

Abstract
Progressive iron overload is the most salient and ultimately fatal complication of β-thalassemia. However, little is known about the relationship among ineffective erythropoiesis (IE), the role of iron-regulatory genes, and tissue iron distribution in β-thalassemia. We analyzed tissue iron content and iron-regulatory gene expression in the liver, duodenum, spleen, bone marrow, kidney, and heart of mice up to 1 year old that exhibit levels of iron overload and anemia consistent with both β-thalassemia intermedia (th3/+) and major (th3/th3). Here we show, for the first time, that tissue and cellular iron distribution are abnormal and different in th3/+ and th3/th3 mice, and that transfusion therapy can rescue mice affected by β-thalassemia major and modify both the absorption and distribution of iron. Our study reveals that the degree of IE dictates tissue iron distribution and that IE and iron content regulate hepcidin (Hamp1) and other iron-regulatory genes such as Hfe and Cebpa. In young th3/+ and th3/th3 mice, low Hamp1 levels are responsible for increased iron absorption. However, in 1-year-old th3/+ animals, Hamp1 levels rise and it is rather the increase of ferroportin (Fpn1) that sustains iron accumulation, thus revealing a fundamental role of this iron transporter in the iron overload of β-thalassemia.
Introduction
β-Thalassemia is the most common congenital hemolytic anemia due to partial or complete lack of synthesis of β-globin chains. Cooley anemia,1 also known as β-thalassemia major, is the most severe form of β-thalassemia, which is characterized by profound ineffective erythropoiesis (IE) requiring regular red blood cell (RBC) transfusions to sustain life. Transfusion therapy leads to excess iron accumulation in many organs resulting in tissue damage. Therefore, iron chelation is essential in the management of this otherwise fatal disease.2 In β-thalassemia intermedia, in which a larger amount of β-globin chains are synthesized, the clinical picture is milder and the patients do not require frequent transfusions. However, progressive iron overload still occurs due to increased gastrointestinal (GI) iron absorption.3–5 Studies in thalassemic patients showed that the rate of iron uptake from the GI tract is approximately 3 to 4 times greater than normal.6 Ferrokinetic studies revealed that 75% to 90% of the iron in donor serum, labeled with 59Fe and injected into healthy subjects, appeared in circulating red cells within 7 to 10 days. In some thalassemic patients, however, only 15% of the 59Fe was incorporated into circulating erythrocytes.7 This discrepancy was attributed to the fact that iron would be sequestered in those organs in which premature destruction of erythroid precursors occurs. In β-thalassemia, it has been suggested that 60% to 80% of erythroid precursors die in the marrow and extramedullary sites.8–10 Therefore, in β-thalassemia erythropoietic organs such as the bone marrow (BM) in humans and the BM and spleen in mice would be expected to show the highest iron concentrations. This hypothesis has never been corroborated, however.
Two mouse models of β-thalassemia intermedia are available, th1/th1 and th3/+. In the th1/th1 mouse model,11 a deletion eliminates one of the 2 mouse β-globin genes, namely βmajor. Mice heterozygous for this mutation do not show anemia, whereas homozygous (th1/th1) mice show hemoglobin (Hb) levels in the range of 9 to 10 g/dL.11–13 The second mouse model (th3/+) harbors a deletion that eliminates both the βminor and βmajor genes in heterozygosity.14,15 The th3/+ mice show Hb levels in the range of 8 to 9 g/dL.13–15 Adult th1/th1 and th3/+ mice have a degree of disease severity (hepatosplenomegaly, anemia, aberrant erythrocyte morphology) comparable to that of patients affected by β-thalassemia intermedia. Unfortunately, mice completely lacking the adult β-globin genes (th3/th3) die late in gestation,14 limiting their use as a model for Cooley anemia. We established the first adult mouse model of β-thalassemia major by transplantation of hematopoietic fetal liver cells (HFLCs) harvested from th3/th3 embryos at 14.5 days of gestation into lethally irradiated syngeneic adult recipients.16 Hematologic analyses of engrafted mice performed 6 to 8 weeks after transplantation revealed severe anemia due to low reticulocyte counts, profound splenomegaly, and extensive hepatic extramedullary hematopoiesis (EMH).16 Whereas IE is characterized by a modest reduction in RBCs and an increase in reticulocytes in th1/th1 and th3/+ mice, in th3/th3 mice IE is extreme and characterized by almost absent reticulocyte and erythrocyte production.16
In recent years remarkable progress has been made in identifying and characterizing genes associated with iron metabolism. Insight has also been gained from studying various forms of hereditary hemochromatosis (HH),17,18 which results in an excess of total body iron and might lead to organ failure due to iron toxicity. HH mutations have now been described in several different genes, including HFE19–23 (encoding the hemochromatosis gene), TFR224–27 (encoding transferrin receptor-2), HJV28,29 (encoding hemojuvelin), HAMP30–33 (encoding hepcidin), and FPN134–41 (encoding ferroportin). FPN1 is a key player in regulating dietary iron absorption in the duodenum, whereas serum HAMP levels control the concentration of FPN1 on the basolateral cell surface of enterocytes.42 Murine Hamp1 is synthesized predominantly in the liver and secreted into the bloodstream. Hamp1 mRNA levels are homeostatically regulated by body iron stores, erythroid iron needs, hypoxia, and inflammation.30,43–48 HAMP targets FPN1, triggering its degradation.42 In all forms of HH described so far, an excess of total body iron is due to 2 factors. In the first case, HH results from low or inappropriately low levels of HAMP expression. In the second case, HH is due to the presence of mutant forms of FPN1 that are insensitive to protein degradation mediated by HAMP association.18,42 Therefore, iron absorption is mostly, if not entirely, mediated by the balance between the levels of HAMP made in the liver and FPN1 produced in the duodenum.
Several gene products have been shown to control Hamp1 expression, such as Smad449 (encoding the transcriptional factor MAD-homolog-4); IL-6 (encoding the inflammatory cytokine IL-6) through IL-6R activation of STAT348; BMP-2, -4, and -950,51 (bone morphogenetic proteins); Cebpa52 (encoding the CCAAT/enhancer binding protein-α); Hjv; Hfe, and Tfr2.49,50,52 Except for recent findings about IL-6 and Stat3, Hjv, and BMPs, however, the precise mechanisms dictating Hamp1 expression are unknown.
Our goal was to correlate longitudinal changes in levels of IE with organ iron distribution and with the expression level of iron-regulatory genes in mouse models of β-thalassemia intermedia (th3/+) and major (th3/th3). Our initial studies on iron absorption indicated that liver Hamp1 mRNA levels are decreased in th3/th3 mice.53–56 On the other hand, in th3/+ and th1/th1 mice we and others observed mixed results,13,53,54 where some animals exhibited low Hamp1 levels and others exhibited those of wild-type (wt) mice. Although the th3/+ and th1/th1 mice are both affected by β-thalassemia intermedia, they show slightly different levels of anemia.13 It is possible that in these conditions, Hamp1 expression, as well as that of other iron regulatory genes, may be determined by the diet and the relative ratio between anemia and iron overload, which may vary depending on genotype and age. Nevertheless, in β-thalassemia intermedia we cannot exclude the possibility that alteration of the expression levels of other iron regulatory genes might also contribute to iron overload. Therefore, we initiated a thorough analysis of tissue iron content, gene expression, and erythropoiesis. We also analyzed mice affected by β-thalassemia major rescued through blood transfusion.
