

Abstract
Although myc and bcl-2 synergize in tumor development, particularly lymphomagenesis, it is not known whether endogenous bcl-2 is required for myc-induced tumorigenesis. To investigate the role of endogenous Bcl-2 in myc-induced lymphomagenesis, we bypassed the early death of Bcl-2–deficient mice by reconstituting lethally irradiated wild-type (wt) mice with a hematopoietic system from fetal liver–derived stem cells of Eμ-myc/bcl-2−/− or control Eμ-myc transgenic embryos. In premalignant (healthy) recipients, loss of Bcl-2 caused a moderate decrease in pre-B and immature B cells, and a dramatic reduction of mature B lymphocytes expressing the Eμ-myc transgene. Furthermore, cultured preneoplastic Eμ-myc/bcl-2−/− mature B cells displayed accelerated apoptosis compared with Eμ-myc B cells. However, despite the striking reduction in B-cell numbers in vivo, ablation of endogenous Bcl-2 did not prevent or even delay development of Eμ-myc lymphoma. Moribund mice presented with similar degrees of splenomegaly, blood leukocyte numbers, and tumor dissemination at death. These findings demonstrate that the initiation, development, continued growth, and severity of Eμ-myc lymphoma do not depend upon endogenous Bcl-2, nor upon the total number of B lymphoid cells driven by the Eμ-myc transgene. These results have implications for the treatment of hematopoietic tumors, particularly those that are not caused by Bcl-2 overexpression.
Introduction
Activation of the c-myc proto-oncogene by the t(8;14) chromosomal translocation is the key transforming event in the etiology of human Burkitt lymphoma, and deregulated myc expression also features in numerous other human cancers, including cervical and small-cell lung carcinoma and myeloid leukemia.1 The transcriptional regulator c-Myc drives diverse cellular processes, including cell volume growth, cell-cycle progression, inhibition of differentiation, and, when growth factors are limiting, apoptotic cell death.2 Apoptosis (programmed cell death) is a genetically controlled process for killing unwanted cells that is essential for the normal development and function of multicellular organisms.3 Abnormalities in cell death control can also promote tumorigenesis.4 The first cell death regulator to be discovered was the bcl-2 gene, which was identified by its frequent translocation in human follicular B-cell lymphoma.5 Its oncogenic potential was revealed when Bcl-2 overexpression was shown to promote the survival (but not proliferation) of pro-B cells deprived of cytokine.6
Experiments using transgenic mice have provided considerable insight into the complexities of neoplastic transformation. Deregulated c-myc expression under the control of the immunoglobulin heavy chain (IgH) gene enhancer (Eμ) causes abnormal growth and proliferation of pre-B and B cells and impedes differentiation, culminating in clonal pre-B or immature B-cell lymphoma.7–9 The stochastic nature of tumor onset in these animals indicates that malignant transformation requires secondary genetic aberrations, such as inactivation of the ARF-Mdm2-p53 pathway,10 loss of proapoptotic BH3-only proteins Bim11 or Puma,12 or deregulated expression of Bcl-2–like prosurvival proteins.13–15 These changes are believed to counter the proapoptotic action of Myc under suboptimal growth conditions.4
Increased cell survival on its own is not a strong impetus for tumorigenesis. Transgenic mice expressing Bcl2 under control of the IgH gene enhancer, mimicking the t(14;18) translocation found in follicular-center B lymphoma, have a 3- to 5-fold increase in B cells,16,17 yet only approximately 5% develop lymphoma within the first year of life.18 Bcl-2 appears to promote tumorigenesis by allowing cells to survive while acquiring additional oncogenic mutations, such as a c-myc translocation. Indeed, Eμ-myc/Eμ-bcl-2 bitransgenic mice succumb to lymphoma more rapidly (100% mortality by 7 weeks of age) than those bearing only the Eμ-myc transgene (less than 40% mortality at this time),13 and progression of human follicular lymphoma from an indolent to a more aggressive state is often associated with acquisition of a c-myc translocation.19,20
Recent work has established that sustained Bcl-2 overexpression is required for the continued growth of lymphomas evoked by the deregulated expression of both Myc and Bcl-2.21 This observation supports the view that Bcl-2 will be an important target for treating those tumors arising from enforced Bcl-2 expression.22 However, most cancers, including hematopoietic neoplasms, do not harbor bcl-2 gene translocations or amplifications, and the current thrust to develop Bcl-2 antagonists highlights the need to determine whether endogenous bcl-2 is required for the initiation and sustained growth of cancers. It also remains unclear whether the inhibition of apoptosis is critical in the selection of an initiating cell or is required only later in tumor progression. Since Bcl-2 is expressed in many lymphoid cell subsets, including early progenitors,23,24 and because Bcl-2 overexpression enhances the survival of B lymphocytes at all stages of development,16,17,25 endogenous Bcl-2 levels may be a crucial determinant of Eμ-myc–induced lymphomagenesis.
