, dried (Na2SO4), and then filtered. The filtrate was concentrated in vacuo to give a solid, 5.30 g, 74%. The solid (3 g, 11.15 mmol) was dissolved in AcOH (300 mL) and hydrogenated (60–65 psi) on Pd/C (10%, 1.5 g) at rt, to afford benzo[b]thieno[3,2-b]quinoline 4b, 2.0 g, 85%. Mp: 172–173 °C. 1H NMR (300 MHz, CDCl3): δ 8.81 (1H, d, J = 7.2 Hz), 8.61 (1H, s), 8.41 (1H, d, J = 8.4 Hz), 7.86 (1H, d, J = 8.1 Hz), 7.76 (2H, m), 7.57 (3H, m). Calcd for: C15H9NS: C 76.57, H 3.86, N 5.95; Found: C 76.54, H 3.90, N 5.88.
Methylation was achieved using method B and starting with compound 4b to afford 5b, 68%. Mp: 215–216 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.00 (1H, s), 9.10 (1H, d, J = 8.5 Hz), 8.86 (1H, d, J = 9.2 Hz), 8.49 (1H, dd, J = 4.8, 7.5 Hz), 8.34 (1H, t, J = 7.5 Hz), 8.05 (2H, m), 7.85 (1H, t, J = 7.8 Hz), 5.09 (3H, s). Calcd for C14H15IN2S: C 50.94, H 3.21, N 3.71; Found: C 51.07, H 3.13, N 3.64.
5-Methyl-benzo[4,5]furo[3,2-b]quinolin-5-ium iodide, 5a
Compound 4a, benzo[4,5]furo[3,2-b]quinoline, was obtained using Method C above; 23%. Mp: 154–155 °C. 1H NMR (300 MHz, CDCl3): δ 8.18 (1H, d, J = 8.1 Hz), 8.09 (1H, d, J = 8.4 Hz), 7.94 (1H, s), 7.73 (1H, d, J = 8.1 Hz), 7.52 (1H, m), 7.40 (2H, m) 7.25 (1H, m), 7.11 (1H, m). Calcd for: C15H9NO: C 82.18, H 4.14, N 6.39; Found: C 82.08, H 4.20, N 6.33. Compound 4a was methylated using the general procedure Method B to obtain 5a; 81%. Mp: 204–205 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.65 (1H, s), 8.90 (1H, d, J = 8.4 Hz), 8.84 (1H, d, J = 9.3 Hz), 8.57 (1H, d, J = 8.4 Hz), 8.30 (1H, t, J = 9.0 Hz), 8.10 (3H, m), 7.81 (1H, t, J = 8.4 Hz), 5.01 (3H, s). Calcd for C16H12INO: C 53.21, H 3.35, N 3.88; Found: C 53.21, H 3.32, N 3.89.
2-Chloro-5-methyl-benzo[b]thieno[3,2-b]quinolin-5-ium iodide, 5c
Using method C and starting with 5-chloroanthranilic acid and phenylthio acetyl chloride, 2-Chloro benzo[b]thieno[3,2-b]quinoline, 4c, was obtained in a yield of 16%. Without further purification, 4c was methylated using method B to yield the desired compound 5c, 15%. Mp 185–186 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.87 (1H, s), 9.07 (1H, d, J = 8.5 Hz), 8.88 (1H, d, J = 9.6 Hz), 8.65 (1H, d, J = 2.3 Hz), 8.47 (1H, d, J = 8.1 Hz), 8.33 (1H, dd, J = 2.4, 9.6 Hz), 8.04 (1H, t, J = 7.5 Hz), 7.85 (1H, t, J = 7.5 Hz), 5.08 (3H, s). Calcd for C16H11ClINS·0.2H2O: C 46.18, H 2.66, N 3.37; Found: C 46.02, H 2.67, N 3.32.
2-Cyano-5-methyl-benzo[b]thieno[3,2-b]quinolin-5-ium iodide, 5e
Using method C and 5-iodoanthranilic acid as starting material, 2-iodo benzo[b]thieno[3,2-b]quinolinone was obtained and subsequently converted to 2-cyano benzo[b]thieno[3,2-b]quinoline 4e, 32%. Mp: 244–245 °C. 1H NMR (300 MHz, CDCl3): δ 8.78 (1H, d, J = 8.1Hz); 8.67 (1H, s); 8.45 (1H, d, J = 10.8 Hz); 8.34 (1H,s); 7.89 (2H, dd, J = 8.1Hz & 2.7Hz); 7.71 (1H, t, J = 8.1Hz); 7.61(1H, t, J = 8.1Hz); Calcd for: C16H18N2S: C, 73.84; H, 3.07; N, 10.77; Found: C, 73.52; H, 3.16; N, 10.51. Compound 5e was obtained from compound 4e in 74% yield. Mp: 204–205 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.35 (1H, s), 9.25 (1H, d, J = 1.8 Hz), 8.96 (1H, d, J = 8.7 Hz), 8.85 (1H, d, J = 8.1Hz), 8.47 (1H, dd, J = 1.8, 8.1Hz), 8.01 (1H, dt, J = 1.8, 8.1Hz), 7.90 (1H, d, J = 8.1Hz), 7.57(1H, dt, J = 1.8, 8.1Hz), 5.05 (3H,s). Calcd for: C17H11IN2S·2.5H2O: C 45.65, H 3.61, N 6.26; Found: C 45.61, H 3.55, N 6.25.