Here we show that organ iron content in β-thalassemia is increased both in young and old mice. Our study reveals potential roles for Hfe and Cebpa in controlling Hamp1 expression in thalassemic animals who had either received a transfusion or not. In addition, we show for the first time that organ iron distribution and the mechanism that leads to iron overload differ in th3/th3 and th3/+ mice. Comparing mice with β-thalassemia major and intermedia, we observed that their increased iron concentration is dictated by the different degrees of IE and organ iron content, and by the relative expression levels of Hamp1 and Fpn1. Whereas low levels of Hamp1 are sufficient to explain the increased iron content observed in young thalassemic animals, in aging mice the level of expression of Hamp1 rises, but the net rate at which the iron content increases is maintained due to a persistent up-regulation of Fpn1 in the duodenum.
Materials and methods
Generation of BM chimeras and tissue harvesting.
Wild-type (wt or +/+), th3/+, and th3/th3 embryos were genotyped at 13.5 to 15.5 days of gestation and the HFLCs harvested as described.16 HFLCs were then used for transplantation into recipient wt syngeneic animals (C57BL/6).16 Adult +/+ and th3/+ mice, obtained either through transplantation or breeding, and adult th3/th3 mice from transplantation were killed at 2, 5 and 12 months of age or after transplantation by cervical dislocation under isoflurane anesthesia.
Blood transfusion
The life span of mouse RBCs is approximately 60 days, the total blood volume of the mice being approximately 2 mL. This corresponds to a weekly turnover of approximately 250 μL blood. Starting 21 days after transplantation, mice were infused weekly via the tail vein or retro-orbital venous plexus with 250 to 300 μL freshly harvested blood from normal healthy C57BL/6 animals. Blood was collected from the retro-orbital plexus into acid citrate dextrose (7 volumes of blood for 1 volume of acid citrate dextrose), under anesthesia. Hb levels were measured 1 day before and 3 days after transfusion. The animals received transfusions for up to 22 weeks (experiment end point). No significant variations in the Hb levels of the mice were observed during the experiment.
Hematologic studies
Blood samples were obtained by retro-orbital puncture under anesthesia. Complete blood counts (CBCs) were measured on an Advia 120 Hematology System (Bayer, Tarrytown, NY). When Hb evaluation was required frequently (ie, in animals given transfusions), a limited amount of blood was collected from the tail to be analyzed by the HemoCue Hemoglobin System (Lake Forest, CA).
Measurement of tissue iron content, serum iron, transferrin saturation, and labile plasma iron
The iron content of the liver, kidney, heart, and spleen was determined by atomic absorption or by nonheme iron analysis, as described.57,58 Serum iron and transferrin (Tf) saturation levels were quantified using the Iron/UIBC kit manufactured by Thermo Electron (Melbourne, Australia). Labile plasma iron (LPI) was quantified using the method of Esposito et al.59
Flow cytometry
For fluorescence-activated cell sorting (FACS) analysis, 1 × 106 BM and spleen cells were washed and incubated on ice for 15 minutes with 0.1 μg of FITC-labeled anti–mouse CD71 and PE-conjugated anti–mouse Ter119 antibodies (BD PharMingen, San Diego, CA) in PBS-1% BSA. Control samples were incubated with 0.1 μg FITC-labeled anti–mouse IgG1 and PE-conjugated rat IgG2b isotype control antibodies (BD PharMingen). Results were analyzed using Flow-Jo software (Tree Star, Ashland, OR).
Pathology and immunochemical analyses
Tissues were fixed in 10% buffered formalin and embedded in paraffin. Longitudinal sections (4-μm) were stained with hematoxylin and eosin. Sections (4 μm) were also stained for iron using Prussian blue (Poly Scientific, Bayshore, NY). Immunohistochemistry was performed on sections of the duodenum from 1-year-old +/+ and th3/+ mice using a previously described antibody against the C terminus of Fpn160 on the BOND-maX Automated Immunostainer (Vision BioSystems, Melbourne, Australia).
Quantitative real-time PCR
RNA samples extracted from the liver, heart, and duodenum of adult mice were retrotranscribed by using the SuperScript II First Strand Kit (Invitrogen, Carlsbad, CA). cDNAs were analyzed by quantitative real-time polymerase chain reaction (Q-PCR) with specific primers for the genes indicated in Table 1Q-PCRs were performed by using the ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA), with TaqMan (TaqMan PCR 2X Master mix; Applied Biosystem) and SYBR Green (iTaq SYBR Green Supermix, Bio-Rad Laboratories, Hercules, CA) chemistry.
Table 1.
Oligo sequences
| Gene product | Gene symbol | Forward primer | Reverse primer | Accession no. |
|---|---|---|---|---|
| CCAAT/enhancer binding protein-α | Cebpa | 5′-agagccgagataagccaaa-3′ | 5′-ggtcaactccagcaccttct-3′ | NM_007678 |
| Ferritin heavy chain 1 | Fth1 | 5′-aagaaaccagaccgtgatga-3′ | 5′-tagccagtttgtgcagttcc-3′ | NM_010239 |
| Ferritin light chain 1 | Ftl1 | 5′-ctcatcaagaagatgggcaa-3′ | 5′-gagatactcgcccagagacc-3′ | NM_010240 |
| Hemochromatosis | Hfe | 5′-gcaatctcaggccatgatta-3′ | 5′-attccaaccaagaagatggc-3′ | NM_010424 |
| Hemochromatosis type 2 (juvenile) | Hjv, Hfe2 | 5′-gctcgagtttgtccattcaa-3′ | 5′-tgttccaatgtaggcagctc-3′ | NM_027126 |
| Hepcidin antimicrobial peptide 1 | Hamp, Hamp1 | 5′-tgagcagcaccacctatctc-3′ | 5′-acagcagaagatgcagatgg-3′ | NM_032541 |
| Solute carrier family 11 | Dmt1, Slc11a2 | 5′-tgtttgattgcattgggtctg-3′ | 5′-cgctcagcaggactttcgag-3′ | BC019137 |
| Solute carrier family 40 | Fpn1, Slc40a1 | 5′-caagaatgagctcctgacca-3′ | 5′-gccacaacaacaatccagtc-3′ | NM_016917 |
| Transferrin | Tf, Trf | 5′-cttctcatgctgttgtggct-3′ | 5′-gttcctgtgccactttgaga-3′ | NM_133977 |
| Transferrin receptor | Tfr1, Tfrc | 5′-cagaccttgcactctttgga-3′ | 5′-gaaagaaggaaagccaggtg-3′ | NM_011638 |
| Transferrin receptor 2 | Tfr2, Trfr2 | 5′-gggttcagacctcgaagaag-3′ | 5′-aggctcacgtacacaacagc-3′ | NM_015799 |
| β-actin | Actb | 5′-ccttccttcttgggtatgga-3′ | 5′-acggatgtcaacgtcacact-3′ | NM_007393 |
Primer design was performed using Primer Express Software version 2.0 (Applied Biosystems), based on the corresponding mouse gene sequences obtained from PubMed. As endogenous controls to normalize gene expression levels, a mouse Gapdh VIC/MGB probe (Applied Biosystems) and mouse β-actin were used. Two endogenous control genes were used to determine if any difference in amplification existed due to the effect of iron overload.
Results
The phenotypic spectrum of β-thalassemia is reproduced in mouse models
To study the mechanisms driving iron absorption and its correlation with IE in β-thalassemia, we have used mice affected by β-thalassemia and monitored age-related changes in various hematologic parameters (Hb, RBC, and reticulocyte counts) and evaluated the iron content of organs involved in RBC production and iron homeostasis. Bred animals (+/+ and th3/+) were analyzed at 2, 5, and 12 months of age. Animals receiving transplants (tp) (+/+)tp-2M, (th3/+)tp-2M, and (th3/th3)tp-2M were killed at 2 months after transplantation because homozygotes (th3/th3)tp only survive for 8 to 10 weeks due to severe anemia. To evaluate the effect of blood transfusion on iron metabolism, groups of animals that underwent transplantation and received transfusions (tx) were studied at 5 months after transplantation, (th3/+)tptx-5M and (th3/th3)tptx-5M.