Here, we have addressed these issues by investigating the role of endogenous Bcl-2 in Myc-induced growth, proliferation, survival, and transformation of B lymphoid cells. To overcome the complication that young Bcl-2–deficient mice, particularly those on a C57BL/6 background, succumb to polycystic kidney disease,26–29 we have studied wild-type mice whose hematopoietic system was reconstituted with either Eμ-myc/bcl-2−/− or, as a control, with Eμ-myc/bcl-2+/+ hematopoietic stem cells. Surprisingly, despite a marked reduction of B cells in Eμ-myc/bcl-2−/− reconstituted mice, lymphoma onset and severity were not altered compared with control Eμ-myc reconstituted animals.
Materials and methods
Mice and hematopoietic reconstitution
All experiments with animals were conducted according to the guidelines of the Melbourne Directorate Animal Ethics Committee. All mouse strains had been backcrossed to the C57BL/6 (Ly5.2+) genetic background more than 10 generations prior to use in the experiments shown here. The Eμ-myc transgenic mice7 and bcl-2 gene targeted mice27 have both been described previously. Polymerase chain reaction (PCR)–based genotyping for Eμ-myc11 and bcl-229 was performed as described.
Maternal transmission of the Eμ-myc transgene has previously been demonstrated to produce a reduction in tumor latency (A. Harris and J.M.A., unpublished results, February 1988). Breeding was therefore undertaken as follows: Eμ-myc transgenic males were first crossed with bcl-2+/− females, and their Eμ-myc/bcl-2+/− male offspring were then mated with bcl-2+/− females. Embryos from these matings were harvested at embryonic day 14.5 (E14.5) and genotyped by PCR on tail-derived DNA. Single-cell suspensions were prepared from fetal liver, and cell viability was determined by trypan blue exclusion. Then, 2 × 106 hematopoietic fetal liver cells were injected into the tail vein of lethally irradiated (2 × 5.5 Gy in a 3-hour interval) C57BL/6-Ly5.1+ mice. Mice that received transplants were maintained on neomycin sulfate–supplemented drinking water for 14 days after irradiation to prevent infection.
Reconstituted mice were monitored daily for signs of malignancy or other signs of ill health. Mice were killed when deemed moribund by an experienced animal technician who was blinded to the genotype of the cells used for hematopoietic reconstitution. Typically, the moribund mice presented with several (but not always all) of the following features: hunched posture, labored breathing, weight loss, enlarged lymph nodes (LNs) and/or spleen, peripheral white blood cell (WBC) cellularity of 20 × 106/mL or greater, a dominant (clonal) donor-derived (Ly5.2+) B lymphoid (B220+ and/or CD19+) population detected by immunofluorescent staining with cell surface marker–specific antibodies and fluorescence-activated cell sorter (FACS) analysis, the ability of leukemic cells from spleen or LNs to form a tumor upon transplantation into nonirradiated histocompatible recipients, and histologic evidence of disseminated lymphoma.
Preneoplastic analysis was performed on mice 8 to 12 weeks following hematopoietic reconstitution. To confirm that these mice were free of tumors at the time of analysis, 106 spleen or LN cells were injected intravenously into 2 or 3 congenic recipient mice as previously described.8,9 Hematopoietic reconstituted mice were deemed tumor-free when all recipients autopsied 80 days after transplantation demonstrated no overt signs of malignancy.
Immunofluorescent staining, flow cytometric analysis, and cell sorting
Analysis of preleukemic cells and primary lymphomas was performed by preparing single-cell suspensions and incubating cells in 2.4G2 (anti-FcγRII) antibody (at approximately 2 mg/mL) plus 2% normal rat serum and staining with rat monoclonal antibodies to: B220 (RA3-6B2), CD19 (ID3), c-Kit (ACK2 or ACK4), IgM (5.1 or 333.12), IgD (11-26C), CD4 (H1289 or YTA321), CD8 (YTS169), Mac-1(MI/70), Gr-1 (RB6-8C5), and erythroid cell–specific marker (Ter119). Host (Ly5.1+) and donor-derived (Ly5.2+) cell populations were discriminated by staining with monoclonal antibodies: anti-Ly5.1 (A201.1) and anti-Ly5.2 (5.450.15.2). Antibodies were conjugated to fluorescein isothiocyanate (FITC; Molecular Probes, Eugene, OR), phycoerythrin (PE; Prozyme, San Leandro, CA), cyanine 5 (Cy5; Amersham Biosciences, Arlington Heights, IL), allophycocyanin (APC; Prozyme) or biotin (Molecular Probes) according to the manufacturers' protocols. Biotinylated antibodies were detected by secondary staining using FITC-, PE- or Tricolor-coupled streptavidin (Caltag, Carlsbad, CA). Dead cells were excluded by propidium iodide (PI) staining (2 μg/mL). Purification of preleukemic donor-derived B lymphoid subpopulations was performed by multiparameter FACS sorting using a MoFlo (Cytomation, Fort Collins, CO) or DiVa (BD Biosciences, Palo Alto, CA) high-speed flow cytometer. Sorting was performed on the basis of forward (FSC) and side light scatter (SSC), by exclusion of Ly5.1+ and PI+ cells and by the following staining: B220+ c-Kit+ sIg− for pro-B cells, B220+ c-Kit− sIg− for pre-B cells, B220+ sIgMhi sIgDlo for immature B cells, and B220+ sIgMlo sIgDhi for mature B cells. Absolute numbers of B lymphoid subpopulations were determined by cell counting and multiplying cell numbers by the percentage of Ly5.2+ cell subsets as determined by FACS.
