

Abstract
T-cell activation is the net product of competing positive and negative signals transduced by regulatory molecules on antigen-presenting cells (APCs) binding to corresponding ligands on T cells. Having previously identified DC-HIL as a receptor expressed by APCs that contains an extracellular immunoglobulin (Ig)–like domain, we postulated that it plays a role in T-cell activation. To probe this function, we created soluble recombinant DC-HIL, which we observed to bind activated (but not resting) T cells, indicating that expression of the putative ligand on T cells is induced by activation. Binding of DC-HIL to naive T cells attenuated these cells' primary response to anti-CD3 antibody, curtailing IL-2 production, and preventing entry into the cell cycle. DC-HIL also inhibited reactivation of T cells previously activated by APCs (secondary response). By contrast, addition of soluble DC-HIL to either allogeneic or ovalbumin-specific lymphocyte reactions augmented T-cell proliferation, and its injection into mice during the elicitation (but not sensitization) phase of contact hypersensitivity exacerbated ear-swelling responses. Mutant analyses showed the inhibitory function of DC-HIL to reside in its extracellular Ig-like domain. We conclude that endogenous DC-HIL is a negative regulator of T lymphocyte activation, and that this native inhibitory function can be blocked by exogenous DC-HIL, leading to enhanced immune responses.
Introduction
T-cell activation is dependent on signals delivered by antigen-presenting cells (APCs) to the antigen (Ag)–specific T-cell receptor (TCR) and accessory receptors on T cells.1 The principal stimulatory accessory signal is transmitted by B7-1 (CD80) or B7-2 (CD86) on APCs to the CD28 receptor on T cells.2 Interestingly, engagement of the same B7-1 or B7-2 ligand to CTLA-4 (CD152) on T cells markedly attenuates T-cell responses.3,4 The importance of CTLA-4 as an inhibitory regulator of T-cell activation is illustrated by death of CTLA-4–deficient mice within 4 weeks of birth because of massive lymphocytic infiltration destroying critical organs.5
More recently, other inhibitory regulators of T-cell activation were identified, including PD-L1 (B7-H1) and PD-L2 (B7-DC) on APCs and PD-1 on T cells,6 BTLA on B cells and T helper 1 (Th1) effector cells and its ligand (herpes virus entry mediator) on T cells,7,8 and Tim-3 on APCs and Th1 effector cells and Tim-3 ligand on CD4+ T cells.9–11 The T-cell ligands possess a single immunoglobulin (Ig)–like variable (IgV) domain, and the APC receptors contain both IgV and Ig constant (IgC) domains.12 Interactions between ligand-receptor pairs are mediated predominantly by residues of Ig-like domains.12 Because of their structural and functional similarities to B7 molecules, these ligands/receptors are considered members of the B7 receptor superfamily.12
Ligation of PD-1 on T cells leads to inhibited T-cell responses that can be rescued by exogenous IL-2 or CD28 costimulation,13–15 although one report showed that binding of PD-L1 (B7-H1) to PD-1 stimulated T-cell proliferation and IL-10 secretion.16–18 PD-1 deficiency leads to exaggerated autoimmunity since PD-1 knockout mice develop splenomegaly, increased numbers of B and myeloid cells, increased serum IgG and IgA, and a lupus erythematosus–like disease with age.19,20 These mice are also markedly susceptible to Ag-induced experimental autoimmune encephalomyelitis (EAE).19,20 BTLA knockout mice do not exhibit developmental T- or B-cell defects, but their lymphocytes have heightened responses to anti-CD3 antibody (Ab) and to anti-IgM Ab;8 these mice are also prone to developing EAE.8 In the case of the Tim-3 pathway, its blockade by monoclonal Ab (mAb), Fc-fused soluble receptor, or gene disruption leads to exacerbated Th1-mediated autoimmune diabetes mellitus in nonobese diabetic (NOD) mice.10,11
T-cell expression of PD-1, BTLA, or Tim-3 resembles CTLA-4 in that it is not constitutive, but is induced by activation.21 Moreover, the costimulation delivered by each appears to be mediated through the TCR.12 By contrast, expression of PD-1, BTLA, Tim-3, or their ligands differ from CTLA-4 in that it is not restricted to T cells, but is expressed more widely to include B cells and APCs. Indeed, some of these ligands (PD-L1 and PD-L2) are also expressed in nonlymphoid tissues.12 Such broad expression profiles suggest that these molecules can modulate immune responses in secondary lymphoid organs and peripheral tissues,6 consistent with the observation that IFN-γ can induce expression of PD-1 ligands on nonlymphoid cells.13
Previously, we identified DC-HIL as a highly glycosylated type I transmembrane protein of 125 and 95 kDa containing an extracellular Ig-like domain.22 We also showed that DC-HIL is expressed constitutively at high levels on the surface of bone marrow–derived dendritic cells (BM-DCs; Figure S1, available on the Blood website; see the Supplemental Figures link at the top of the online article) and at lower levels on macrophages,22 and that its expression can be induced in nonlymphoid cells (keratinocytes) following IFN-γ treatment (Figure S2). Herein we present new evidence that DC-HIL is a negative regulator of T-cell activation. Our findings indicate that its putative ligand is expressed on activated (but not resting) T cells, and that binding of DC-HIL to this ligand attenuates T-cell activation triggered by anti-CD3 Ab or by APCs in a manner resembling the inhibitory function of PD-L1/PD-L2.
Materials and methods
Mice and cell culture
Female BALB/c and C57BL/6 (5-8 weeks old) mice were obtained from the Animal Breeding Center at The University of Texas Southwestern Medical Center (Dallas, TX). BALB/cTac-TgN(DO11.10)-Rag2tm1 mice23 were purchased from Taconic (Hudson, NY). Following National Institutes of Health guidelines, these animals were housed and cared for in the pathogen-free facility, and all animal studies were approved by the Institutional Animal Care Use Center of the same institution.
Production of DC-HIL-Fc protein
The Fc-fusion proteins (DC-HIL-Fc, its mutants, and Fc alone) were produced in COS-1 cells and purified as described previously.22
Isolation of T cells and binding of DC-HIL
Following manufacturer's recommendations, CD3+, CD4+, and CD8+ T cells were purified from spleen cells of BALB/c mice using pan–T-cell, CD4+, and CD8+ T-cell isolation kits (Miltenyi Biotec, Auburn, CA), respectively.