Extension of the life span of thalassemic animals by blood transfusion
Because one of our goals was to study the effect of blood transfusion on iron overload and the levels of Hamp1 and other iron-regulatory genes in thalassemic animals, we compared (th3/+)tptx-5M and (th3/th3)tptx-5M mice to (+/+)tp-5M, (th3/+)tp-5M and (th3/th3)tp-2M mice generated at the same time. The mice given transplants with th3/th3 HFLCs and not transfusions exhibited very low Hb levels within 10 weeks of transplantation (Figure 1) associated with profound splenomegaly (not shown). The (th3/+)tptx-5M and (th3/th3)tptx-5M mice showed moderately higher Hb levels (Figure 1 inset) and splenomegaly in comparison to (th3/+)tp-5M and (th3/th3)tp-2M mice that did not receive transfusions. In addition, the spleens of mice given transfusions showed normal mature RBCs and larger areas of lymphopoiesis (white pulp) due to less extramedullary hematopoiesis (not shown).
Figure 1.

Mice affected by β-thalassemia intermedia and major show different levels of anemia. The mice have been divided according to their age and treatment (transplantation, tp; blood transfusion, tx). 2M indicates 2-month-old no transplant; 2MTP, 2 months after transplantation; 5M, 5-month-old no transplant; 5MTP, 5 months after transplantation; 12M, 12-month-old no transplant; 5MTPTX, 5 months after transplantation and blood transfusion; ■, Hb (g/dL); ▩, RBC (× 106/μL); □, reticulocytes (× 105/μL). The inset shows Hb levels of (th3/+)tptx-5M and (th3/th3)tptx-5M mice. Error bars represent SD performed on at least 3 animals per group. Sex: female. The age of the animals at transplantation was 8 to 10 weeks.
IE leads to a dramatic expansion of the immature erythroid population in animals affected by β-thalassemia
In patients affected by β-thalassemia, the consequences of IE include anemia and increased production of immature erythroid cells. These parameters were carefully analyzed in our animals. Comparing th3/+ to +/+ mice, irrespective of treatment or age, the total number of reticulocytes was increased, whereas the total number of erythrocytes was slightly decreased (Figure 1). In contrast, the (th3/th3)tp-2M animals, which do not produce any Hb (fetal or adult), showed a dramatic reduction in reticulocytes as well as of RBCs (Figure 1), indicating that RBC loss in (th3/th3)tp-2M animals happens before or during reticulocyte formation. FACS analysis also indicated that in th3/+, and even more so in th3/th3 mice, there was an expansion of the immature erythroid cell pool and a simultaneous decrease in the amount of terminal erythroid cells, a hallmark of IE (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article).
Organ iron content increases over time and is exacerbated in β-thalassemia intermedia
The iron content of the spleen, liver, kidney, and heart was analyzed by atomic absorption in mice at different ages (2, 5, and 12 months) and subjected to different treatments (transplant versus no transplant and transfusion versus no transfusion). Nonheme iron quantification58 was also performed on the liver and spleen tissues, showing a similar trend in iron content (not shown). Iron was also detected by Prussian blue staining of the liver (Figure S2), spleen, and BM sections (not shown). Our first analysis focused on th3/+ and +/+ mice. At 2, 5, and 12 months, the iron content was higher in the spleen, liver, and kidney of th3/+ mice compared to the corresponding organs of controls (Figure 2A ▩). This indicates that iron accumulates in these organs over time in the absence of blood transfusions. In th3/+-2M mice the total amount of iron in the spleen was approximately twice that of the liver (Figure 2C), whereas in th3/+-12M mice the total spleen and liver iron content was greater (Figure 2C), but their ratio was close to 1. The trend of iron accumulation in the heart was different from that of the other organs. In th3/+-2M mice, heart iron levels were approximately twice those in +/+-2M animals. Over time, the th3/+ mice failed to show a further increase in iron concentration, whereas that of +/+ animals slowly increased to the level of the th3/+ mice. Therefore, the pattern of iron accumulation in the heart may differ between mice and humans or the underlying disease may enhance the effect of the small increases in heart iron content.
Figure 2.
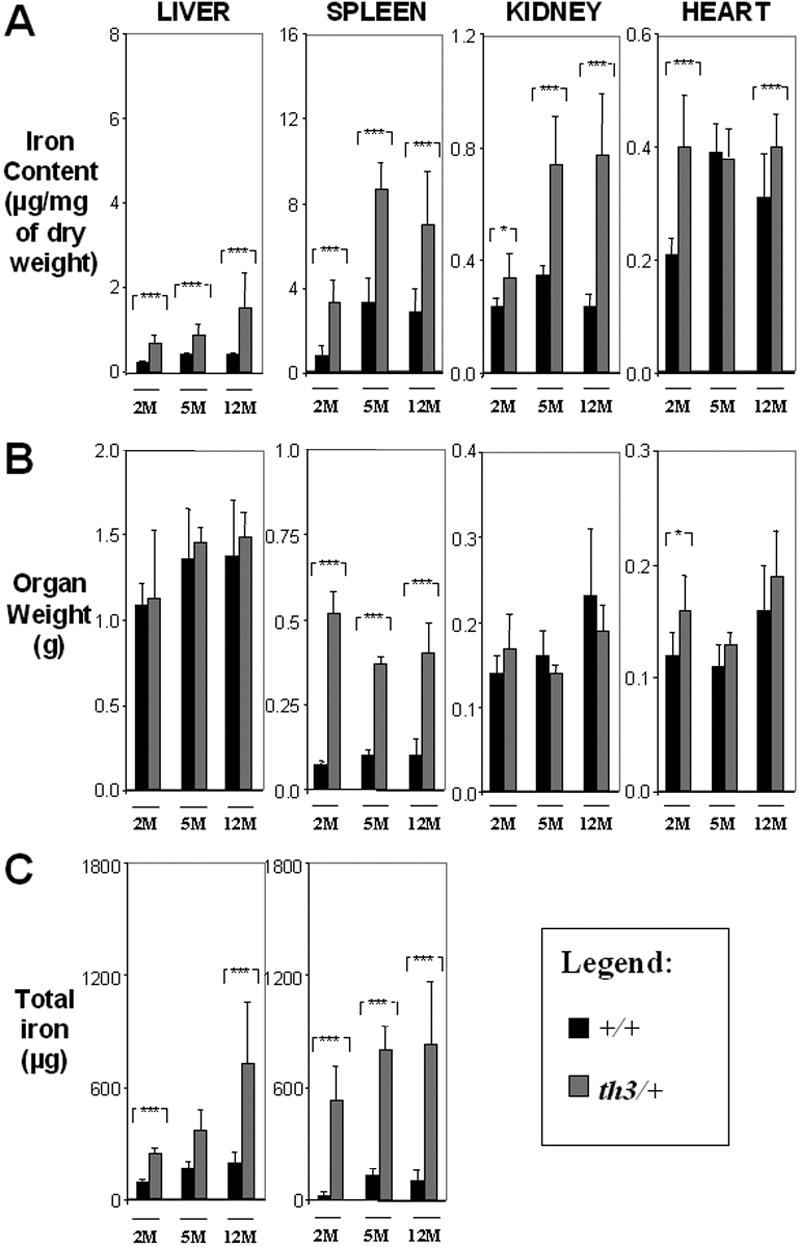
Organ iron content increases over time in β-thalassemia intermedia. (A) Iron content (μg/mg dry weight), (B) organ weight (g), and (C) total iron (μg) of liver, spleen, kidney, and heart. 2M indicates 2-month-old; 5M, 5-month-old; 12M, 12M-month-old; ■, +/+; ▩, th3/+. Error bars represent SD performed on 3 to 11 independent mice/tissues in duplicate for a total of 90 mice sampled. *P < .05, **P < .01, and ***P < .001 relative to control using the Dunnett multiple comparison test.