Cell culture and cell-survival assays
FACS-purified B lymphocyte populations were cultured at 2 to 5 × 104 cells (per time point) in the high-glucose version of Dulbecco modified Eagle medium (DMEM) supplemented with 250 μM L-asparagine, 50 μM 2-mercaptoethanol, and 10% heat-inactivated fetal calf serum (JRH Biosciences, Kansas City, MO). Cell viability was determined by staining with PI (2 μg/mL) and FITC-labeled Annexin V followed by analysis in a FACScan (Becton Dickinson, San Jose, CA).
Cell-cycle analysis
Purified B lymphocyte populations (105 cells) were washed with balanced salt solution (containing Ca2+ and Mg2+) and resuspended in 200 μL of ice-cold PI staining solution (10 mM Tris (pH 8.0), 1 mM NaCl, 0.1% Nonidet P40, 50 μg/mL PI, and 10 μg/mL RNaseA). Samples were vortexed briefly and incubated on ice for 10 minutes. DNA content was determined by analyzing an equal number of events (104 cells/sample) using a FACScan (Becton Dickinson, San Jose, CA) with linear amplification settings. Percentages of cells in G0/G1 and S/G2/M were determined by manual gating using WEASEL (Water and Eliza Hall Institute) analysis software.
Statistical analysis
Statistical significance was assessed using the Student t test, and Kaplan-Meier curves were constructed using Graph Pad Prism software (version 3.0; Cary, NC). Statistical evaluation of tumor-free survival data was undertaken using a log-rank test or a 1-way ANOVA test using Graph Pad Prism (version 3.0). For more information, see the Supplemental Materials, available on the Blood website (click the Supplemental Materials link at the top of the online article).
Results
Abnormally low numbers of Eμ-myc/bcl-2−/− leukocytes in reconstituted mice
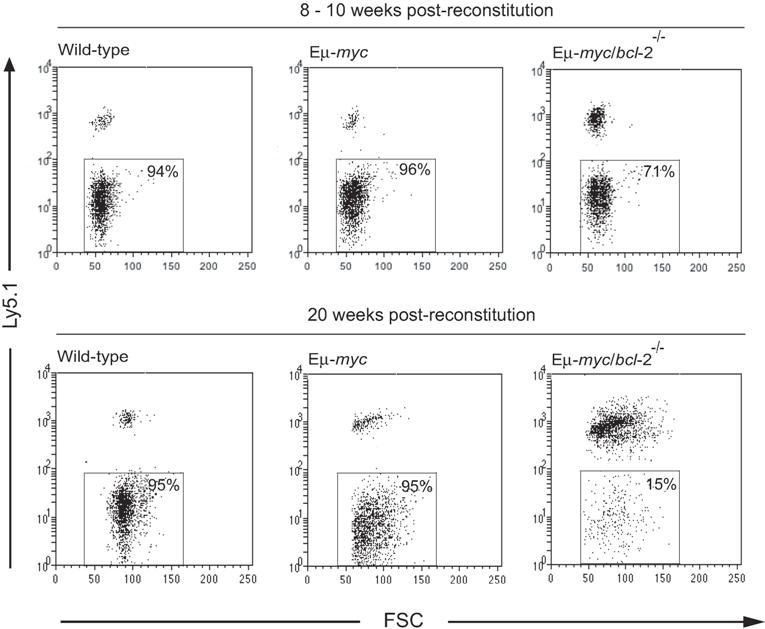
To investigate how endogenous Bcl-2 affects myc-induced lymphomagenesis, we reconstituted lethally irradiated C57BL/6-Ly5.1+(wild-type [wt]) mice with fetal liver hematopoietic stem cells from Ly5.2 Eμ-myc/bcl-2−/− or, as controls, from Eμ-myc/bcl-2+/+, Eμ-myc/bcl-2+/−, bcl-2+/−, bcl-2−/−, and wt embryos (E14.5). Hereafter, these reconstituted animals are referred to as Eμ-myc/bcl-2−/−, Eμ-myc/bcl-2+/−, Eμ-myc, bcl-2−/−, bcl-2+/−, or wt mice. The efficacy of hematopoietic reconstitution was examined initially 8 to 10 weeks after transplantation by staining peripheral blood leukocytes with monoclonal antibodies to Ly5.1 (host) and Ly5.2 (donor). Mice that received transplants of Eμ-myc, Eμ-myc/bcl-2+/−, bcl-2+/−, or wt cells routinely exhibited 85% or more donor-derived hematopoietic cells, whereas recipients of Eμ-myc/bcl-2−/− or bcl-2−/− cells consistently showed lower percentages (less than 70%) and overall numbers of leukocytes (Figure 1A). Subsequent analysis of peripheral blood at 20 weeks after transplantation revealed that while the percentages (and total numbers) of donor-derived leukocytes remained the same (or even increased) for wt or Eμ-myc mice, those in Eμ-myc/bcl-2−/− and bcl-2−/− recipients were frequently reduced even further (Figure S1, available on the Blood website; see the Supplemental Figures link at the top of the online article). The reduced numbers of Bcl-2–deficient leukocytes in the blood is consistent with previous evidence