Splenic CD3+ T cells (1 × 106) were activated by immobilized anti-CD3 Ab (1 or 3 μg/mL) or concanavalin A (10 μg/mL; Sigma, St Louis, MO) for 3 days. Freshly isolated and activated CD3+ T cells were then treated with 5 μg/mL Fc blocker (BD Pharmingen, San Diego, CA) on ice for 30 minutes to block Fc-binding activity of Fc receptors on T cells and incubated with 5 μg/mL PE–anti-CD3 Ab (BD Pharmingen) and 10 μg/mL DC-HIL-Fc or control human IgG (hIgG) plus 5 μg/mL FITC–anti-human IgG (both from Jackson ImmunoResearch, West Grove, PA). In some experiments, activated and Fc-blocked CD3+ T cells were doubly stained with 5 μg/mL FITC-labeled anti-CD4 or anti-CD8 Ab (BD Pharmingen) and Fc proteins/PE–anti-human IgG (BD Pharmingen). The treated cells were also stained with anti-CD69 Ab or isotypic control IgG to evaluate activation levels. After staining, binding of Fc proteins to T cells and expression of marker molecules were analyzed by fluorescence-activated cell sorting (FACS).
T-cell proliferation and IL-2 assay
Purified CD4+ or CD8+ T cells (2 × 105/well) were cultured for 2 days in enzyme-linked immunosorbent assay (ELISA) wells (in triplicate) precoated with indicated doses of anti-CD3 Ab and Fc proteins or anti-CD28 mAb (BD Pharmingen). After pulsing with 3H-thymidine (1 μCi/well [0.037 MBq/well]) for 20 to 22 hours, cells were collected and evaluated for 3H radioactivity. Culture supernatant was used to measure IL-2 production using the mouse IL-2 ELISA kit (BD Pharmingen). To examine the effects of DC-HIL on reactivation of previously activated T cells, spleen cells (1 × 106/mL) isolated from Tac-TgN(DO11.10)-Rag2tm1 mice were cultured for 3 days in the presence of the ovalbumin OVA323-339 peptide (1 μg/mL).24 After purifying CD4+ T cells from the culture, cells (1 × 106/mL) were cultured for another day without stimuli and then subjected to T-cell proliferation and IL-2 assays.
Cell-cycle analyses
Cell cycles of CD4+ T cells treated with anti-CD3 Ab and Fc protein were examined using carboxy-fluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR). Purified CD4+ T cells (1 × 106) were labeled by 1 μM CFSE/DPBS for 15 minutes at 37°C. After another 30 minutes of incubation in culture medium, labeled T cells were cultured in ELISA wells coated with anti-CD3 Ab (0.3 μg/mL) and control IgG or DC-HIL-Fc (5 μg/mL). At different time points, cells were harvested to examine asynchronous cell division by FACS. Cell cycles of treated T cells were also analyzed using FITC-BrdU flow kit (BD Pharmingen), following the manufacturer's recommendations. BrdU incorporation and DNA content on a per-cell basis were analyzed by FACS and presented as dot plots.
MLR
BALB/c T cells and C57BL/6 spleen cells served as responders and stimulators, respectively. C57BL/6 spleen cells (5 × 104) were γ-irradiated (2000 Gy) and mixed with CD4+ T cells (2 × 105) purified from BALB/c spleen in 96-microwell plates. Fc fusion protein or control hIgG was added to the mixed leukocyte reaction (MLR) culture and incubated for varying periods. After 3H-thymidine pulsing for 20 hours, cells were harvested and the cell-incorporated 3H radioactivity was measured. T-cell proliferation was expressed as radioactivity left after subtracting background counts per minute (cpm; 3H cpm of control culture in which γ-irradiated responders and stimulators were mixed) from experimental cpm.
In vitro antigen presentation by DCs
BM cells were prepared from the femurs of BALB/c mice and cultured with 10 ng/mL of granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech, Rocky Hill, NJ).25 After culturing for 6 days, DCs were harvested, seeded on 96-well plates at a density of 5 × 105 cells/well, and cultured with OVA323-339 peptide (2 μg/mL)24 synthesized by the Protein Chemistry Technology Center, UT Southwestern. After 6 hours of antigen pulsing, DC cultures (5 × 104 cells) were added with CD4+ T cells (1 × 105/well) purified from the spleens of BALB/cTac-TgN(DO11.10)-Rag2tm1 mice.23 After coculture (2 days), cells and supernatant were harvested: cells were stained with FITC–anti-CD4 and PE–anti-CD69 Ab and determined by FACS for frequency of CD69+ cells in CD4+ T cells. The supernatant was assayed by ELISA for IL-2 production.
Knockdown of DC-HIL expression
A total of 2 different DC-HIL–targeted siRNAs (21 nucleotides long) were synthesized and purified by Qiagen (Valencia, CA): DC-HIL siRNA no. 3, sense 5′-r(AACUUGUCUGAUGAGAUCU)dTdT-3′ and antisense 5′-r(AGAUCUCAUCAGACAAGUU)dTdT-3′; DC-HIL siRNA no. 10, sense 5′-r(GCGUACAAGCCAAUAGGAA)dTdT-3′ and antisense, 5′-r(UUCCUAUUGGCUUGUACGC)dTdT-3′. Shuffled sequences of these 2 siRNAs were used as controls. A mixture of siRNA no. 3 and no. 10 (each 1.5 μg) or of the shuffled siRNAs was treated with 5 μL of Gene Silencer (Genlantis, San Diego, CA) in the serum-free RPMI for 20 minutes at room temperature and then added to BM-DCs (2 × 106 cells). After culturing for 4 hours at 37°C, 20% FCS-RPMI was added to the culture and allowed to incubate for another 24 hours. These transfected DCs were harvested and examined by Western blotting for protein expression of DC-HIL or mixed with OVA-specific T cells as described in “In vitro antigen presentation by HSCs.”
CH assays
BALB/c mice (n = 5) were sensitized for contact hypersensitivity (CH) on day 0 by painting 2% oxazolone (Ox; Sigma) in acetone–olive oil (4:1 in volume) on shaved abdominal skin (sensitization).26 Mice were challenged on day 6 by painting 1% Ox and solvent control onto right and left ears, respectively (elicitation). Thereafter, CH was assessed daily through day 12 by measuring ear thickness and calculating changes in ear swelling (thickness of right ear minus thickness of control left ear).27 Different panels of mice were injected intraperitoneally with DC-HIL-Fc or the control hIgG (10 mg/kg each) or DPBS on days −1, 1, and 3 (before and after sensitization) or on days 5, 7, and 9 (before and after elicitation). The Student t test was used to determine statistically significant differences in ear-swelling responses.