2-Methoxy-5-methyl-benzo[b]thieno[3,2-b]quinolin-5-ium iodide, 5f
Using method C and 5-iodoanthranilic acid as starting material, 2-iodo benzo[b]thieno[3,2-b]quinolinone was obtained and subsequently converted to 2-methoxy benzo[b]thieno[3,2-b]quinoline 4f, 32%. 1H NMR (300 MHz, CDCl3): δ8.60 (1H, d, J = 6.9 Hz), 8.44 (1H, s), 8.16 (1H, d, J = 9.3 Hz), 7.81 (1H, d, J = 7.2 Hz), 7.58 (1H, dt, J = 2.7, 7.2 Hz), 7.53 (1H, dt, J = 2.7, 7.2 Hz), 7.41 (1H, dd, J = 2.7, 9.3 Hz), 7.11 (1H, d, J = 2.7 Hz), 3.95 (3H, s). Starting from compound 4f, and using method B, 5f was obtained in 51% yield. Mp: 216–217 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.77 (1H, s), 9.02 (1H, d, J = 8.4 Hz), 8.78 (1H, d, J = 9.8 Hz), 8.43 (1H, d, J = 8.1 Hz), 7.96 (2H, m), 7.82 (2H, m), 5.06 (3H, s), 4.04 (3H, s). Calcd for C17H14INOS·0.5H2O: C 49.05, H 3.63, N 3.36; Found: C 49.01, H 3.60, N 3.30.
2-Carboxy-5-methyl-benzo[b]thieno[3,2-b]quinolin-5-ium iodide, 5g
A mixture of 2-cyano benzo[b]thieno[3,2-b]quinoline, 4e (0.302 g, 1.16 mmol), conc. H2SO4 and H2O (1:1 mixture, 4 mL) was stirred at 55 °C overnight. After cooling to rt, H2O (80 mL) was added. A solid was collected, and washed with water. Further purification was conducted on silica gel to afford a solid 4g, 200 mg, 65%; eluent: CH2Cl2:EtOAc (4/2). The crude product was used for alkylation without further purification. Using Method B, compound 4g was converted to 5g, 64%. Mp: decomposes at 203–204 °C. 1H NMR (300 MHz, DMSO-d6): δ 13.08 (1H, brs), 9.08 (1H, d, J = 9.0Hz), 8.95 (1H, d, J = 3.0 Hz), 8.84 (1H, d, J = 9.0 Hz), 8.65 (1H, dd, J = 9.0 Hz & 3.0 Hz), 8.04 (1H, t, J = 9.0 Hz), 7.91 (1H, d, J = 3.0 Hz), 7.86(2H, m), 5.10 (3H, s). Calcd for. C17H12INO2S: C 48.47, H 2.87, N 3.32; Found: C 48.72, H 2.61, N 3.22.
2-Carbamoyl-5-methyl-benzo[b]thieno[3,2-b]quinolin-5-ium iodide, 5h
A mixture of 2-cyano benzo[b]thieno[3,2-b]quinoline, 4e (0.2 g, 0.76 mmol) and polyphophoric acid (30 g) was heated at 110 °C for 3 h. After cooling to rt, the mixture was poured onto ice/water (100 mL) and neutralized with saturated solution of NaHCO3. A solid was obtained, washed with H2O and EtOAc to yield 180 mg, 84%. The crude product, 4h was used for alkylation without further purification. Using Method B, compound 4h was converted to 5h, 53%. Mp: 234–235 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.05 (1H, s), 9.12 (1H, d, J = 9.0 Hz), 8.96 (1H, d, J = 3.0 Hz), 8.93 (1H, s), 8.55 (1H, dd, J = 3.0, 9.0, Hz), 8.54 (1H, s), 8.51 (1H, d, J = 3.0 Hz), 8.05 (1H, t, J = 3.0 Hz), 7.87 (2H, m); 5.12 (3H, s). Calcd for: C17H13IN2OS·1.25H2O: C 46.11, H 3.53, N 6.33; Found: C 46.40, H 3.16, N 6.19.