of attrition of bcl-2−/− cells in peripheral lymphoid organs.29,30 To determine whether the lowered leukocyte cellularity of Eμ-myc/bcl-2−/− and bcl-2−/− mice reflected a decreased proportion of B cells (sIg+), we stained leukocytes from peripheral blood of Eμ-myc/bcl-2−/−, Eμ-myc, bcl-2−/− and wt mice with monoclonal antibodies to B220, IgM, IgD, and Ly5.2. Indeed, bcl-2−/− mice contained significantly fewer donor-derived sIg+ B cells in their blood than their wt counterparts (Figure 1A). Moreover, Eμ-myc/bcl-2−/− mice exhibited a notable decrease (P < .05) when compared with bcl-2−/− mice, and a profound decline (P < .005) relative to Eμ-myc animals (Figure 1A).
Figure 1.

Impaired B lymphopoiesis in Eμ-myc/bcl-2−/− reconstituted mice. Analysis of total numbers of leukocytes and B lymphocyte populations in peripheral blood (A) and bone marrow (B) from lethally irradiated mice reconstituted 8 to 12 weeks earlier with wt, bcl-2−/−, Eμ-myc, or Eμ-myc/bcl-2−/− fetal liver–derived stem cells. (A) Blood leukocytes were counted in an automated blood analyzer and percentages of donor-derived sIg+ B cells determined by staining with fluorochrome-conjugated monoclonal antibodies to B220, IgM, IgD, and Ly5.2 followed by FACS analysis. (B) Leukocytes from bone marrow (2 femurs per mouse) were counted in a hemocytometer and donor derived pro-B (Ly5.2+ B220+ c-Kit+ sIg−), pre-B (Ly5.2+ B220+ c-Kit− sIg−), and B cells (Ly5.2+ B220+ c-Kit− sIg+) quantified by staining with surface marker–specific monoclonal antibodies followed by FACS analysis. Data represent means ± SEM from 4 to 6 mice of each genotype. Statistically significant differences: *P < .05; **P < .005.
Preneoplastic Eμ-myc/bcl-2−/− mice display a marked reduction in mature B cells
The reduced numbers of B cells in the blood of Eμ-myc/bcl-2−/− mice led us to determine the specific developmental stage at which B lymphopoiesis was perturbed in these animals. As expression of the Eμ-myc transgene commences very early in B lineage commitment,8 we examined B-cell development from the pro-B to the mature B-cell stage in the reconstituted mice, all of which were verified to be lymphoma-free by demonstrating that injection of 106 spleen or LN cells into non-irradiated C57BL/6 mice did not result in tumor formation within 80 days. We quantified pro-B (B220+ or CD19+ c-Kit+ sIg−), pre-B (B220+ or CD19+ c-Kit− sIg−), immature B (B220+ or CD19+ sIgMhi sIgDlo), and mature B cells (B220+ or CD19+ sIgMlo sIgDhi) in bone marrow, spleen, and LNs by immunofluorescent staining with surface marker–specific monoclonal antibodies and FACS analysis.
In the marrow (Figure 1B), the bcl-2−/− recipients had numbers of pro-B and pre-B cells similar to that of wt recipients; however, bcl-2−/− reconstituted mice displayed an approximately 3-fold drop in sIg+ B cells (P < .05). Compared with wt recipients, the Eμ-myc marrow had an approximately 5-fold excess of large pre-B cells and an approximately 3-fold reduction in the more differentiated sIg+ B cells.8 Compared with the Eμ-myc animals, the marrow of Eμ-myc/bcl-2−/− mice had similar numbers of pro-B cells, but approximately 2- to 3-fold lower numbers (P < .05) of both pre-B and sIg+ B cells (Figure 1B).