Histologic examination of skin and phenotyping of LN cells
After painting Ox on ears of Ox-sensitized mice treated with Fc protein (2 days), ear skin and draining lymph nodes (LNs) were procured. Ear skin was embedded in paraffin, thin-sectioned, and stained with hematoxylin-eosin (Sigma). Histologic examination was carried out under light microscopy using an Olympus BH2 microscope (Olympus, Center Valley, PA) at a magnification of × 10. In independent experiments, unsensitized mice were treated similarly and their draining LNs excised 2 days after ear challenge. LN cells were counted and examined for spontaneous proliferation and frequency of CD69+ cells.
For proliferation, LN cells (4 × 105/well) from untreated or treated mice were cultured without stimulation for 3 days and pulsed with 3H-thymidine (1 μCi/well [0.037 MBq/well]) for 20 hours. For CD69 expression, LN cells (5 × 105) were stained with FITC–anti-CD69 mAb (eBioscience, San Diego, CA) or FITC–isotypic control hamster IgG (BD Pharmingen) (2.5 μg/mL each) in the presence or absence of Ab directed at T-cell or B-cell surface markers (CD4, CD8, and B220; 2.5 μg/mL each) and examined by FACS for surface expression of CD69 in each leukocyte subpopulation.
Generation of mutants DC-HIL-Fc carrying the RAA mutant (replacement of RGD sequence with RAA) was generated as before.22 PRR and PKD mutants (lacking a region between amino acids 301 to 334 and 230 to 355, respectively) were produced by polymerase chain reaction (PCR)–based mutagenesis. Resulting nucleotides coding extracellular domains of DC-HIL mutants were inserted in-frame to the coding sequence of the human IgG1 Fc in pSecTagA plasmid (Invitrogen, Carlsbad, CA) using 3 restriction enzyme sites (from the 5′ end, HindIII, EcoRI, and XbaI). The mutant DC-HIL-Fc proteins were produced as described previously.22 The yield for each mutant was very similar to the wild-type and all preparations showed a single band reactive to anti–human IgG Ab.
Results
Activated T cells express ligands of DC-HIL
To study the function of DC-HIL, we created soluble DC-HIL receptors (DC-HIL-Fc) consisting of the extracellular domain fused with the Fc portion of human IgG1 (hIgG),22 and used FACS analysis to examine binding to T cells. DC-HIL-Fc did not bind to T cells freshly isolated from spleen of naive mice, but did so after the T cells were activated by concanavalin A (Figure 1A) or by immobilized anti-CD3 Ab (Figure 1B). Binding was noted as early as a day after stimulation and lasted for at least 3 days. We also examined binding to T-cell subsets (Figure 1B) and observed that activated CD4+ and CD8+ T cells were bound at frequencies of 35.4% and 10.2%, respectively. These results indicated that T cells express putative ligands of DC-HIL (DC-HIL-L) after activation. Involvement of Fc receptors in binding was excluded by failure of Fc blocker to inhibit binding of DC-HIL-Fc to T cells and inability of recombinant Fc alone to bind T cells (data not shown).
Figure 1.

DC-HIL-Fc binds to activated (but not resting) T cells. (A) Purified splenic CD4+ T cells were cultured with concanavalin A (10 μg/mL) for 3 days. After blocking the binding activity of Fc receptors on cells with a Fc blocker, treated T cells were stained with DC-HIL-Fc/ FITC-anti-human IgG Ab (open histograms), or corresponding control Abs (gray histograms). Binding of DC-HIL-Fc to T cells was analyzed by FACS. (B) Purified CD3+ T cells were treated with immobilized anti-CD3 Ab (1 μg/mL) for 3 days and then stained with FITC-labeled anti-CD4 or anti-CD8 Ab and with DC-HIL-Fc or hIgG/PE-anti-human IgG. Numbers in each quadrant represent percentages of the total cell population. Data shown are representative of 3 independent experiments.
Immobilized DC-HIL inhibits T-cell activation triggered via the TCR
We next used immobilized anti-CD3 Ab as a surrogate stimulator of TCR-dependent T-cell activation. CD4+ T cells were cultured in microculture wells precoated with increasing concentrations of anti-CD3 Ab and a constant concentration of DC-HIL-Fc or control hIgG. T-cell activation was assessed by proliferative capacity measured by 3H-thymidine incorporation. Treatment with immobilized anti-CD3 Ab led to activation of CD4+ T cells in a dose-dependent manner (Figure 2A). Coimmobilization of DC-HIL-Fc with anti-CD3 Ab attenuated CD4+ T-cell activation at all doses of anti-CD3 Ab tested; inhibition was most marked at suboptimal doses (0.1 and 0.3 μg/mL) of anti-CD3 Ab, and was counteracted by increasing doses of anti-CD3 Ab (1 and 3 μg/mL). By contrast, coimmobilization of control hIgG with anti-CD3 Ab had almost no effect on CD4+ T-cell activation. An irrelevant Fc fusion protein dectin-2-Fc,28 a C-type lectin receptor, had little to no effect on T-cell activation (Figure S3). The ability of DC-HIL to inhibit T-cell activation was also documented by little to no IL-2 secreted by T cells treated with DC-HIL-Fc (Figure 2B). Similar outcomes were observed for CD8+ T cells, although the degree of DC-HIL–induced inhibition was less than for CD4+ T cells (Figure 2C-D). These results indicate that binding of immobilized DC-HIL-Fc to its putative ligand on T cells inhibits TCR-dependent proliferation and IL-2 production. Indeed, addition of exogenous IL-2 rescued inhibition induced by DC-HIL (data not shown). We next titrated the inhibitory capacity of DC-HIL against the stimulatory ability of anti-CD3 Ab by keeping the dose of the latter constant (0.3 μg/mL) while adding the former in incremental doses (Figure 2E). Doses of DC-HIL-Fc greater than 5 μg/mL were required to inhibit T-cell activation.
Figure 2.