5-Methyl-benzo[b]thieno[3,2-b]quinolin-5-ium triflate, 6
A mixture of benzo[b]thieno[3,2-b]quinoline 4b (80 mg, 0.34 mmol) in toluene (5 mL) and MeOTf (0.2 mL) was stirred for 12h at rt. A yellow solid appeared and was collected by filtration. Recrystalization from MeOH-Et2O afforded 5-Methyl benzo[b]thieno[3,2-b]quinolin-5-ium triflate 6, 114 mg, 84%. Mp: 237–238 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.99 (1H, s), 9.10 (1H, d, J = 8.4 Hz), 8.87 (1H, d, J = 9.0 Hz), 8.50 (1H, d, J = 3.6 Hz), 8.48 (1H, d, J = 3.6 Hz), 8.34 (1H, t, J = 8.4 Hz), 8.09 (1H, d, J = 7.8 Hz), 8.02 (1H, d, J = 7.8 Hz), 7.86 (1H, t, J = 8.1 Hz), 5.10 (3H, s). Calcd for C17H12F3NO3S2·0.2H2O: C 50.66, H 3.00, N 3.49; Found: C 50.58, H 2.89, N 3.52.
5-(5′-Cyclohexyl-pentyl)-benzo[b]thieno[3,2-b]quinolin-5-ium iodide, 7
Using Method B, compound 4b was converted to 7 in 30% yield. Mp: 182–184 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.02 (1H, s), 8.64 (1H, d, J = 9.0 Hz), 8.65 (1H, d, J = 8.4 Hz), 8.51 (2H, d, J = 8.2 Hz), 8.36 (1H, t, J = 7.5 Hz), 8.10 (1H, d, J = 7.2 Hz), 8.04 (1H, d, J = 8.0 Hz), 7.92 (1H, t, J = 7.2 Hz), 5.50 (2H, t, J = 7.8 Hz), 2.19 (2H, m), 1.66 (7H, m), 1.44 (2H, m), 1.21 (6H, m), 0.86 (2H, m). Calcd for C26H30INS·2.5 H2O: C 55.71, H 6.29, N 2.50; Found: C 55.68, H 5.88, N 2.29.
Synthesis of Benzothieno[3,2-b]pyridines 10a–d
3-Bromo-1-(4-nitro-phenoxy)-pyridinium tetrafluoroborate, 8
To a solution of 3-bromopyridine N-oxide in (4 g, 23 mmol) in anhydrous CH3CN (20 mL) was added 4-nitrobenzene diazonium tetrafluoroborate (5.5 g, 23 mmol) with vigorous stirring. The mixture was allowed to stir at rt for 48 hours. EtOAc (50 mL) was added, and the solution was allowed to sit for an additional 24 hours, the salt proceeded to crystallize out of solution. The crystals where collected by filtration and washed with MeOH to yield pure 3-bromo-1-(4-nitro-phenoxy)-pyridinium tetrafluoroborate, 8, 6.1 g, 68%. Mp: 149–150 °C (lit. 155–156 °C). 1H NMR (DMSO-d6): δ 10.27 (1H, s), 9.77 (1H, d, J = 6.4 Hz), 9.13 (1H, d, J = 8.3 Hz), 8.39 (1H, dd, J = 6.4, 8.4 Hz), 8.36 (2H, d, J = 9.27 Hz), 7.52 (2H, d, J = 8.2 Hz).
3-Bromo-2-(2-hydroxy-5-nitrophenyl) pyridine, 9a
A solution of 3-bromo-1-(4-nitrophenoxy)-pyridinium tetrafluoroborate, 8 (20 g, 0.052 mol) in anhydrous CH3CN was refluxed for 30 min, after which Et3N (5 mL) was added dropwise. The mixture was allowed to reflux for an additional hour, after which the solvent was removed in vacuo. The remaining residue was purified by chromatography on silica gel with 20–50 % EtOAc/Hexane to yield 2-(3-bromo-pyridin-2-yl)-5-nitrophenol 9a, 10 g, 65%. mp: 157–159 °C (lit. 165 °C); 1H NMR (DMSO-d6): δ 11.40 (1H, s), 8.63 (1H, dd, J = 1.0, 4.4 Hz), 8.27 (1H, dd, J = 2.9, 9.0 Hz), 8.17 (1H, d, J = 8.4 Hz), 8.05 (1H, d, J = 2.8 Hz), 7.39 (1H, dd, J = 4.8, 8.0 Hz), 7.10 (1H, d, J = 7.90 Hz).
O-[2-(3-bromopyridine)-4-nitrophenyl] dimethylthiocarbamate, 9b
To a solution of 3-bromo-2-(2-hydroxy-5-nitrophenyl) pyridine, 9a (5g, 0.017 mol) and KOH (2.54 g) in H2O (90 ml) cooled to 5 °C, was added dropwise, a solution of dimethylthiocarbamoyl chloride (3.86 g, 0.031 mol) in anhydrous THF (40 mL) over a period of 15 minutes. The mixture was allowed to stir at rt for 45 minutes, after which it was poured into an aqueous solution of 10% KOH and extracted with EtOAc (5 x 200 ml). The extracts were combined, washed with brine, dried with MgSO4, and evaporated in vacuo. The crude product was poured unto a column of silica gel and eluted with EtOAc/Hexane (1:4) to give o-[2-(3-bromopyridine)-5-nitrophenyl] dimethylthiocarbamate, 9b, as a light brown solid, 4.82 g, 75%. mp: 102–103 °C (lit. 107–108 °C). 1H NMR (300 MHz, DMSO-d6): 8.64 (1H, dd, J = 1.3, 4.7 Hz), 8.37 (1H, dd, J = 3.2, 8.8 Hz), 8.31 (1H, d, J = 2.83 Hz), 8.22 (1H, d, J = 1.3 Hz), 7.55 (1H, d, J = 8.8 Hz), 7.42 (1H, dd, J = 4.5, 8.1 Hz), 3.14 (3H, s), 3.10 (3H, s).