In the spleens of the recipients, flow cytometric analysis (Figure 2A) revealed that the Eμ-myc/bcl-2−/− recipients exhibited a modest decline in the proportion of immature B cells (R1 window) and a marked reduction in mature B cells (R2 window). This reduction in the percentage of mature B cells translated into a more than 10-fold reduction (P < .005) in their total number compared to that in Eμ-myc animals (Figure 2A). Similarly, the LNs (Figure 2B) of Eμ-myc/bcl-2−/− mice were almost completely devoid of mature B lymphocytes (approximately 40-fold less than in Eμ-myc mice; P < .001). Comparison of all B lymphoid subsets between Eμ-myc and Eμ-myc/bcl-2+/− mice revealed that loss of a single bcl-2 allele did not significantly reduce the size of any of these populations (Figure S2). Taken together, these results indicate that endogenous Bcl-2 is critical for the survival of mature Eμ-myc B cells but is less important for the maintenance of pro-B, pre-B, and immature B cells.
Figure 2.

Preleukemic Eμ-myc/bcl-2−/− reconstituted mice have abnormally low numbers of mature B cells. Spleen and LNs were harvested from lethally irradiated wt mice 8 to 12 weeks after reconstitution with fetal liver cells from wt, bcl-2−/−, Eμ-myc, and Eμ-myc/bcl-2−/− (E14.5) embryos. Single-cell suspensions were prepared, stained with fluorochrome-conjugated monoclonal antibodies to B220, IgM, IgD, and Ly5.2, and analyzed by FACS. (A) Representative FACS profiles illustrating the abundance of immature (R1: B220+ sIgMhi sIgDlo) and mature (R2: B220+ sIgMlo sIgDhi) splenic B cells, and total numbers in spleens from wt, bcl-2−/−, Eμ-myc/bcl-2+/+, and Eμ-myc/bcl-2−/− fetal liver reconstituted mice. Profiles are gated on Ly5.2+ B220+ cells. (B) Total numbers of donor-derived mature B cells (B220+ sIgMlo sIgDhi) in LNs from wt, bcl-2−/−, Eμ-myc, and Eμ-myc/bcl-2−/− reconstituted mice. Graphs represent means ± SEM from 5 to 8 mice of each genotype. Statistically significant differences: *P < .05; **P < .001.
Loss of bcl-2 accelerates Eμ-myc–induced apoptosis of cytokine-deprived mature B cells in culture
To determine whether the reduced numbers of mature B cells in Eμ-myc/bcl-2−/− mice was a reflection of increased apoptosis, we purified donor-derived (Ly5.2+) pro-B (B220+ c-Kit+ sIg−), pre-B (B220+ c-Kit− sIg−), immature B (B220+ sIgMhi sIgDlo), and mature B cells (B220+ sIgMlo sIgDhi) from bone marrow or spleen by FACS sorting and monitored their survival in culture following cytokine deprivation. Upon culture in simple medium (without added growth factors), bcl-2−/− pro-B, pre-B, and immature B cells died at comparable rates to that of control wt cells (Figure 3). In contrast, as described previously,29 loss of Bcl-2 significantly (eg, at 24 hours: P < .001) enhanced the death of mature B cells in culture (Figure 3).
Figure 3.

Abnormally accelerated apoptosis of Eμ-myc/bcl-2−/− B cells in culture. Survival assays were performed on purified populations of preneoplastic bone marrow–derived pro-B (Ly5.2+ B220+ sIg− c-Kit+) or pre-B cells (Ly5.2+ B220+ sIg− c-Kit−) and splenic immature (Ly5.2+ B220+ sIgMhi sIgDlo) or mature B lymphocytes (Ly5.2+ B220+ sIgMlo sIgDhi) from mice 8 to 12 weeks following reconstitution with fetal liver cells from wt (▴), bcl-2−/− (▵), Eμ-myc (■), and Eμ-myc/bcl-2−/− (□) E14.5 embryos. The purified donor-derived B-cell populations were cultured in the absence of cytokines for the indicated time periods, and cell viability was measured by staining with PI plus annexin V and FACS analysis. Data represent means ± SEM from 4 to 8 independent experiments for each cell subset and genotype.
Deregulated myc expression enhances cytokine withdrawal-induced apoptosis of many cell types,31 including B lymphoid cells.32 Accordingly, Eμ-myc B lymphoid cells at all developmental stages died more rapidly than their wt (nontransgenic) counterparts (Figure 3). The Eμ-myc/bcl-2−/− and Eμ-myc pre-B cells died at a similar rate, whereas immature B cells of Eμ-myc/bcl-2−/− origin showed moderately enhanced death, most evident at the latest time point (48 hours). Notably, a significant acceleration of apoptosis (eg, at 16 hours; P < .001) was evident in Eμ-myc/bcl-2−/− mature B cells across all time points examined; indeed, by 24 hours, all of the Eμ-myc/bcl-2−/− mature B cells, yet only 55% of their Eμ-myc counterparts had died (Figure 3). These observations demonstrate that endogenous Bcl-2 is critical for countering the proapoptotic effects of deregulated myc expression in mature B cells, and has a minor role in immature B cells, but appears to be dispensable for pro-B and pre–B-cell survival.