Iron distribution and content in the liver and spleen differs in β-thalassemia intermedia compared to β-thalassemia major
The highest total body iron content was observed in (th3/th3)tp-2M mice, indicating an extremely rapid iron absorption, which correlates with the lowest levels of Hb (Figure 1) and highest levels of IE, (as shown by the increase of early erythroid cells, CD71+/Ter119+; Figure S1). Surprisingly, the average iron concentration in the spleen was higher in (th3/+)tp-2M and th3/+-2M mice. This may be due to the high number of reticulocytes produced in (th3/+)tp-2M mice compared to (th3/th3)tp-2M mice (Figure 1). In addition, because th3/th3 mice produce fewer RBCs, this reduces the accumulation of iron in macrophages resulting from the breakdown of senescent RBCs. Consequently, a significant amount of the iron absorbed in (th3/th3)tp-2M mice may not be used for erythroid production, instead being diverted to the liver. Comparing animals with or without transfusions, the iron content was clearly reduced in the livers of (th3/th3)tptx-5M mice (Figure 3A). The spleen (Figure 3A) and the BM (not shown) were the only organs that showed an increase in iron content. In addition to the difference in the iron content of (th3/+)tp-2M and (th3/th3)tp-2M livers, there was also a difference in its cellular localization. In (th3/+)tp-2M (and th3/+-2M) mice, excess iron was located almost exclusively in the Kupffer cells, whereas in (th3/th3)tp-2M mice it was located primarily in parenchymal cells (Figure S2). In the subset of β-thalassemia major mice with higher Hb levels (Table S1, group 1), liver iron levels were similar to those observed in older th3/+-12M animals (Figure 2A), which have lower Hb levels and more extensive splenomegaly than younger th3/+ mice. The previous observation suggests that not only does iron absorption increases with decreasing levels of Hb, but also that the liver is the primary organ where iron accumulates in conditions of severe anemia and a high degree of IE. Thus, infusing normal RBCs into thalassemic mice might reverse this IE-driven phenomenon. In fact, the iron content of the spleen was increased in mice given transfusions, whereas that of the liver was reduced (Figure 3A). In iron overload, the plasma nonheme iron concentration may rise significantly, leading to the appearance of non–transferrin-bound iron (NTBI). NTBI would further contribute to hepatic iron deposition, because it has been suggested that NTBI is rapidly assimilated by parenchymal cells.61,62 NTBI, unlike transferrin-bound iron, has been assumed to be chemically labile in terms of redox activity. A component of NTBI, labile plasma iron (LPI), modifies the balance between pro-oxidant and antioxidant activities in the serum. In thalassemic patients, it has been shown that accumulation of plasma NTBI correlates with the appearance of oxidation products and a decrease in plasma antioxidant capacity.59,63 Therefore, we evaluated total serum iron levels, Tf saturation, and the amount of LPI in our animals. As shown in Figure 4, the total amount of serum iron, Tf saturation, and LPI levels were elevated in (th3/th3)tp-2M mice compared to +/+ and th3/+ mice.
Figure 3.
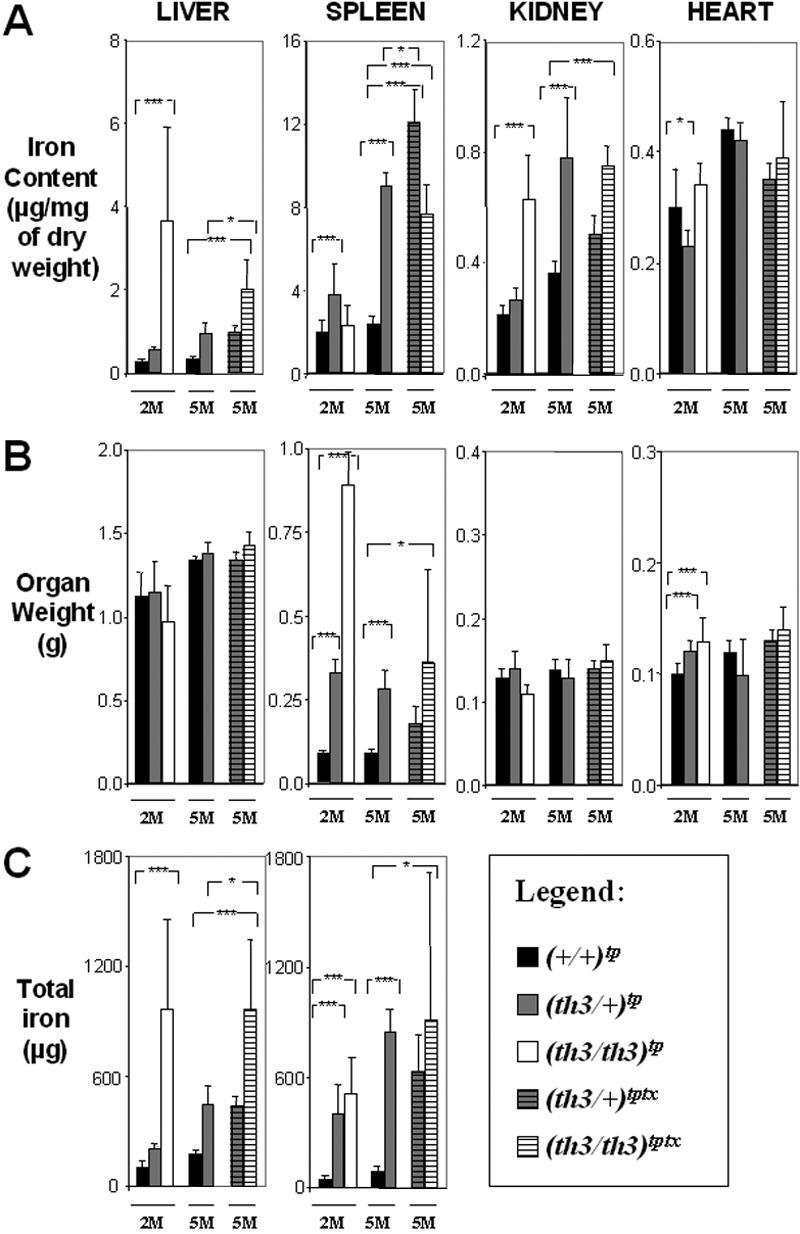
Organ iron content differs in β-thalassemia intermedia versus major. (A) Iron content (μg/mg dry weight), (B) organ weight (g), and (C) total iron (μg) of liver, spleen, kidney, and heart. The mice have been categorized according to their age as indicated in Figure 2. Mice receiving transplants are ■, +/+; ▩, th3/+; □, th3/th3; gray striped, th3/+ transfusion; white striped, th3/th3 transfusion. Error bars represent SD performed on 3 to 11 independent mice/tissue in duplicate per group for 90 mice sampled. *P < .05, **P < .01, and ***P < .001 relative to control using the Dunnett multiple comparison test. In the heart, the iron content did not increase in mice given a transplant compared to those not so treated (Figure 2A) and comparing (th3/th3)tp-2M to (th3/+)tp-2M. This indicates that, at least in mice, increasing the body iron content by augmented intestinal absorption or by indirect administration of iron through blood transfusion does not lead to increased accumulation of iron in this organ over time. This was also confirmed by Prussian blue staining (not shown).