8-Nitro-benzo[4,5]thieno[3,2-b]pyridine, 10a
Compound 9b (2 g, 5.23 mmol) was heated under nitrogen for 1 hour at 190–195 °C. After cooling to room temperature, a solution of potassium hydroxide (1.10 g) in water (3 mL) and ethylene glycol (15 mL) was added and the resulting solution was refluxed for 45 minutes, and then allowed to cool to room temperature. On cooling, 8-nitro-benzo[4,5]thieno[3,2-b]pyridine 10a precipitated out of solution and was collected by filtration, washed with EtOAc and dried under vacuum, 1 g, 83%. mp: 256–258 °C (lit.# 258–260 °C). 1H NMR (DMSO-d6): δ 9.07 (1H, s), 8.85 (1H, d, J = 4.4 Hz), 8.65 (1H, d, J = 8.2 Hz), 8.45 (2H, m), 7.65 (1H, m).
8-Amino-benzo[4,5]thieno[3,2-b]pyridine, 10b
To a solution of SnCl2 (7.2 g, 38 mmol) in 6N HCl (72 mL) was added 7-nitro benzo[4,5]thieno[3,2-b]pyridine, 10a (1.2 g, 5 mmol), and the solution was heated to boiling under reflux for 3.5 hours. On cooling, the solution was brought to pH 10 with 30% NaOH and extracted with EtOAc (3 x 200 ml). The combined extracts was dried with MgSO4 and concentrated in vacuo. The residue was purified on a column of silica gel eluted with EtOAc to yield 7-amino-benzo[4,5]thieno[3,2-b]pyridine, 10b, 0.8 g, 80%. mp: 183–185 (lit. 186–187 °C); 1H NMR (DMSO-d6): δ 8.65 (1H, m), 8.37 (1H, d, J = 8.2 Hz), 7.66 (1H, d, J = 8.5 Hz), 7.56 (1H, d, J = 2.2 Hz), 7.42 (1H, dd, J = 4.5, 8.0 Hz), 6.93 (1H, dd, J = 2.4, 8.5 Hz), 5.83 (2H, brs).
Benzo[4,5]thieno[3,2-b]pyridine, 10c
To a mixture of conc. HCl (5 mL) and H2O (5 mL) was added 8-amino-benzo[4,5]thieno[3,2-b]pyridine, 10b (1 g, 5 mmol). The mixture was cooled to 0 °C, and a cold solution of NaNO2 (1 g, 14.5 mmol) was added slowly with the temperature being kept at 0–5 °C. The diazotized solution was filtered and cooled to 0 °C, and a cold solution of H3PO2 (30 ml) was added at 0 °C. The solution was allowed to stir at rt for 48 hours, after which the pH was made alkaline (9–12) using KOH pellets and the solution was extracted with EtOAc (3 x 100 mL). The combined extracts was dried with MgSO4 and concentrated in vacuo. The residue was then dried under vacuum to yield benzo[4,5]thieno[3,2-b]pyridine, 10c as a light brown solid, 610 mg, 65%. mp: 80–81 °C (lit. 81–82 °C); 1H NMR (CDCl3): δ 8.74 (1H, dd, J = 1.4, 4.7 Hz), 8.52 (1H, m), 8.17 (1H, dd, J = 1.4, 8.2 Hz), 7.6 (3H, m), 7.37 (1H, dd, J = 4.7, 8.2 Hz).
General procedure for the N-alkylation of benzothieno[3,2-b]pyridine
1-Methyl-benzo[4,5]thieno[3,2-b]pyridin-1-ium iodide 11c: (Method D)
To a solution of benzothieno[3,2-b]pyridine, 10c (200 mg, 1.08 mmol) in TMS (0.2 mL) was added CH3I (0.2 mL). The mixture was heated at 110 °C for 12 hours in a sealed pressure tube. After cooling to rt, EtOAc (10 mL) was added to precipitate a solid. The precipitate was collected, washed with additional EtOAc and recrystalized from MeOH-Et2O as a yellow powder, 1-methyl-benzo[4,5]thieno[3,2-b]pyridin-1-ium iodide, 11c, 252 mg, 71%. mp 194–196 °C; 1H NMR (300 MHz, DMSO-d6): δ 9.3 (1H, d, J = 8.0 Hz), 9.19 (1H, d, J = 6.0 Hz), 8.85 (1H, d, J = 8.4 Hz), 8.45 (1H, d, J = 8.0 Hz), 8.18 (1H, dd, J = 6.0, 8.1 Hz), 7.94 (1H, t, J = 7.5 Hz), 7.81 (1H, t, J = 7.5 Hz), 4.91 (3H, s). Calcd for C12H10INS: C 44.05, H 3.08, N 4.29; Found: C 44.30, H 3.07, N 4.13.