Deregulated cycling of Eμ-myc transgenic pro-B and pre-B cells is unaffected by loss of endogenous Bcl-2
Preleukemic Eμ-myc transgenic mice have a 4- to 5-fold expansion in cycling pro-B and pre-B cells.8 Since lymphocytes overexpressing Bcl-2 exhibit delayed cell-cycle entry following mitogenic stimulation,33–35 we examined whether loss of endogenous Bcl-2 affected the cycling of Eμ-myc or nontransgenic B cells. Donor-derived (Ly5.2+) pro-B and pre-B cells purified from the bone marrow of preleukemic reconstituted animals by FACS sorting were subjected to DNA content analysis by PI staining and flow cytometry. Quantification of the cells residing in the S/G2/M phases revealed that loss of endogenous Bcl-2 did not affect cycling of pro-B or pre-B cells, regardless of whether or not they expressed the Eμ-myc transgene (Figure 4). Moreover, flow cytometric analysis of forward light scatter revealed no difference in size between Eμ-myc and Eμ-myc/bcl-2−/− pro-B or pre-B cells (Figure S3). These results show that endogenous Bcl-2 is not required for the myc-induced growth or cycling of B lymphoid cells.
Figure 4.

Loss of endogenous Bcl-2 does not affect the enhanced proliferation of Eμ-myc transgenic B lymphoid cells. Donor-derived pro-B cells (Ly5.2+ B220+ sIg− c-Kit+) and pre-B cells (Ly5.2+ B220+ sIg− c-Kit−) were purified by multiparameter cell sorting from the bone marrow of mice reconstituted with wt, bcl-2−/−, Eμ-myc, and Eμ-myc/bcl-2−/− cells. The purified cells were then permeabilized and stained with PI, and their DNA content was analyzed by flow cytometry. The average percentages of cells in the G0/G1 and S/G2/M phases, obtained by manual gating, are indicated in the top right corners of the histograms. Results are representative of 3 to 4 independent experiments from 3 to 5 mice of each genotype.
Loss of endogenous bcl-2 does not affect Eμ-myc–induced lymphomagenesis
To examine the role of endogenous Bcl-2 in Eμ-myc–induced lymphoma development, cohorts of Eμ-myc (n = 18), Eμ-myc/bcl-2+/− (n = 27), and Eμ-myc/bcl-2−/− (n = 26) reconstituted mice were monitored daily for general health, LN enlargement, and splenomegaly. To serve as tumor-free controls, lethally irradiated mice were also reconstituted with wt, bcl-2+/−, or bcl-2−/− fetal liver cells, and, as expected, none of these mice developed lymphoma over the course of 18 months. As previously observed,36,37 tumor latency is longer in mice reconstituted with Eμ-myc cells than in nonmanipulated Eμ-myc transgenic animals (compare data in Figure S4 with those in Figure 5 and Figure S5). Nevertheless, mice reconstituted with Eμ-myc fetal liver cells reproducibly developed lymphoma with a median onset of 49 weeks and 70% mortality by 18 months (Figure 5). Remarkably, loss of both alleles of bcl-2 did not retard Eμ-myc–induced lymphoma development, yielding a median tumor latency of 46 weeks (Figure 5). Statistical analysis revealed no significant difference in tumor latency or overall mortality between these cohorts of animals (Figure S5).
Figure 5.

Loss of endogenous Bcl-2 does not delay Eμ-myc–induced B lymphoma development. Kaplan-Meier analysis of tumor latency in lethally irradiated wt (Ly5.1+) mice reconstituted with fetal liver cells from Ly5.2+ Eμ-myc (■) and Eμ-myc/bcl-2−/− (□) (E14.5) embryos. Mice were killed when deemed moribund by an animal technician who was blinded to the genotype of the mice. Cause of death was classified as Eμ-myc lymphoma according to the criteria detailed in “Materials and methods.”