Figure 4.

Serum iron levels, percentage of Tf saturation, and LPI levels are elevated in mice affected by β-thalassemia major. Mice affected by β-thalassemia major showed a predominant iron deposition in hepatic parenchymal cells. To evaluate whether mice affected by β-thalassemia major had elevated levels of NTBI, we investigated mice at 2 months, given transplants or not so treated. These mice showed the same serum iron, percentage of Tf saturation, and LPI values. Therefore, to simplify our graphs, we combined the animals given transplants and not given transplants into a single group for each genotype. Error bars represent SD performed on at least 5 animals per group. *P < .05, **P < .01, and ***P < .001 relative to controls using the Dunnett multiple comparison test.
In young thalassemic animals Hamp1 is responsible for increased iron absorption
To evaluate the role of Hamp1 in increased iron absorption in β-thalassemia, we first analyzed the liver of 2-month-old animals given transplants or not, aware that the mRNA data might not reflect the amount of protein or its activity, but only RNA expression or stability. Our data suggested that decreased Hamp1 expression could only explain the increased iron absorption in th3/+-2M and (th3/+)tp-2M mice. However, all animals given transplants showed low Hamp1 levels at 2 months, indicating that transient anemia due to the transplantation procedure was sufficient to decrease Hamp1 expression in the liver (Figure 5A). Nonetheless, the observation that Hamp1 levels were lowest in the livers of (th3/th3)tp-2M mice suggests an inversely proportional correlation between the level of IE and Hamp1 expression.
Figure 5.

In older thalassemic mice, Hamp1 expression increases to the level observed in normal mice, whereas Fpn1 is up-regulated. (A) Mice at 2 months of age. We used the average gene expression value of +/+-2M animals to normalize the values of th3/+-2M mice and of (+/+)tp-2M, (th3/+)tp-2M, and (th3/th3)tp-2M mice, as shown by the horizontal line in the figure. We decided to use this value because our iron data clearly indicated that (+/+)tp-2M mice show increased iron content in their spleen compared to +/+-2M animals. Bars represent the average fold change in mRNA expression compared to +/+-2M mice. (B) Mice at 5 months of age and given transfusions. +/+-5M and (+/+)tp-5M mice showed the same iron values (Figures 2–3) indicating that at this point the effects of the transplantation on erythropoiesis and iron metabolism had disappeared. The same was observed comparing th3/+-5M and (th3/+)tp-5M mice. Therefore, to simplify our graphs, we combined the mice given transplants and not so treated into a single group for each genotype (the light gray bars designating, only in this case, both groups receiving transplants and those that did not). Bars represent the average fold change in mRNA expression when compared with the group of (+/+)tp-5M plus +/+-5M mice. Blood transfusion did not appear to have any effect on the expression level of the genes analyzed in the duodenum, but it increased Hamp1 and normalized and normalized Cebpa liver expression levels in (th3/+)tptx-5M and (th3/th3)tptx-5M mice. Accordingly, in the liver of (th3/+)tptx-5M and (th3/th3)tptx-5M mice, Tfr1 decreased compared to the (th3/+)tp-5M - th3/+-5M mice pool and (th3/th3)tp-2M animals not given transfusions. Ftl1 and Fth1 expression levels were also assessed in the heart, but no differences were observed between thalassemic animals receiving transfusions, those not receiving transfusions, and control animals at 2, 5, and 12 months (not shown). (C) Mice at 12 months. Bars represent the average fold change in mRNA expression when compared with control +/+-12M mice. All the expression levels were normalized using oligonucleotides for mouse Gapdh or β-actin RNA. Ftl1 indicates ferritin-light chain; Fth1, ferritin-heavy chain, Tf, transferrin. Hamp1, Hjv, Hfe, Tfr1, Tfr2, and Tf were extremely low or undetectable in duodenum. For the 60 mice sampled, the Q-PCRs were performed on 3 to 7 independent mice/tissues in duplicate for each gene. Error bars represent SD performed on at least 3 animals per group. *P < .05, **P < .01, and ***P < .001 relative to controls using the Dunnett multiple comparison test. The complete list of genes analyzed is indicated in Table 1. Only genes whose expression was statistically different between control and thalassemic organs are described in this figure.
In young thalassemic animals low levels of Hamp1 are associated with low levels of Hfe and Cebpa
To investigate whether increased organ iron content in thalassemic animals was associated with dysregulation of additional iron-related genes, we evaluated the expression of several such genes in the liver of all animals generated (Table 1 provides the complete list of genes analyzed). Hfe mRNA was reduced in the liver of the animals that expressed low Hamp1 levels. Hfe-KO mice show hemochromatosis and have inappropriately low levels of Hamp1 expression.21,64 Therefore, our data suggest that Hfe could play a direct role in Hamp1 regulation in β-thalassemia (Figure 5A). In addition, low levels of Cebpa mRNA were observed in the liver of animals that exhibited further decreases in Hamp1 expression, namely, (th3/th3)tp-2M mice. Of note, Cebpa-KO mice show low levels of Hamp1 expression and are iron overloaded.52 Therefore our analysis suggests that low amounts of Cebpa might contribute to further decreases in Hamp1 synthesis (Figure 5A). Ftl1 and Fth1, which play a role in iron storage, were also reduced, both in the liver and in the duodenum of thalassemic mice. In (th3/+)tp-2M and th3/+-2M mice, Tfr1 and Fpn1, which are respectively responsible for internalization and egress of iron, were not altered in the liver and duodenum (Figure 5A). In contrast, Tfr1 and Fpn1 were highly up-regulated in the liver of (th3/th3)tp-2M mice. Tfr1 is expressed in immature or nucleated erythroid cells and up-regulated in thalassemic erythroid cells (Raffaella Schiró et al, “Ineffective erythropoiesis in beta-thalassemia is characterized by increased cell proliferation and attenuated cell death,” manuscript in preparation). In addition, EMH is clearly visible in the liver of th3/th3 mice (Figure S2). Therefore, we evaluated whether up-regulation of Tfr1 in the liver of (th3/th3)tp-2M mice was due to increased hepatocyte Tfr1 expression or due to EMH, by determining the levels of α-globin and Tfr1 expression in our liver specimens (Figure S3). Our analysis was consistent with the interpretation that increased Tfr1 mRNA was attributable to EMH.
In older thalassemic mice, increased iron content is dictated by the relative expression of Hamp1 in the liver and Fpn1 in the duodenum
Both (+/+)tp-5M and (th3/+)tp-5M mice showed the same iron (Figure 3A) and gene expression values (not shown) as 5-month-old animals not given transplants (Figure 2A), indicating that, at this point, the effects of the transplantation on iron metabolism disappeared. Therefore, the gene expression levels of 5-month-old animals given transplants or not were combined into a single group for each genotype (Figure 5B). At 5 and 12 months, the expression levels of all the genes analyzed in the liver of th3/+ mice, except for Fpn1, were similar to those of the +/+ control mice (Figure 5B-C). We were surprised to find that the levels of Hamp1 mRNA were similar to those of wt animals. Because iron levels were increased in all tested organs of aging th3/+ mice (5 and 12 months), these data indicate that in conditions of relatively modest anemia (Figure 1), Hamp1 may still be partially responsive to augmented iron content. Thus, another mechanism may contribute to increased iron absorption in these mice. In the duodenum, Fpn1 mRNA levels were slightly up-regulated in th3/+ mice at 5 months and clearly augmented at 12 months (Figure 5B-C). Similar findings were observed at the protein level by immunohistochemical analysis (Figure 6). This indicates that Fpn1 may play a major role in increased iron absorption, especially in chronic anemias, such as in β-thalassemia intermedia, that worsen over time. In the livers of th3/+ mice at 5 and 12 months, Hamp1, Ftl1, and Fth1 mRNA showed normal or increased levels of expression compared to th3/+-2M and (th3/+)tp-2M mice, indicating that these genes might share a common regulatory pathway (Figure 5). In addition, blood transfusion normalized or increased the expression levels of Hamp1 in the liver, as shown in Figure 5B. It has been postulated that erythropoiesis controls iron absorption via a hypothetical erythroid regulator, whose function would be to communicate the erythroid demand for iron resulting in modulation of Hamp1 expression. As the marrow iron requirement for erythropoiesis increases, more erythroid regulator would be produced thereby limiting Hamp1 expression and augmenting iron absorption. Therefore, in our mice that received transfusions, augmented Hamp1 levels might be due to the combined effect of reduced anemia, increased iron content, and suppression of erythropoiesis (Figure 5B).