Immobilized DC-HIL-Fc inhibits T-cell activation triggered by anti-CD3 Ab. CD4+ (A-B) or CD8+ (C-D) T cells (2 × 105 each) purified from BALB/c spleens were cultured for 48 hours in microculture wells precoated with increasing doses of anti-CD3 Ab and with a constant dose (10 μg/mL) of DC-HIL-Fc (●), control hIgG (○), or neither (none; ▵). After pulsing with 3H-thymidine, cells and culture supernatant were harvested. 3H-thymidine incorporation into cells (A,C) and IL-2 production (B,D) were determined, and values (cpm or ng/mL) plotted at a logarithmic scale, respectively. (E) Titration of inhibitory function of DC-HIL-Fc. CD4+ T cells were cultured for 48 hours in microculture wells precoated with a constant dose (0.3 μg/mL) of anti-CD3 Ab and increasing doses of DC-HIL-Fc or control hIgG. (F) CD28 costimulation rescues DC-HIL-Fc–induced inhibition of CD4+ T-cell activation. Purified CD4+ T cells were cultured in wells precoated with anti-CD3 Ab (0.3 μg/mL), hIgG or DC-HIL-Fc (5 μg/mL), and anti-CD28 mAb (increasing doses). (G-H) Previously activated T cells were prepared from Tac-TgN(DO11.10)-Rag2tm1mice and reactivated by immobilized anti-CD3 Ab (varying doses) and DC-HIL-Fc or hIgG (constant dose). (I-J) Inhibitory function of DC-HIL-Fc was titrated against reactivation of previously activated T cells by anti-CD3 Ab (1 μg/mL). Proliferation (G,I) and IL-2 production (H,J) were measured. Results are expressed as mean values ± SDs. Data shown are representative of 6 (A-B), 3 (G-J), and 2 (C-F) experiments, respectively.
Because ligation of CD28 on T cells amplifies TCR-mediated T-cell activation,29 we questioned whether CD28 costimulation can rescue DC-HIL–induced inhibition (Figure 2F). Anti-CD28 mAb (in increasing doses) was immobilized on microwells precoated with DC-HIL-Fc or hIgG (10 μg/mL each) and anti-CD3 Ab (0.3 μg/mL). Purified CD4+ T cells were cultured in coated wells, and activation status was determined by 3H-thymidine incorporation. In the absence of anti-CD28 mAb, DC-HIL-Fc again markedly inhibited T-cell activation (Figure 2F). However, such inhibition was overcome by addition of anti-CD28 mAb in a dose-dependent manner, with 3 μg/mL anti-CD28 mAb completely rescuing DC-HIL-Fc–induced inhibition.
We also examined whether DC-HIL exerts inhibitory effects on previously activated T cells (Figure 2G-J). Spleen cells isolated from BALB/cTac-TgN(DO11.10)-Rag2tm1mice23 bearing a transgene encoding TCR specific for the ovalbumin peptide (OVA323-339)25 were activated by adding the OVA peptide to the culture. Activated CD4+ T cells were isolated and reactivated with immobilized anti-CD3 Ab and DC-HIL-Fc or control Ig. DC-HIL markedly inhibited T-cell proliferation (Figure 2G) in response to anti-CD3 Ab and abrogated IL-2 production (Figure 2H). We also titrated the inhibitory capacity of DC-HIL against the reactivation, as described previously (Figure 2I-J). Similar to effects on naive T cells (Figure 2E), DC-HIL-Fc strongly inhibited T-cell proliferation at 5 μg/mL (Figure 2I), and reduction of IL-2 production was even greater (Figure 2J). These results indicate that the putative T-cell ligand for DC-HIL mediates a potent signal that can inhibit activation of primary as well as secondary T-cell responses.
Binding of DC-HIL to T cells induces cell-cycle arrest
Since PD-1–mediated signals prevented anti-CD3 Ab-treated T cells from entering the cell cycle,13,15 we questioned whether inhibition of T-cell activation by DC-HIL-Fc is similarly achieved. CD4+ T cells were cultured in wells coated with anti-CD3 Ab plus DC-HIL-Fc or control hIgG; at different time points thereafter, the T cells were assayed for asynchronous cell division using CFSE labeling and FACS analysis (Figure 3A). For anti-CD3 Ab and hIgG-treated cells, the number of cell divisions increased 6-fold and the frequency of divided cells increased up to 51%. DC-HIL-Fc–treated T cells also divided several times, but at a lesser frequency (Figure 3A). To more precisely analyze cell-cycle effects, T cells at 48 hours after activation were labeled with 7-AAD (to stain chromosomal DNA in all cells) and immunofluorescent BrdU (to stain proliferating cells) (Figure 3B). Based on a ratio of 7-AAD–staining to BrdU-staining intensities, T cells treated with anti-CD3 Ab and control hIgG sorted into S phase (9.5%), G2/M phase (0.5%), G0 /G1 phase (85.6%), and apoptotic cells (3.7%). For cells treated with anti-CD3 Ab and DC-HIL-Fc, similar portions of cells sorted into G2/M phase (0.2%), G0 /G1 phase (96.4%), and apoptotic cells (2.2%), but markedly fewer were sorted into S phase (0.5%). These results indicate that signaling through the putative DC-HIL-L leads to cell-cycle arrest (rather than apoptosis).
Figure 3.

Cell-cycle analyses of CD4+ T cells treated with DC-HIL-Fc. (A) T cells (6 × 106) were labeled with CFSE (1 μM) and then cultured in microwells precoated with anti-CD3 Ab (0.3 μg/mL) plus hIgG or DC-HIL-Fc (5 μg/mL). At the indicated time points, cells were harvested and analyzed by FACS for fluorescence intensity. The frequency (%) of divided cells is shown in histograms. (B) T cells from 48-hour culture similarly treated were analyzed for incorporation of BrdU (using FITC–anti-BrdU Ab) and total DNA content (stained with 7-AAD) by FACS; data shown as dot plots of BrdU versus 7-AAD. Data shown are representative of 3 (A) and 2 (B) independent experiments.
Soluble DC-HIL enhances T-cell activation triggered by APCs
We next examined the effect of soluble DC-HIL-Fc on the MLR, in which alloreactive T cells are activated by APCs. In this MLR, spleen cells from C57BL/6 mice served as stimulators, CD4+ T cells from BALB/c mice served as responders, and the proliferative capacity of responder cells was measured by 3H-thymidine incorporation (Figure 4A-B). To our surprise, the addition of DC-HIL-Fc (but not hIgG) enhanced the MLR in a dose- and time-dependent manner. Control hIgG or an irrelevant Fc fusion protein dectin-2-Fc had no effect on MLR (Figure S4).