The predominant immunophenotype of the lymphomas arising in the Eμ-myc/bcl-2−/− reconstituted animals was that of pro/pre-B (B220+ CD19+ sIg−), and to a lesser extent, of immature B (B220+ CD19+ sIgM+ sIgD−)–cell origin (Figure 6A-B; Table S1). Although moribund Eμ-myc/bcl-2−/− mice frequently presented with smaller LNs than lymphomatous Eμ-myc and Eμ-myc/bcl-2+/− mice, no overall reduction in severity of lymphoma was conferred by loss of Bcl-2, as evidenced by comparable spleen size (Eμ-myc = 0.32 ± 0.1 g vs Eμ-myc/bcl-2−/− = 0.28 ± 0.05 g) and similarly elevated leukocyte cellularity in the blood (Eμ-myc = 46 ± 14 × 106/mL vs Eμ-myc/bcl-2−/− = 39 ± 21 × 106/mL; Figure 7A). In addition, histologic analysis revealed similar pathology in moribund Eμ-myc/bcl-2−/− and Eμ-myc mice, including extensive disruption of the normal architecture of LNs and bone marrow by large lymphoma cells and frequent lymphoma infiltration of the liver, lung, and kidney (Figure 7B; data not shown). Moreover, transplantation tests established that all Eμ-myc/bcl-2−/− (n = 8) and control Eμ-myc (n = 5) lymphomas tested were transplantable, producing lymphoma and leukemia in (nonirradiated) histocompatible recipients within 30 days.
Figure 6.

Eμ-myc/bcl-2−/− reconstituted mice develop characteristic Eμ-myc lymphoma. (A) Representative immunophenotyping FACS plots of lymphomas arising in Eμ-myc and Eμ-myc/bcl-2−/− mice. Single-cell suspensions were prepared from fresh tumor samples and stained with monoclonal antibodies to Ly5.2, Ly5.1, B220, CD19, IgM, IgD, CD4, CD8, Gr-1, Mac-1, and Ter119 antibodies. All lymphomas were confirmed as donor-derived by expression of Ly5.2 antigen. (B) The overall proportion of pro/pre-B and immature B lymphomas arising in moribund Eμ-myc and Eμ-myc/bcl-2−/− reconstituted mice.
Figure 7.

Loss of endogenous Bcl-2 does not reduce the severity of Eμ-myc lymphoma. (A) Comparison at time of death of spleen weights (g) from moribund Eμ-myc (n = 8), Eμ-myc/bcl-2−/− (n = 10), and normal control (wt) reconstituted mice (n = 4), and of peripheral leukocyte cellularity (× 106 cells/mL) from moribund Eμ-myc (n = 8), Eμ-myc/bcl-2−/− (n = 9), and normal control (wt) (n = 9) reconstituted mice. Bars indicate the mean values. (B) Hematoxylin/eosin staining of representative LN (×40), bone marrow (×20), and liver (×20) sections taken from terminally ill Eμ-myc and Eμ-myc/bcl-2−/− reconstituted animals. Images were visualized using an Olympus BX50 microscope (Olympus, Tokyo, Japan) equipped with a 40×/0.85 NA or a 20×/0.5 NA objective. Images were photographed with an Olympus U-HAD3 digital camera and recorded using Axiovision 4.2 software (Carl Zeiss, Thornwood, NY). Images were processed with Adobe Photoshop CS2 software, version 9.0 (Adobe Systems, San Jose, CA).
To begin to explore whether loss of endogenous Bcl-2 had selected for development of lymphomas expressing abnormal levels of anti- or proapoptotic members of the Bcl-2 family or other apoptosis regulators, we subjected lysates from 3 Eμ-myc/bcl-2−/− and 3 control Eμ-myc lymphomas to Western blot analysis. The expression levels of Bcl-xL and Mcl-1 varied among the lymphomas tested, but there was no obvious correlation with the presence or absence of endogenous Bcl-2. The proapoptotic BH3-only proteins Bim, Puma, Bad, and Bid were expressed at comparable levels in all lymphomas, as was the multi-BH domain proapoptotic Bcl-2 family member Bax, as well as Apaf-1, and caspase-1, caspase-2, caspase-6, caspase-8, and caspase-9 (Figure S6). Curiously, caspase-3 was not detectable in all the tumors analyzed (Figure S6), but there was no obvious difference between the Eμ-myc and Eμ-myc/bcl-2−/− lymphomas, since 1 in 3 of each appeared to lack caspase-3 expression. These results suggest that the Eμ-myc/bcl-2−/− lymphomas appear not to have selected for any consistent abnormality in expression levels of pro- or antiapoptotic Bcl-2 family members or downstream effectors of apoptosis.
Discussion
Despite current efforts to develop Bcl-2 antagonists for antitumor therapy.22 the contribution of endogenous bcl-2 to lymphomagenesis has not previously been investigated. Since Bcl-2 is expressed at multiple stages of B-cell development,23,24 and inhibition of apoptosis is considered an essential requirement for tumorigenesis,4,38 we reasoned that loss of endogenous bcl-2 might impair Eμ-myc–induced B lymphomagenesis. To explore this issue, we circumvented the early postnatal lethality caused by germline Bcl-2 deficiency26,29 by studying lymphoma development in wt mice reconstituted with Eμ-myc/bcl-2−/− or Eμ-myc/bcl-2+/+ (Eμ-myc) fetal liver stem cells. This system has proven very effective for revealing how genetic changes can accelerate or delay Eμ-myc–induced lymphomagenesis.36,37 Moreover, the protracted tumor-free period in these animals allowed us to determine the consequences of Bcl-2 loss on nontransformed Eμ-myc–expressing B lymphoid cells.