Figure 6.

Fpn1 is increased in the duodenum of 12-month-old mice affected by β-thalassemia intermedia. Duodenum of (A) +/+-12M and (B) th3/+-12M mice. This assay confirmed our Q-PCR data that indicated up-regulation of Fpn1 in 1-year-old th3/+ mice compared to +/+ animals. (immunoperoxidase; original magnification, ×66). Images were captured on a Nikon Eclipse E800 microscope (Nikon, Melville, NY) with a Retiga Exi camera (Qimaging, Burnaby, BC, Canada) and a Plan Fluor 20x/0.75 NA objective, then acquired using IP Lab 3.65a software (Scanalytics, Fairfax, VA). Brightness/contrast and color balance were adjusted using Adobe Photoshop 7.0.1 (Adobe Systems, San Jose, CA).
Discussion
Our goals were to investigate the role of Hamp1 and other iron-regulatory genes in iron overload and to evaluate their correlation with IE in mouse models of β-thalassemia intermedia and major. In particular, the th3/th3 mouse model represents the most severe form of β-thalassemia, since in these animals the remaining globin genes are of embryonic origin and are expressed at a very low level in adult erythroid tissue.65 The degree of IE in th3/th3 mice is extremely severe, as shown by the low levels of Hb, the decreased reticulocyte counts, and the abundance of CD71+/Ter119+ early erythroid cells in the BM and spleen. Therefore, the th3/+ and th3/th3 mouse models are invaluable when it comes to elucidating the underlying mechanisms of iron absorption and distribution when red cell production varies from being reduced (th3/+) to almost absent (th3/th3).
Our first conclusion is that IE dictates the pattern of iron distribution. Our data show that iron accumulates progressively in the spleen of th3/+ mice and at a lower pace in the Kupffer cells of the liver, whereas in th3/th3 mice iron overload occurs rapidly and involves predominantly liver parenchymal cells. The highest liver iron content is observed in th3/th3 mice that show the lowest Hb levels and the highest degree of IE. In contrast, blood transfusion reduces IE and iron deposition in the livers of the same animals. Altogether these observations indicate that in conditions of extreme IE, there is relatively little peripheral destruction of red cells and iron accumulates more rapidly in the liver than in the spleen, consistent with the interpretation that iron loading results primarily from increased intestinal absorption. In fact, the iron concentration does not increase in the spleens of th3/th3 mice, although IE is more severe in β-thalassemia major than in β-thalassemia intermedia. There are at least 2 possible explanations for the lower splenic iron concentration in th3/th3 mice, as compared to th3/+ mice. One possibility is that less mature cells predominate in the erythron of th3/th3 mice and that the CD71+/Ter119+ early erythroid cells contain less heme-iron than the mature ones. Our nonheme iron assay excluded this hypothesis, however. Alternatively, the iron absorbed in β-thalassemia major might exceed the capacity of erythroid precursors to make use of it. Our analysis indicates that th3/th3 mice have significantly more iron in their serum, 4 to 5 times more than the +/+ and th3/+ mice analyzed at the same time point. Iron exceeding the erythroid demand would be deposited in parenchymal cells. NTBI might further contribute to the iron overload of these cells.61,62 Our data indicate that NTBI is elevated in th3/th3 mice, supporting this notion. Regardless, in th3/th3 mice, extreme IE seems to override iron overload, resulting in low levels of Hamp1 expression leading to the increased iron content of the liver. In contrast, in states of relatively mild anemia, iron absorption would be lower and the erythroid organs, spleen and BM, would use the absorbed iron, as is observed in th3/+ animals.
Our second major conclusion is that the mechanisms, which maintain a net positive increase in organ iron content, are different in th3/th3 versus th3/+ mice and in young versus old thalassemic mice. In th3/th3 mice, extreme IE and hypoxia are able to override the expected increase in Hamp1 due to high liver iron concentration, whereas in conditions of relatively mild anemia (th3/+ mice), the level of expression of Hamp1 is likely determined by both the relative degree of iron load and the erythropoietic rate, as Hamp1 might be expected to respond to iron overload in these conditions. In fact, whereas Hamp1 levels are low in 2-month-old th3/+ mice, in 12-month-old animals Hamp1 levels are similar to those of wt mice, although relatively low considering their iron stores. Because the total amount of iron in the spleen and liver increases progressively in th3/+ mice over time, as does the level of Hamp1 expression, other factors may contribute to the increased iron content observed. Our data suggest a possible alternative mechanism involving up-regulation of Fpn1 in the duodenum of older th3/+ mice. Our finding is even more intriguing considering that it has been reported that Fpn1 mRNA expression or protein levels are increased in HH.20,66 Compared to younger animals, old thalassemic mice might accumulate or produce some factors at a higher level resulting in increased production or stability of Fpn1 in the liver and duodenum, augmenting iron absorption in the latter case.
Our third conclusion is that Hfe, and to some extent Cebpa, may play a role in mediating Hamp1 expression in β-thalassemia. We have previously shown that Hamp1 expression is down-regulated in the liver of mice affected by β-thalassemia.53,54 This study confirms and extends this observation comparing young versus old animals and showing that Hfe, and to some extent Cebpa, are down-regulated in thalassemic animals that express low Hamp1 levels, while being up-regulated in mice undergoing transfusion, showing normal or increased Hamp1 levels. Obviously, the potential role of these proteins in the regulation of Hamp1 expression in β-thalassemia will need to be confirmed by protein analyses and genetic experiments.
The final picture that emerges from our study is that extreme IE in β-thalassemia major leads to excessively low Hamp1 expression resulting in more iron being absorbed than that required by red cell production, despite an increasing liver iron content. In contrast, iron absorption in β-thalassemia intermedia responds to the relative degree of IE, the body iron content, and other as yet undefined factors that accumulate or appear in aging thalassemic mice and regulate Hamp1 and Fpn1 expression.
Recently, De Franceschi et al13 analyzed the gene expression levels of several iron-regulatory genes in the liver and quantified the serum levels of cytokines that might control Hamp1 expression in 2 models of β-thalassemia intermedia (th1/th1 and th3/+). Apart from the cytokine analyses, which we did not investigate, our work confirms some of their data and extends the analysis to th3/+ mice at earlier and later time points (2 and 12 months). Although De Franceschi and colleagues did not see any reduction of Hamp1 in the mice affected by β-thalassemia intermedia at 4, 8, and 10 months of age, we show that 2-month-old th3/+ mice have a lower level of Hamp1 than their normal counterparts +/+-2M. This difference may be due to the fact that at 2 months of age the body iron levels of th3/+ mice are still too low to counterbalance the level of anemia. In addition, we analyzed both mice given transfusions and those not receiving transfusions affected by β-thalassemia major (th3/th3), which where not used by De Franceschi.13 Furthermore, we have shown, for the first time, the distribution and quantification of iron in several organs of thalassemic mice, including the heart, spleen, and kidney. We also correlated the level of IE with the distribution of body iron and have discovered a potential role of Fpn1 in the iron overload seen in β-thalassemia.