1-Methyl-8-nitro-benzo[4,5]thieno[3,2-b]pyridin-1-ium iodide, 11a
Using method D and 10a, compound 11a, was obtained in 78% yield. Mp: 254–255 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.49 (1H, d, J = 8.2 Hz), 9.46 (1H, d, J = 1.5 Hz), 9.34 (1H, d, J = 6.0 Hz), 8.760 (1H, d J = 9.0 Hz), 8.71 (1H, dd, J = 1.8, 9.0 Hz), 8.31 (1H, dd, J = 6.0. 8.3 Hz), 5.03 (3H, s). Calcd for C13H9IN2O2S: C 38.73, H 2.44, N 7.53; Found: C 38.70, H 2.47, N 7.50.
8-Dimethylamino-1-methyl-benzo[4,5]thieno[3,2-b]pyridin-1-ium iodide, 11b
Using method D and starting material 10b (100 mg, 0.5 mmol), a mixture of compounds 8-amino-1-methyl-benzo[4,5]thieno[3,2-b]pyridin-1-ium iodide and 8-methylamino-1-methyl-benzo[4,5]thieno[3,2-b]pyridin-1-ium iodide was obtained. The above mixture was treated with MeI (0.2 mL) and NaHCO3 (1.0 g, 11.9 mmol) with stirring for 12 h at room temperature. Solid was filtered off, and filtrate was concentrated in vacuo, to dry. The residue was crystallized from MeOH-Et2O to give compound 11b, 120 mg, 65%. Mp: 207–208 °C. 1H NMR (300 MHz, DMSO-d6): δ9.26 (1H, d, J = 8.1 Hz), 9.13 (1H, d, J = 6.0 Hz), 8.19 (1H, d, J = 9.0 Hz), 8.08 (1H, dd, J = 6.0, 8.1 Hz), 7.76 (1H, d, J = 2.1 Hz), 7.47 (1H, dd, J = 2.1, 9.0 Hz), 4.94 (3H, s), 3.32 (3H, s), 3.25 (3H, s). Calcd for C14H15IN2S· 3.8H2O: C 38.33, H 3.45, N 6.39; Found: C 38.27, H 3.77, N 6.24.
1-(5′-Phenyl-pentyl)-benzo[4,5]thieno[3,2-b]pyridin-1-ium iodide, 11d
Using method D, a mixture of compound 10c (70 mg, 0.38 mmol), 5-phenylpentyl iodide (0.2 ml) produced 11d, 80 mg, 82%. Mp: 158–160 °C. 1H NMR (300 MHz, DMSO-d6): δ 9.40 (1H, d, J = 8.1 Hz), 9.22 (1H, d, J = 5.7 Hz), 8.57 (1H, d, J = 8.4 Hz), 8.47 (1H, d, J = 7.9 Hz), 8.20 (1H, dd, J = 6.0, 8.1 Hz), 7.94 (1H, t, J = 7.8 Hz), 7.84 (1H, t, J = 7.8 Hz), 7.23 (2H, m), 7.14 (3H, m), 5.24 (2H, t, J = 7.1 Hz), 2.55 (2H, t, J = 7.6 Hz), 2.07 (2H, m), 1.64 (2H, m), 1.51 (2H, m). Calcd for C22H22INS·1.1H2O: C 55.14, H 4.63, N 2.92; Found: C 55.19, H 4.69, N 2.99.
Biological Testing
Antifungal and Antibacterial Testing
All organisms were obtained from the American Type Culture Collection (Manassas, VA) and include Candida albicans ATCC 90028, Cryptococcus neoformans ATCC 90113, Aspergillus fumigatus ATCC 90906 and Mycobacterium intracellulare ATCC 23068. Susceptibility testing was performed using a modified version of the NCCLS methods.19 M. intracelluare is tested using a modified method of Franzblau, et al.20 DMSO solutions of samples were serially-diluted in saline, and transferred in duplicate to 96 well microplates. Microbial suspensions were diluted in broth to afford desired colony forming units/mL according to the 0.5 McFarland Standard [C. albicans: either Saboraud Dextrose broth (SDB) or RPMI 1640, C. neoformans: SDB, A. fumigatus: either YM broth (for MICs) or RPMI-1640 + 5% Alamar Blue (for IC50 determination), M. intracellulare: Middlebrook 7H9 broth with OADC enrichment + 5% Alamar Blue.] After adding microbial cultures to the samples affording a final volume of 200μL and final test concentration starting with 20μg/mL, plates were read prior to and after incubation using either fluorescence at 544ex/590em (M. intracellulare, A. fumigatus) using the Polarstar Galaxy Plate Reader (BMG LabTechnologies, Germany) or optical density at 630nm using the EL-340 Biokinetics Reader (Bio-Tek Instruments, Vermont). Growth (saline only), solvent and blank (media only) controls were included on each test plate. Drug controls [Ciprofloxacin (ICN Biomedicals, Ohio) for bacteria and Amphotericin B (ICN Biomedicals, Ohio) for fungi] are included in each assay. Percent growth is calculated and plotted versus test concentration to afford the IC50 (sample concentration that affords 50% growth of the organism). The minimum inhibitory concentration (MIC) was determined by visually inspecting the plate, and is defined as the lowest test concentration that allows no detectable growth (for Alamar Blue assays, no color change from blue to pink).