Figure 4.

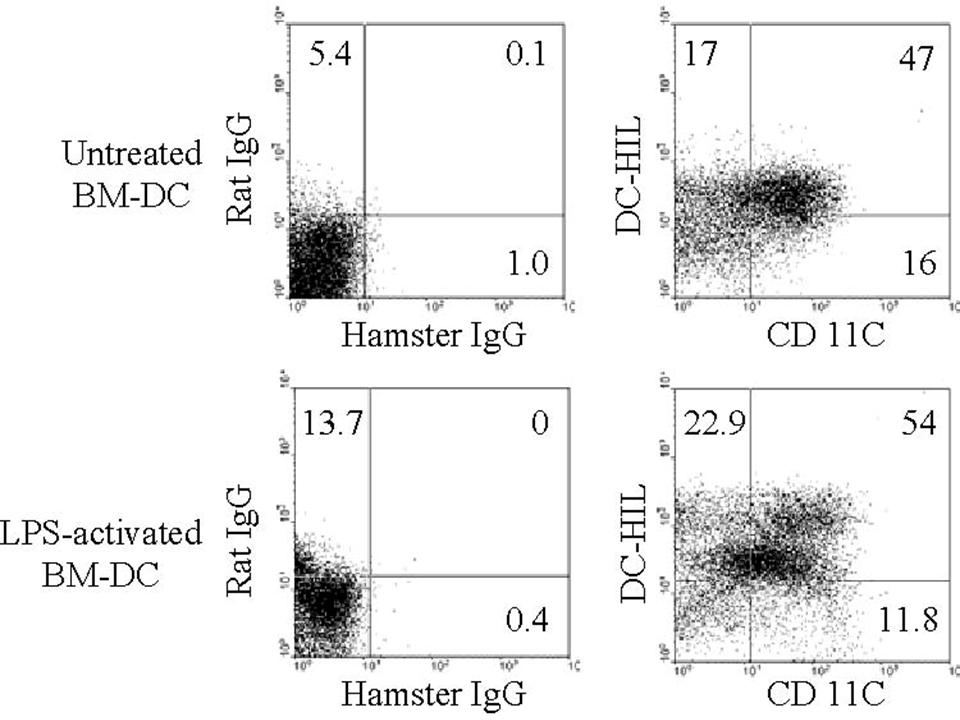
Soluble DC-HIL-Fc enhances responses of CD4+ T cells by APCs. Effects of soluble DC-HIL-Fc on T-cell activation were examined in MLR (A-B), anti-CD3 response (C), or in OVA-specific antigen presentation (D-E). MLR: C57BL/6 spleen cells (5 × 104) were γ irradiated and mixed with CD4+ T cells (2 × 105) purified from BALB/c mouse spleens. (A) Increasing doses of hIgG or DC-HIL-Fc were added to the MLR culture and incubated for 2 days prior to 3H-thymidine pulsing. Proliferative response of T cells was assayed by incorporation of 3H-thymidine. (B) MLR was incubated in the absence/presence of hIgG or DC-HIL-Fc (20 μg/mL) for 1, 2, or 3 days before pulsing. (C) Soluble (Sol) DC-HIL-Fc does not inhibit T-cell activation triggered by immobilized (Im) anti-CD3 Ab. CD4+ T cells were cultured in microwells precoated with anti-CD3 Ab (0.3 μg/mL) and 5 μg/mL of DC-HIL-Fc. In some wells, soluble hIgG or DC-HIL-Fc in increasing doses was added to culture in wells coated with the same amount of anti-CD3 Ab. T-cell activation was expressed as proliferative capacity. (D-E) OVA-specific response: CD4+ T cells purified from the spleens of BALB/cTac-TgN(DO11.10)-Rag2tm1 mice were cocultured without (No) or with BM-DCs (from BALB/c mice) previously pulsed with OVA peptide. T-cell activation was assayed by IL-2 production (D) and by FACS for frequency of CD69+/CD4+ T cells (E). Control staining was performed with FITC-rat IgG (rIgG) and PE-hamster IgG (haIgG). (F) siRNA-mediated knockdown of DC-HIL. At 1 day after transfection of DCs with control (Ctrl; shuffled) siRNA or DC-HIL–targeted siRNA, cells were harvested and assayed by immunoblotting for protein expression of DC-HIL or β-actin. (G) Increasing numbers of transfected DCs were pulsed with OVA peptide and cocultured with a constant number of OVA-specific CD4+ T cells. Activation was measured by IL-2 production. (H) At 2 days after coculturing, frequency of CD69+ in the CD4+ T cells was determined by FACS. *Statistical significance (P < .001) compared with T-cell responses treated with hIgG control. Data shown are representative of at least 3 independent experiments.
Because the enhancing effect of soluble DC-HIL on the MLR contrasted with the inhibitory effect of immobilized DC-HIL-Fc on anti-CD3 Ab–induced T-cell proliferation (Figure 2), we compared effects of soluble versus immobilized DC-HIL-Fc in parallel, using in vitro T-cell proliferation assays in which immobilized anti-CD3 Ab (rather than APCs) served as the primary stimulator (Figure 4C). Immobilized DC-HIL-Fc (5 μg/mL) abrogated T-cell activation, whereas soluble DC-HIL-Fc (final concentration of 20 μg/mL) had no effect on activation of purified T cells. Taken together, these results indicate that soluble DC-HIL-Fc is capable of binding to T cells, but fails to transduce signals in T cells; more than likely, it interferes with binding of DC-HIL (on APCs) to DC-HIL-L (on T cells), thereby neutralizing the inhibitory function of endogenous DC-HIL, leading to an enhanced MLR.
To more rigorously examine the effects of soluble DC-HIL-Fc on T-cell activation, we used an in vitro antigen presentation assay in which BM-DCs were pulsed with OVA peptide (OVA323-339)24 and allowed to activate CD4+ T cells prepared from unprimed BALB/cTac-TgN(DO11.10)-Rag2tm1 mice. Different doses of DC-HIL-Fc or control hIgG were added to the assay, and T-cell activation was measured by IL-2 production and frequency of CD69+/CD4+ cells (Figure 4D-E). DC-HIL-Fc augmented T-cell activation in a dose-dependent manner (up to 3-fold increase in IL-2 production); control hIgG had very little effect (binding of hIgG to DCs was very weak; Figure S5). DC-HIL-Fc–treated T cells showed an increased frequency (88%) of activated (CD69+) cells (versus 68% for controls). Having shown previously that both immature and mature DCs express DC-HIL on the surface (Figure S1), we interpreted these results to mean that soluble DC-HIL blocked endogenous DC-HIL function, leading to augmented T-cell responses.