The results could not be simply transferred in thalassemic patients who have several interacting and modifier factors playing a role in iron homeostasis. However, the finding of decreased levels of HAMP in the urine of thalassemic patients67 corroborates our murine data on Hamp1. If these data are supported by preclinical studies wherein Hamp1 is administrated to thalassemic mice, new therapeutic approaches to preventing iron overload based on administration of HAMP or agents with HAMP-like activity may hold great promise.
Supplementary Material
Acknowledgments
This work was supported by grants from the Carlo and Micòl Schejola Foundation, the Cooley's Anemia Foundation, the Children's Cancer and Blood Foundation, and NIH-R21DK065169 (S.R.), R01DK55463 (R.W.G.), Associazione per la Lotta alla Talassemia di Rovigo (S.G. and L.B.), Associazione Regionale Sarda per la Lotta Contro la Thalassemia e per l'Assistenza dei Talassemici (M.F.M.), the Roche Foundation for Anemia Research (N.C.A.), National Institutes of Health training grant T32 HL07623 (D.M.W.), and the American Portuguese Biomedical Fund (APBRF, USA)/Inova grant (M.S. and P.R.). S.G. and L.B. are fellows of the Associazione per la Lotta alla Talassemia di Rovigo (AVLT); M.F.M. is a fellow of the Associazione Regionale Sarda per la Lotta Contro la Thalassemia e per l'Assistenza ai Thalassemici; and P.R. is a fellow of American Portuguese Biomedical Fund (APBRF/USA/Inova).
The authors would like to thank Dr Rosandra Kaplan, Dr Kristian Jensen, and Kimberly Young for reading the manuscript and for helpful discussions, as well as Scott A. Kerns for technical support.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.G. analyzed the mice, performed the gene expression analysis in the liver and heart, and collected and analyzed data; M.F.M. performed gene expression analysis in the duodenum and the non-EMH iron analysis; P.R. performed the FACS analysis; E.G. performed the bone marrow transplantations and CBC analysis; L.B. maintained the mouse colony and collected the mouse organs and RNA samples; A.C. performed the pathologic examination of the tissue samples; Y.L. performed the tissue and the immunohistochemistry stainings; N.A., G.R., and E.A.R. provided some of the information to perform the gene expression analysis and analyzed data; W.B. and Z.I.C. performed the LPI quantification; D.M.W. and N.C.A. provided some of the reagents to perform the iron analysis and the immunohistochemistry staining and analyzed the data; M.d.S. and P.J.G. provided vital reagents and analyzed the data; R.W.G. performed the atomic absorption iron analysis and analyzed the data; and S.R. analyzed the data and wrote the manuscript.
Conflict-of-interest disclosure. The authors declare no competing financial interests.
Correspondence: Stefano Rivella, Department of Pediatric Hematology-Oncology, Weill Medical College of Cornell University, 515E 71st St S702, New York, NY 10021; e-mail: str2010@med.cornell.edu.
References
- 1.Cooley TB, Lee P. A series of cases of splenomegaly in children with anemia and peculiar bone changes. Trans Am Pediat. Soc. 1925;37:29–33. [Google Scholar]
- 2.Giardina PJ, Grady RW. Chelation therapy in beta-thalassemia: an optimistic update. Semin Hematol. 2001;38:360–366. doi: 10.1016/s0037-1963(01)90030-7. [DOI] [PubMed] [Google Scholar]
- 3.Bannerman RM, Keusch G, Kreimer-Birnbaum M, Vance VK, Vaughan S. Thalassemia intermedia, with iron overload, cardiac failure, diabetes mellitus, hypopituitarism and porphyrinuria. Am J Med. 1967;42:476–486. doi: 10.1016/0002-9343(67)90276-8. [DOI] [PubMed] [Google Scholar]
- 4.Heinrich HC, Gabbe EE, Oppitz KH, et al. Absorption of inorganic and food iron in children with heterozygous and homozygous beta-thalassemia. Z Kinderheilkd. 1973;115:1–22. doi: 10.1007/BF00438987. [DOI] [PubMed] [Google Scholar]
- 5.Cossu P, Toccafondi C, Vardeu F, et al. Iron overload and desferrioxamine chelation therapy in beta-thalassemia intermedia. Eur J Pediatr. 1981;137:267–271. doi: 10.1007/BF00443255. [DOI] [PubMed] [Google Scholar]
- 6.Fiorelli G, Fargion S, Piperno A, Battafarano N, Cappellini MD. Iron metabolism in thalassemia intermedia. Haematologica. 1990;75:89–95. [PubMed] [Google Scholar]
- 7.Finch CA, Deubelbeiss K, Cook JD, et al. Ferrokinetics in man. Medicine. 1970;49:17–53. doi: 10.1097/00005792-197001000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Schrier SL. Pathophysiology of thalassemia. Curr Opin Hematol. 2002;9:123–126. doi: 10.1097/00062752-200203000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Mathias LA, Fisher TC, Zeng L, et al. Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol. 2000;28:1343–1353. doi: 10.1016/s0301-472x(00)00555-5. [DOI] [PubMed] [Google Scholar]
- 10.Centis F, Tabellini L, Lucarelli G, et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood. 2000;96:3624–3629. [PubMed] [Google Scholar]
- 11.Skow LC, Burkhart BA, Johnson FM, et al. A mouse model for beta-thalassemia. Cell. 1983;34:1043–1052. doi: 10.1016/0092-8674(83)90562-7. [DOI] [PubMed] [Google Scholar]
- 12.Curcio MJ, Kantoff P, Schafer MP, Anderson WF, Safer B. Compensatory increase in levels of beta minor globin in murine beta-thalassemia is under translational control. J Biol Chem. 1986;261:16126–16132. [PubMed] [Google Scholar]
- 13.De Franceschi L, Daraio F, Filippini A, et al. Liver expression of hepcidin and other iron genes in two mouse models of beta-thalassemia. Haematologica. 2006;91:1336–1342. [PubMed] [Google Scholar]
- 14.Yang B, Kirby S, Lewis J, Detloff PJ, Maeda N, Smithies O. A mouse model for beta 0-thalassemia. Proc Natl Acad Sci U S A. 1995;92:11608–11612. doi: 10.1073/pnas.92.25.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ciavatta DJ, Ryan TM, Farmer SC, Townes TM. Mouse model of human beta zero thalassemia: targeted deletion of the mouse beta maj- and beta min-globin genes in embryonic stem cells. Proc Natl Acad Sci U S A. 1995;92:9259–9263. doi: 10.1073/pnas.92.20.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rivella S, May C, Chadburn A, Riviere I, Sadelain M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101:2932–2939. doi: 10.1182/blood-2002-10-3305. [DOI] [PubMed] [Google Scholar]
- 17.Andrews NC. Molecular control of iron metabolism. Best Pract Res Clin Haematol. 2005;18:159–169. doi: 10.1016/j.beha.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Camaschella C, Merlini R. Inherited hemochromatosis: from genetics to clinics. Minerva Med. 2005;96:207–222. [PubMed] [Google Scholar]
- 19.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 20.Bridle KR, Frazer DM, Wilkins SJ, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361:669–673. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 21.Zhou XY, Tomatsu S, Fleming RE, et al. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc Natl Acad Sci U S A. 1998;95:2492–2497. doi: 10.1073/pnas.95.5.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy CN, Penny DM, Feder JN, Enns CA. The hereditary hemochromatosis protein, HFE, specifically regulates transferrin-mediated iron uptake in HeLa cells. J Biol Chem. 1999;274:9022–9028. doi: 10.1074/jbc.274.13.9022. [DOI] [PubMed] [Google Scholar]