Screening for in vitro Antimalarial Activity
The screening for antimalarial activity was performed as described earlier 21. For the assay a suspension of red blood cells infected with D6 or W2 strains of P. falciparum (200 μL, with 2% parasitemia and 2% hematocrit in RPMI 1640 medium supplemented with 10% human serum and 60 μg/mL amikacin) is added to the wells of a 96-well plate containing 10 μl of test samples diluted in medium at various concentrations. The plate is incubated at 37 °C, for 72 h in a modular incubation chamber flushed with a gas mixture of 90% N2, 5% O2, and 5% CO2 . Parasitic LDH activity is determined by using Malstat™ reagent (Flow Inc., Portland, OR). Briefly, 20 μl of the incubation mixture is mixed with 100 μl of the Malstat™ reagent and incubated at room temperature for 30 min. 20 μl of a 1:1 mixture of NBT/PES (Sigma, St. Louis, MO) is then added, and the plate is further incubated in the dark for 1 h. The reaction is stopped by the addition of 100 μl of a 5% acetic acid solution. The plate is read at 650 nm on an EL-340 Biokinetics Reader (Bio-Tek Instruments, Vermont). Percent growth inhibition is calculated and IC50 values are computed from the dose response curves. Artemisinin and chloroquine are included as the drug controls. DMSO (0.25%) is used as vehicle control.
Assay for in vitro antileishmanial activity
Antileishmanial activity was tested in vitro against a culture of Leishmania donovani promastigotes, grown in RPMI 1640 medium supplemented with 10% fetal calf serum (Gibco Chem. Co.) at 26 ° C. A 3 day-old culture was diluted to 5×105 promastigotes/mL. Drug dilutions were prepared directly in cell suspension in 96-well plates. Plates were incubated at 26 ° C for 48 h and growth of leishmania promastigotes was determined by Alamar Blue assay as described earlier21. Fluorescence was measured on a Fluostar Galaxy plate reader (BMG Lab Technologies) at excitation wavelength of 544 nm and emission wavelength of 590 nm. Pentamidine and amphotericin B were used as the standard antileishmanial agents. IC50 and IC90 values were computed from dose curves generated by plotting percent growth versus drug concentration.
Cytotoxicity Assay
The in vitro cytotoxicity was determined against mammalian kidney fibroblast (VERO) cells. The assay is performed in 96-well tissue culture-treated microplates and compounds were tested up to a highest concentration of 23.8 μg/ml as described earlier.22 In brief, cells (25,000 cells/well) were seeded to the wells of the plate and incubated for 24 h. Samples were added and plates were again incubated for 48 h. The number of viable cells was determined according to neutral red assay as previously described.22 IC50 values were determined from dose curves of growth inhibition versus concentration. Doxorubicin was used as a positive control, while DMSO was used as the negative (vehicle) control.
Scheme 3.
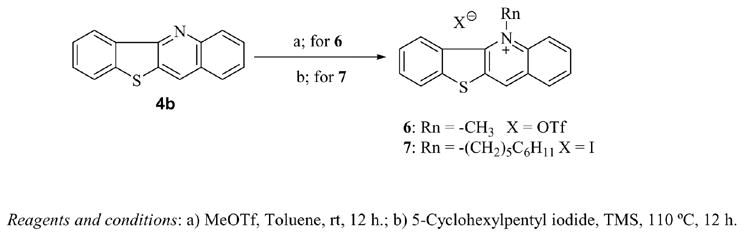
N-5 Alkylation of Benzothieno[3,2-b]quinoline.
Scheme V.