DCs with knockdown DC-HIL display enhanced immunostimulatory capacity
To evaluate the function of DC-associated DC-HIL, we prepared DCs with genetically modified (knockdown) expression of DC-HIL and examined immunostimulatory capacity. Transfection of DC-HIL siRNA inhibited most of the DC-HIL protein expression in DCs when compared with DCs with control siRNA (Figure 4F). Increasing numbers of these transfected DCs were pulsed with OVA peptide and then allowed to activate a constant number of CD4+ T cells purified from Tac-TgN(DO11.10)-Rag2tm1mice. T-cell activation was assayed by IL-2 production (Figure 4G). At every dose tested, DC-HIL siRNA–transfected DCs were more potent activators of OVA-specific T cells, compared with control siRNA DCs. Enhanced activation was also affirmed by a higher frequency of CD69+ T cells (92.6% vs 81.7%) in coculture (Figure 4H).
In vivo injection of soluble DC-HIL augments CH responses
To ascertain the biological significance of the DC-HIL/DC-HIL-L pathway, we examined the effects of soluble DC-HIL on CH, an experimental model of delayed-type, T-cell–mediated skin inflammation. BALB/c mice were sensitized by topical application of the hapten Ox at a dose of 2% on abdominal skin (day 0) and then challenged/elicited by painting ears with 1% Ox (day 6) (Figure 5). Mice in different panels were injected intraperitoneally with DC-HIL-Fc, hIgG, or PBS every other day on 3 occasions either starting a day before sensitization (Figure 5A) or a day before elicitation (Figure 5B). Ear swelling was measured and change in ear thickness was calculated. Ear swelling in mice treated with DC-HIL-Fc just before and after hapten sensitization was no different from that of mice injected with control hIgG (Figure 5A). By contrast, mice injected with DC-HIL-Fc just before and after hapten elicitation displayed significantly greater ear swelling that persisted longer (up to 6 days after elicitation), compared to those of control mice (Figure 5B). Histologic analysis of Ox-painted ear skin revealed a marked increase in skin thickness and number of skin-infiltrating leukocytes in mice treated with DC-HIL-Fc, but not with control hIgG (Figure 5C).
Figure 5.

Soluble DC-HIL-Fc enhances elicitation of Ox-induced contact hypersensitivity in mice. Sensitization of BALB/c mice (n = 5) with Ox for CH (A-B): on day 0, mice were painted with 2% Ox on abdominal skin (Senst). On day 6, CH was elicited in sensitized mice by painting 1% Ox or solvent control to right and left ears, respectively (Challenge). CH was assessed daily through day 9 or 12 by measuring ear thickness (▴). Mice were injected intraperitoneally with PBS, hIgG, or DC-HIL-Fc (10 mg/kg each) on days −1, 1, and 3 (before and after sensitization) (A) or on days 5, 7, and 9 (before and after challenge) (B). Daily change in ear thickness was plotted for each panel during sensitization (A) or elicitation (B). *P < .003; **P < .05 compared with ear thickness of mice treated with hIgG. (C) Ear skin was excised from mice treated without (None) or with Ox and Fc protein (2 days after elicitation) and examined histologically (10×/10 objective lens). Data shown (A-B) are representative of 4 independent experiments.
To determine the activation status of T cells in Ox-sensitized mice injected with DC-HIL-Fc, we compared draining LNs and LN cells of DC-HIL-Fc–treated versus control mice 2 days after Ox challenge, which was the time of greatest ear swelling. LNs of DC-HIL–treated mice were 3 times larger than hIgG-treated mice, contained 3 times the number of cells (Figure 6A), and displayed 3-fold greater T-cell proliferation in the absence of stimuli (Figure 6B). We also compared the frequency of CD4+, CD8+, or B220+ LN cells (Figure 6C). Draining LN cells of DC-HIL–treated mice had greater portions of CD4+ T cells (44% vs 33%) and of B cells (25% vs 18%) compared with those of hIgG-treated mice. Finally, we examined the frequency of CD69+ (activated) cells (Figure 6D). DC-HIL treatment was associated with an increase in the activation phenotype for each of the 3 leukocyte subpopulations examined: 15% versus 12% for CD4+ T cells, 10.7% versus 7.9% for CD8+ T cells, and 6.3% versus 4% for B cells. These findings constitute in vivo support for previous in vitro observations that (1) DC-HIL-L are expressed on activated (but not resting) T cells, (2) DC-HIL is a negative regulator of T-cell activation, and (3) soluble DC-HIL enhances T-cell–mediated responses.
Figure 6.

Ox/DC-HIL-Fc–treated LN cells display hyperactivation phenotypes. In an independent experiment, draining LN (DLN) cells prepared from BALB/c mice treated similarly (as in Figure 5) were examined. (A) DLN cells were counted. (B) Spontaneous activation was measured by 3H-thymidine incorporation of DLN cells (4 × 105/well) cultured for 3 days without stimuli. (C-D) frequency of leukocytes: DLN cells were stained with FITC-Ab against CD4, CD8, or B220 alone (C) or doubly stained with PE–anti-CD69 (D), and then analyzed by FACS. CD69 expression (D) is shown in LN cells stained positively with the surface marker Ab. Results (A and B) are shown as mean values ± SDs; *P < .001 compared with LN responses treated with hIgG control. Data shown are representative of 3 independent experiments.