- 23.de Almeida SF, Carvalho IF, Cardoso CS, et al. HFE cross-talks with the MHC class I antigen presentation pathway. Blood. 2005;106:971–977. doi: 10.1182/blood-2004-12-4640. [DOI] [PubMed] [Google Scholar]
- 24.Kawabata H, Fleming RE, Gui D, Moon SY, et al. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood. 2005;105:376–381. doi: 10.1182/blood-2004-04-1416. [DOI] [PubMed] [Google Scholar]
- 25.Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105:1803–1806. doi: 10.1182/blood-2004-08-3042. [DOI] [PubMed] [Google Scholar]
- 26.Kawabata H, Yang R, Hirama T, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274:20826–20832. doi: 10.1074/jbc.274.30.20826. [DOI] [PubMed] [Google Scholar]
- 27.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–28498. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 28.Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 29.Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115:2187–2191. doi: 10.1172/JCI25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33:21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 32.Camaschella C, Roetto A, De Gobbi M. Juvenile hemochromatosis. Semin Hematol. 2002;39:242–248. doi: 10.1053/shem.2002.35635. [DOI] [PubMed] [Google Scholar]
- 33.Papanikolaou G, Tzilianos M, Christakis JI, et al. The molecular basis of ferroportin-linked hemochromatosis. Proc Natl Acad Sci U S A. 2005;102:8955–8960. doi: 10.1073/pnas.0503804102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drakesmith H, Schimanski LM, et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood. 2005;106:1092–1097. doi: 10.1182/blood-2005-02-0561. [DOI] [PubMed] [Google Scholar]
- 36.Schimanski LM, Drakesmith H, Merryweather-Clarke AT, et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood. 2005;105:4096–4102. doi: 10.1182/blood-2004-11-4502. [DOI] [PubMed] [Google Scholar]
- 37.Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32:131–138. doi: 10.1016/j.bcmd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Corradini E, Montosi G, Ferrara F, et al. Lack of enterocyte iron accumulation in the ferroportin disease. Blood Cells Mol Dis. 2005;35:315–318. doi: 10.1016/j.bcmd.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 40.McKie AT, Marciani P, Rolfs A, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 41.Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 42.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 43.Fleming RE, Sly WS. Hepcidin: a putative iron-regulatory hormone relevant to hereditary hemochromatosis and the anemia of chronic disease. Proc Natl Acad Sci U S A. 2001;98:8160–8162. doi: 10.1073/pnas.161296298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 45.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ganz T. Hepcidin—a peptide hormone at the interface of innate immunity and iron metabolism. Curr Top Microbiol Immunol. 2006;306:183–198. doi: 10.1007/3-540-29916-5_7. [DOI] [PubMed] [Google Scholar]
- 47.Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108:3730–3735. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204–3209. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang RH, Li C, Xu X, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 50.Babitt JL, Huang FW, Wrighting DM, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38:531–539. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 51.Truksa J, Peng H, Lee P, Beutler E. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci U S A. 2006;103:10289–10293. doi: 10.1073/pnas.0603124103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Courselaud B, Pigeon C, Inoue Y, et al. C/EBPalpha regulates hepatic transcription of hepcidin, an antimicrobial peptide and regulator of iron metabolism. Cross-talk between C/EBP pathway and iron metabolism. J Biol Chem. 2002;277:41163–41170. doi: 10.1074/jbc.M202653200. [DOI] [PubMed] [Google Scholar]
- 53.Weizer-Stern O, Adamsky K, Amariglio N, et al. mRNA expression of iron regulatory genes in beta-thalassemia intermedia and beta-thalassemia major mouse models. Am J Hematol. 2006;81:479–483. doi: 10.1002/ajh.20549. [DOI] [PubMed] [Google Scholar]
- 54.Adamsky K, Weizer O, Amariglio N, et al. Decreased hepcidin mRNA expression in thalassemic mice. Br J Haematol. 2004;124:123–124. doi: 10.1046/j.1365-2141.2003.04734.x. [DOI] [PubMed] [Google Scholar]
- 55.Breda L, Gardenghi S, Guy E, et al. Exploring the role of hepcidin, an antimicrobial and iron regulatory peptide, in increased iron absorption in beta-thalassemia. Ann N Y Acad Sci. 2005;1054:417–422. doi: 10.1196/annals.1345.069. [DOI] [PubMed] [Google Scholar]
- 56.Weizer-Stern O, Adamsky K, Amariglio N, et al. Downregulation of hepcidin and haemojuvelin expression in the hepatocyte cell-line HepG2 induced by thalassaemic sera. Br J Haematol. 2006;135:129–138. doi: 10.1111/j.1365-2141.2006.06258.x. [DOI] [PubMed] [Google Scholar]
- 57.Graziano JH, Grady RW, Cerami A. The identification of 2, 3-dihydroxybenzoic acid as a potentially useful iron-chelating drug. J Pharmacol Exp Ther. 1974;190:570–575. [PubMed] [Google Scholar]
- 58.Torrance JD, Bothwell TH. Iron Methods in Hematology. Vol. 1. New York, NY: Churchill Livingstone; 1980. Tissue iron stores. pp. 90–115. [Google Scholar]
- 59.Esposito BP, Breuer W, Sirankapracha P, Pootrakul P, Hershko C, Cabantchik ZI. Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood. 2003;102:2670–2677. doi: 10.1182/blood-2003-03-0807. [DOI] [PubMed] [Google Scholar]
- 60.Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 61.Edwards CQ, Griffen LM, Kaplan J, Kushner JP. Twenty-four hour variation of transferrin saturation in treated and untreated haemochromatosis homozygotes. J Intern Med. 1989;226:373–379. doi: 10.1111/j.1365-2796.1989.tb01411.x. [DOI] [PubMed] [Google Scholar]
- 62.Craven CM, Alexander J, Eldridge M, Kushner JP, Bernstein S, Kaplan J. Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: a rodent model for hemochromatosis. Proc Natl Acad Sci U S A. 1987;84:3457–3461. doi: 10.1073/pnas.84.10.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pootrakul P, Breuer W, Sametband M, Sirankapracha P, Hershko C, Cabantchik ZI. Labile plasma iron (LPI) as an indicator of chelatable plasma redox activity in iron-overloaded beta-thalassemia/HbE patients treated with an oral chelator. Blood. 2004;104:1504–1510. doi: 10.1182/blood-2004-02-0630. [DOI] [PubMed] [Google Scholar]
- 64.Nicolas G, Viatte L, Lou DQ, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34:97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 65.Geiger H, Sick S, Bonifer C, Muller AM. Globin gene expression is reprogrammed in chimeras generated by injecting adult hematopoietic stem cells into mouse blastocysts. Cell. 1998;93:1055–1065. doi: 10.1016/s0092-8674(00)81210-6. [DOI] [PubMed] [Google Scholar]
- 66.Adams PC, Barbin YP, Khan ZA, Chakrabarti S. Expression of ferroportin in hemochromatosis liver. Blood Cells Mol Dis. 2003;31:256–261. doi: 10.1016/s1079-9796(03)00136-0. [DOI] [PubMed] [Google Scholar]
- 67.Papanikolaou G, Tzilianos M, Christakis JI, et al. Hepcidin in iron overload disorders. Blood. 2005;105:4103–4105. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.