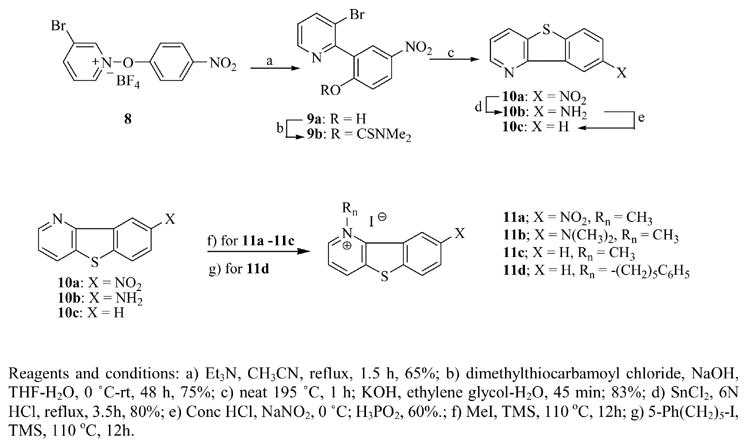
Synthesis of Benzothieno[3,2-b]pyridinium Iodides 11a–d
Acknowledgments
We gratefully acknowledge research support from the National Institutes of Health, NIAID, through AREA grant number R15 Al37976-01, RCMI grant number G12 RR 03020, MBRS grant # GM 08111 and Title III grant to Florida A&M University. The authors would also like to thank Ms Marsha Wright and Mr John Trott for conducting the antimicrobial and antimalarial testing, which was supported by the NIH, NIAID, Division of AIDS, Grant No. AI 27094, and the USDA Agricultural Research Service Specific Cooperative Agreement No. 58-6408-2-0009. We also gratefully acknowledge John Puziss of Bristol-Myers Squib and Margaret Shaw of Eli Lilly for the activity and toxicity data in Tables 1 and 3. This work was supported in part by the Pharmaceutical Research Center NIH/NCRR 1 C06-RR12512-01 grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Ablordeppey SY, Fan P, Ablordeppey JH, Mardenborough L. Curr Med Chem. 1999;6:1151–1195. [PubMed] [Google Scholar]
- 2.UNAIDS Report, 2006 at www.unaids.org
- 3.a) Ablordeppey SY, Hufford CD, Borne RF, Dwuma-Badu D. Planta Med. 1990;56:416. doi: 10.1055/s-2006-960998. [DOI] [PubMed] [Google Scholar]; b) Singh M, Singh MP, Ablordeppey SY. Drug Dev Ind Pharm. 1996;22:379–383. [Google Scholar]; c) Oyekan AO, Ablordeppey SY. Gen Pharmacol. 1993;24:1285–1290. doi: 10.1016/0306-3623(93)90382-8. [DOI] [PubMed] [Google Scholar]; d) Oyekan AO, Ablordeppey SY. Gen Pharmacol. 1993;24:461–469. doi: 10.1016/0306-3623(93)90333-s. [DOI] [PubMed] [Google Scholar]; e) Oyekan AO, Ablordeppey SY. Med Chem Res. 1996;6:602–610. [Google Scholar]
- 4.Ablordeppey SY, Fan P, Clark AM, Nimrod A. Bioorg Med Chem. 1999;7:343–349. doi: 10.1016/s0968-0896(98)00244-2. [DOI] [PubMed] [Google Scholar]
- 5.Dwuma-Badu D, Ayim JS, Fiagbe NI, Knapp JE, Schiff PL, Jr, Slatkin DJ. J Pharm Sci. 1978;67:433–434. doi: 10.1002/jps.2600670350. [DOI] [PubMed] [Google Scholar]; b) Boakye-Yiadom K, Heman-Ackah SM. J Pharm Sci. 1979;68:1510–1514. doi: 10.1002/jps.2600681212. [DOI] [PubMed] [Google Scholar]; c) Paulo A, Pimentel M, Viegas S, Pires I, Duarte A, Cabrita J, Gomes ET. J Ethnopharmacol. 1994;44:73–77. doi: 10.1016/0378-8741(94)90071-x. [DOI] [PubMed] [Google Scholar]; d) Sawer IK, Berry MI, Brown MW, Ford JL. Journal of Applied Bacteriology. 1995;79:314–321. doi: 10.1111/j.1365-2672.1995.tb03143.x. [DOI] [PubMed] [Google Scholar]; d) Paulo A, Duarte A, Gomes ET. J Ethnopharmacol. 1994;44:127–130. doi: 10.1016/0378-8741(94)90079-5. [DOI] [PubMed] [Google Scholar]; e) Yang SW, Abdel-Kader M, Malone S, Werkhoven MCM, Wisse JH, Bursuker I, Neddermann K, Fairchild C, Raventos-Suarez C, Menendez AT, Lane K, Kingston DGI. J Nat Prod. 1999;62:976–983. doi: 10.1021/np990035g. [DOI] [PubMed] [Google Scholar]; f) Cimanga K, De Bruyne T, Lasure A, Van Poel B, Pieters L, Claeys M, Vanden Berghe D, Kambu K, Tona L, Vlietinck AJ. Planta Medica. 1996;62:22–27. doi: 10.1055/s-2006-957789. [DOI] [PubMed] [Google Scholar]; g) Noamesi BK, Bamgbose SO. Planta Med. 1980;39:51–56. doi: 10.1055/s-2008-1074902. [DOI] [PubMed] [Google Scholar]; h) Noamesi BK, Bamgbose SO. Planta Med. 1982;44:241–245. doi: 10.1055/s-2007-971458. [DOI] [PubMed] [Google Scholar]
- 6.Mardenborough LG, Zhu XY, Fan P, Jacob MR, Khan SI, Walker LA, Ablordeppey SY. Bioorg Med Chem. 2005;13:3955–3963. doi: 10.1016/j.bmc.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 7.Ablordeppey SY, Fan P, Li S, Clark AM, Hufford CD. Bioorg Med Chem. 2002;10:1337–1346. doi: 10.1016/s0968-0896(01)00401-1. [DOI] [PubMed] [Google Scholar]