PKD domain is required for the inhibitory function of DC-HIL on T-cell activation
We reported previously that the extracellular domain of DC-HIL contains an RGD motif required for integrin-mediated cell adhesion; an Ig-like polycystic kidney disease (PKD) domain,30 and a proline-rich region (PRR) involved in protein-protein interactions.31 To determine whether some or all are required for the inhibitory function of DC-HIL on T-cell activation, we created extracellular domains of mutant DC-HIL lacking PKD or PRR, or containing RAA (instead of RGD), and fused these mutants to the IgG-Fc (Figure 7A). Purity of the mutants was quite high (similar to the wild-type) as judged by SDS-PAGE/Coomassie blue staining (Figure 7B). We then examined the capacity of mutants to inhibit T-cell activation by titrating a given dose of mutant (or wild-type) DC-HIL-Fc to increasing doses of anti-CD3 Ab (Figure 7C). The inhibitory activity of wild-type DC-HIL-Fc (5 μg/mL) decreased progressively with increasing doses of anti-CD3 Ab. The titration curve of the RAA mutant was similar to the wild-type. The PRR-deficient mutant showed only minimally reduced inhibitory activity at a dose of 0.3 μg/mL anti-CD3 Ab. By contrast, the PKD-deficient mutant almost completely lost inhibitory activity at each dose of anti-CD3 Ab tested. We also compared mutants versus wild-type with respect to T-cell–binding capacity (Figure 7D). RAA and PRR mutants bound to T cells as efficiently as the wild-type, whereas the PKD mutant failed to bind to T cells (correlating with results of inhibitory function). These findings indicate that the Ig-like PKD domain is required for binding of DC-HIL to T cells and for its inhibitory function. By contrast, neither proline-mediated interaction nor RGD-dependent cell adhesion appears necessary for DC-HIL's inhibitory function.
Figure 7.

Mutant analyses of DC-HIL-Fc function. (A) Protein structures of DC-HIL-Fc wild-type (WT) and mutants are represented schematically. Extracelluar domains (ECDs) of mutants, RAA (replacement of RGD sequence with RAA; ▴) and deletion mutants lacking PRR (amino acid [aa] 301-334) and PKD230-355 were linked to a Fc portion of hIgG and produced in COS-1 cells. (B) After purifying mutant DC-HIL-Fc proteins, a small aliquot (2 μg/lane) was run on SDS-PAGE and then stained with Coomassie Blue to visualize protein bands. (C) T-cell activation. Highly purified DC-HIL-Fc WT or mutants (5 μg/mL each) and anti-CD3 Ab (increasing doses) were coated on microwells for culture with CD4+ T cells for 2 days and pulsed with 3H-thymidine for 20 hours. Results are shown as mean values ± SDs. (D) Binding of DC-HIL mutants to T cells. Activated CD4+ T cells were incubated with WT and mutants of DC-HIL (10 μg/mL) and analyzed for binding by FACS. Histograms of T cells stained with hIgG (filled) and a mutant (open) are overlaid. Data (C-D) shown are representative of 3 experiments.
Discussion
Subtractive cDNA cloning of mouse XS52 DCs minus J774 macrophages22 led to our discovery of DC-HIL, also known as human nmb glycoprotein or gpnmb,32,33 and rat osteoactivin.34 The extracellular domain of DC-HIL contains a putative heparin-binding site,35 many N-glycosylation sites, an RGD cell-adhesion motif,36 a PRR (involved in O-glycosylation37 and/or protein-protein interactions31), and an Ig-like PKD domain conserved among 14 repeats in the extracellular region of the PKD-susceptible gene product, polycystin-1.30,38 Moreover, we showed that DC-HIL is highly N-glycosylated, that it recognizes heparan sulfate (especially on small-vessel endothelial cells [SVECs]),22 and that its RGD motif is responsible for integrin-mediated cell adhesion.22 Our present study uncovered a new function for DC-HIL, as a potent inhibitor of TCR-induced T-cell activation for both primary and secondary responses.
Compared with known pairs of inhibitory regulators of T-cell activation, DC-HIL and its putative ligand (DC-HIL-L) best resemble of PD-L1/PDL2 and its ligand, PD-1. For example, unlike BTLA8 or Tim-3,10,11 whose expressions are restricted strictly to leukocytes, DC-HIL (Figure S2) or PD-L1/PD-L2 expression can be induced in nonleukocytes by proinflammatory stimuli like IFN-γ.15 Moreover, in contrast to B7-1/B7-2, whose T-cell ligand CD28 is present on resting T cells, the T-cell ligand of DC-HIL or of PD-L1/PD-L2 is not expressed constitutively, requiring activation of the T cells for expression. Engagement of DC-HIL or of PD-L1/PD-L2 with their respective T-cell ligand attenuates T-cell activation, suppresses IL-2 secretion, and arrests T-cell proliferation, all of which can be rescued by costimulation of CD28. By contrast, interference with binding of DC-HIL to DC-HIL-L or of PD-L1/PD-L2 to PD-139 leads to enhanced T-cell responses in MLR and Ag-specific reactions in vitro and in CH in vivo. This antagonism by soluble DC-HIL is not unusual, since other Fc-tagged recombinant proteins have been used to block the endogenous functions of CD200, TREM-1, PD-1, DIgR2, and other receptors, respectively.39–42
As cited previously, all known pairs of T-cell inhibitory regulators on APCs and their ligands on T cells (including CD80 and CD86, which bind to CTLA-4,4,43 and PD-L1/PD-L2, which bind to PD-113,16) possess Ig domains that allow their categorization as members of the B7 receptor superfamily. Indeed, the Ig domains are responsible for ligation of each pair. DC-HIL differs from these inhibitors in not possessing an Ig domain typical of the B7 receptor family, instead containing a PKD domain that can fold into an Ig-like tertiary structure critical to its binding and inhibitory functions.
An important task ahead is the identification of the DC-HIL-L on activated T cells. Given our previous finding that DC-HIL recognizes heparin sulfate on endothelial cells,22 we speculate that heparan sulfate is involved in the binding of DC-HIL to DC-HIL-L. However, heparan sulfate alone is not likely to be the complete ligand since the PKD-deficient DC-HIL mutant we tested bound heparan sulfate but not activated T cells. Rather, we hypothesize the putative ligand to bear heparan sulfate plus a peptide with affinity for the PKD domain.
In summary, DC-HIL is a newly identified inhibitor of T-cell activation that shares many common functional properties with PD-L1/PD-L2. Our discovery should prompt greater investigation on the role of DC-HIL/DC-HIL-L relative to other pathways regulating T-cell activation since better understanding of these relationships and their biologic consequences may lead to the development of more effective T-cell–based therapeutic applications.
Supplementary Material
Acknowledgments
We are grateful to Drs Chandra Mohan and Michael Bennett for critical reading of this manuscript, and Susan Milberger for administrative assistance.