- 8.Mardenborough LG, Fan PC, Ablordeppey SY, Nimrod A, Clark AM. Med Chem Res. 1999;9:118–132. [Google Scholar]
- 9.Arzel E, Rocca P, Grellier P, Labaeid M, Frappier F, Gueritte F, Gaspard C, Marsais F, Godard A, Queguiner G. J Med Chem. 2001;44:949–960. doi: 10.1021/jm0010419. [DOI] [PubMed] [Google Scholar]
- 10.a) Bierer DE, Fort DM, Mendez CD, Luo J, Imbach PA, Dubenko LG, Jolad SD, Gerber RE, Litvak J, Lu Q, Zhang P, Reed MJ, Waldeck N, Bruening RC, Noamesi BK, Hector RF, Carlson TJ, King SR. J Med Chem. 1998;41:894–901. doi: 10.1021/jm9704816. [DOI] [PubMed] [Google Scholar]; b) Bierer DE, Dubenko LG, Zhang P, Lu Q, Imbach PA, Garofalo AW, Phuan P-W, Fort DM, Litvak J, Gerber RE, Sloan B, Luo J, Cooper R, Reaven GM. J Med Chem. 1998;41:2754–2764. doi: 10.1021/jm970735n. [DOI] [PubMed] [Google Scholar]
- 11.Yamato Mastoshi. Eur Pat Appl. 1988:EP 264124. [Google Scholar]
- 12.Radl S, Konvicka P, Vachal P. J Heterocyclic Chem. 2000;37:855–862. [Google Scholar]
- 13.Abramovitch RA, Inbasekaran MN, Miller AL, Hanna JM., Jr J Heterocyclic Chem. 1982;19:509. [Google Scholar]
- 14.Bartsch RA, Yang W., II J Heterocycl Chem. 1984;21:1063–1064. [Google Scholar]
- 15.a) Feldmesser M, Tucker S, Casadevall A. Trends Microbiol. 2001;9:273–278. doi: 10.1016/s0966-842x(01)02035-2. [DOI] [PubMed] [Google Scholar]; b) Casadevall A, Perfect JR. Cryptococcus neoformans. ASM Press; Washington, DC, USA: 1998. pp. 351–380. [Google Scholar]; c) Mitchell AP. J Clin Invest. 2006;116:1481–1483. doi: 10.1172/JCI28842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.http://www.who.int/mediacentre/factsheets/fs116/en/
- 17.Cimanga K, De Bruyne T, Pieters L, Vlietinck AJ, Turger CA. J Nat Prod. 1997;60:688–691. doi: 10.1021/np9605246. [DOI] [PubMed] [Google Scholar]
- 18.Lisgarten JN, Pous J, Coll M, Wright CW, Aymami J. Acta Crystallographica, Section D: Biological Crystallography. 2002;D58:312–313. doi: 10.1107/s0907444901018960. [DOI] [PubMed] [Google Scholar]
- 19.National Committee for Clinical Laboratory Standards. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: M7-A5. 2 Vol. 20. National Committee for Clinical Laboratory Standards; Wayne, PA: 2000. [Google Scholar]; b) National Committee for Clinical Laboratory Standards. Reference method for broth dilution antifungal susceptibility testing of yeasts: Approved standard M27-A2. 15 Vol. 22. National Committee for Clinical Laboratory Standards; 2002. [Google Scholar]; c) National Committee for Clinical Laboratory Standards. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard, M38-A. 16 Vol. 22. National Committee on Clinical Laboratory Standards; 2002. [Google Scholar]; d) National Committee for Clinical Laboratory Standards. Tentative standard M24-T2. 2. 26 Vol. 20. National Committee for Clinical Laboratory Standards; Wayne, PA: 2000. Susceptibility testing of mycobacteria, nocardia, and otheraerobic actinomycetes. [Google Scholar]
- 20.Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. J Clin Microbiol. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain M, Khan SI, Tekwani BL, Jacob MR, Singh S, Singh PP, Jain R. Bioorg Med Chem. 2005;13:4458–4466. doi: 10.1016/j.bmc.2005.04.034. [DOI] [PubMed] [Google Scholar]
- 22.Mustafa J, Khan SI, Ma G, Walker LA, Khan IA. Lipids. 2005;40:375–382. doi: 10.1007/s11745-006-1397-x. [DOI] [PubMed] [Google Scholar]