Supported by a research grant (A164927-01) from the National Institutes of Health and a Pilot and Feasibility Study Grant from Galderma (Fort Worth, TX).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Author contributions: Most of the work was done by J.-S.C.; K.S. contributed to initial studies, and I.I.D. conducted the animal studies. The entire study was conceptualized and supervised by K.A. in collaboration with P.D.C.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kiyoshi Ariizumi, 5323 Harry Hines Blvd, Dallas, TX 75390-9069; e-mail: kiyoshi.ariizumi@utsouthwestern.edu.
References
- 1.Chambers CA, Allison JP. Co-stimulation in T cell responses. Curr Opin Immunol. 1997;9:396–404. doi: 10.1016/s0952-7915(97)80087-8. [DOI] [PubMed] [Google Scholar]
- 2.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3:939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 3.Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. [PubMed] [Google Scholar]
- 4.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 6.Okazaki T, Iwai Y, Honjo T. New regulatory co-receptors: inducible co-stimulator and PD-1. Curr Opin Immunol. 2002;14:779–782. doi: 10.1016/s0952-7915(02)00398-9. [DOI] [PubMed] [Google Scholar]
- 7.Sedy JR, Gavrieli M, Potter K, G, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe N, Gavrieli M, Sedy JR, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 9.Monney L, Sabatos CA, Gaglia JL, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 10.Sabatos CA, Chakravarti S, Cha E, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4:1102–1110. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez-Fueyo A, Tian J, Picarella D, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4:1093–1101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 12.Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. 2002;20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806. [DOI] [PubMed] [Google Scholar]
- 13.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 14.Tseng SY, Otsuji M, Gorski K, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–846. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 17.Dong H, Chen L. B7-H1 pathway and its role in the evasion of tumor immunity. J Mol Med. 2003;81:281–287. doi: 10.1007/s00109-003-0430-2. [DOI] [PubMed] [Google Scholar]
- 18.Subudhi SK, Zhou P, Yerian LM, et al. Local expression of B7-H1 promotes organ-specific autoimmunity and transplant rejection. J Clin Invest. 2004;113:694–700. doi: 10.1172/JCI19210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 20.Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol. 1998;10:1563–1572. doi: 10.1093/intimm/10.10.1563. [DOI] [PubMed] [Google Scholar]
- 21.Liang L, Sha WC. The right place at the right time: novel B7 family members regulate effector T cell responses. Curr Opin Immunol. 2002;14:384–390. doi: 10.1016/s0952-7915(02)00342-4. [DOI] [PubMed] [Google Scholar]
- 22.Shikano S, Bonkobara M, Zukas PK, Ariizumi K. Molecular cloning of a dendritic cell-associated transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J Biol Chem. 2001;276:8125–8134. doi: 10.1074/jbc.M008539200. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh CS, Macatonia SE, Tripp CS, et al. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 24.Demotz S, Barbey C, Corradin G, Amoroso A, Lanzavecchia A. The set of naturally processed peptides displayed by DR molecules is tuned by polymorphism of residue 86. Eur J Immunol. 1993;23:425–432. doi: 10.1002/eji.1830230219. [DOI] [PubMed] [Google Scholar]
- 25.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cruz PDJ, Nixon-Fulton J, Tigelaar RE, Bergstresser PR. Local effects of UV radiation on immunization with contact sensitizers. I. Down-regulation of contact hypersensitivity by application of TNCB to UV-irradiated skin. Photodermatol. 1988;5:126–132. [PubMed] [Google Scholar]
- 27.Cruz PDJ, Bergstresser PR. The low-dose model of UVB-induced immunosuppression. Photodermatol. 1988;5:151–161. [PubMed] [Google Scholar]
- 28.Sato K, Yang XL, Yudate T, et al. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor chain to induce innate immune responses. J Biol Chem. 2006;281:38854–38866. doi: 10.1074/jbc.M606542200. [DOI] [PubMed] [Google Scholar]
- 29.Linsley PS, ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. 1993;11:191–212. doi: 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- 30.Bycroft M, Bateman A, Clarke J, et al. The structure of a PKD domain from polycystin-1: implications for polycystic kidney disease. EMBO J. 1999;18:297–305. doi: 10.1093/emboj/18.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- 32.Anderson MG, Smith RS, Hawes NL, et al. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat Genet. 2002;30:81–85. doi: 10.1038/ng794. [DOI] [PubMed] [Google Scholar]
- 33.Weterman MA, Ajubi N, van Dinter I, et al. nmb, a novel gene, is expressed in low-metastatic human melanoma cell lines and xenografts. Int J Cancer. 1995;60:73–81. doi: 10.1002/ijc.2910600111. [DOI] [PubMed] [Google Scholar]
- 34.Safadi FF, Xu J, Smock SL, et al. Cloning and characterization of osteoactivin, a novel cDNA expressed in osteoblasts. J Cell Biol. 2001;84:12–26. doi: 10.1002/jcb.1259. [DOI] [PubMed] [Google Scholar]
- 35.Cardin AD, Weintraub HJ. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- 36.Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 37.Wilson IB, Gavel Y, von Heijne G. Amino acid distributions around O-linked glycosylation sites. Biochem J. 1991;275:529–534. doi: 10.1042/bj2750529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ponting CP, Hofmann K, Bork P. A latrophilin/CL-1-like GPS domain in polycystin-1. Curr Biol. 1999;9:R585–R588. doi: 10.1016/s0960-9822(99)80379-0. [DOI] [PubMed] [Google Scholar]
- 39.Brown JA, Dorfman DM, Ma FR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 40.Gorczynski R, Chen Z, Kai Y, et al. CD200 is a ligand for all members of the CD200R family of immunoregulatory molecules. J Immunol. 2004;172:7744–7749. doi: 10.4049/jimmunol.172.12.7744. [DOI] [PubMed] [Google Scholar]
- 41.Gibot S, Kolopp-Sarda MN, Bene MC, et al. A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J Exp Med. 2004;200:1419–1426. doi: 10.1084/jem.20040708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi L, Luo K, Xia D, et al. DIgR2, dendritic cell-derived immunoglobulin receptor 2, is one representative of a family of IgSF inhibitory receptors and mediates negative regulation of dendritic cell-initiated antigen-specific T-cell responses. Blood. 2006;108:2678–2686. doi: 10.1182/blood-2006-04-015404. [DOI] [PubMed] [Google Scholar]
- 43.Tsushima F, Iwai H, Otsuki N, et al. Preferential contribution of B7-H1 to programmed death-1-mediated regulation of hapten-specific allergic inflammatory responses. Eur J Immunol. 2003;33:2773–2782. doi: 10.1002/eji.200324084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}