Mice reconstituted with wt or bcl-2−/− fetal liver cells had similar numbers of pro-B cells and pre-B cells in their bone marrow, and these cells died at comparable rates in culture. Similarly, premalignant Eμ-myc/bcl-2−/− and control Eμ-myc mice had comparable numbers of pro-B and pre-B cells that survived equally well in culture. Bcl-2 is probably not essential for the survival of these cells because pre-B cells normally express only low levels of Bcl-2.23,24 This indicates that other prosurvival Bcl-2 family members, such as Bcl-xL, which is highly expressed in pre-B lymphocytes,39 are essential for inhibiting myc-induced apoptosis in these cells.40 Moreover, since pro-B cells express relatively high levels of Bcl-2,23,24 their survival may be guaranteed by the redundant action of Bcl-2 and at least one of its homologs.
Mice deficient for bcl-2 have been reported to display high levels of spontaneous lymphocyte apoptosis in vivo but, surprisingly, cell culture experiments indicated that bcl-2−/− splenic B cells died at a rate comparable with their wt counterparts.30 Those studies, however, were carried out with total splenic B220+ cells, most of which in a bcl-2−/− reconstituted mouse are of immature (sIgMhi sIgDlo) B-cell origin. When we analyzed the immature (B220+ sIgMhi sIgDlo) and mature (B220+ sIgMlo sIgDhi) B cells separately, we found that loss of Bcl-2 accelerated the death of the mature B cells in culture and reduced their number in vivo much more than that of immature B cells. These observations, which held both for control (nontransgenic) B cells and those expressing the Eμ-myc transgene, are consistent with the finding that mature B cells express high levels of Bcl-2, whereas immature B cells express only low levels.23 Pertinently, experiments with cells from Bcl-x–deficient embryos have indicated that Bcl-xL is the prosurvival Bcl-2 family member critical for blocking apoptosis in immature sIgMhi sIgDlo B cells.41
The reduced repopulation potential we observed with Eμ-myc/bcl-2−/− fetal liver–derived stem cells, like that noted previously for Bcl-2–deficient cells,30 probably reflects the prevalence of Bcl-2 expression in hematopoietic stem and progenitor cells.23,24 Nevertheless, despite the reduced reconstitution efficacy and the marked mature B-cell deficit in preleukemic Eμ-myc/bcl-2−/− mice, their progression to malignancy occurred with an onset and incidence similar to those observed in Eμ-myc reconstituted control animals. The comparable lymphoma onset kinetics, taken together with the acceleration of Eμ-myc/bcl-2−/− mature B-cell apoptosis both in vivo and in vitro, demonstrate that the survival of the more mature B cells is not essential for Eμ-myc–induced lymphomagenesis or sustained lymphoma growth.
These findings support the notion that a more immature cell type—one for which Bcl-2 is dispensable for survival—is the likely origin of early neoplastic clones in Eμ-myc transgenic mice. An analogous observation has been made with preleukemic Eμ-myc/Eμ-max41 bitransgenic mice, which have less than 1% of the normal number of circulating peripheral blood B lymphocytes, yet develop lymphoma at a rate comparable with that of Eμ-myc control mice.42 Conversely, Eμ-myc/Eμ–bcl-2 bitransgenic animals exhibit prodigious numbers of cycling mature (sIg+) B lymphoid cells, but all the tumors that arise in these animals have a very primitive (“stem cell”–like) phenotype, and the more mature bitransgenic B cells do not elicit tumors on transplantation.13 Taken together, these observations suggest that even though many Eμ-myc lymphomas express surface immunoglobulin, indicative of a mature phenotype, lymphomagenesis most likely initiates in a primitive lymphoid progenitor or stem cell. It may be relevant that Burkitt lymphomas, although they have a very different cellular origin (germinal-center B cells) contain very little Bcl-2.43 However, whether Bcl-2 was present during their development remains unknown.
Our finding that loss of endogenous Bcl-2 accelerated apoptosis of preneoplastic mature Eμ-myc B cells, together with the demonstration that removing Bcl-2 decimated the tumors arising in Eμ-myc/Eμ–bcl-2 mice,21 supports the concept that inactivating the function (or expression) of Bcl-2 and closely related prosurvival proteins will prove an important new approach to therapy.44 Indeed, in preclinical studies, the compound ABT-737, which inactivates several Bcl-2 family members, has shown considerable promise with chronic lymphocytic leukemia as well as certain other tumors.22 Our finding that endogenous Bcl-2 is not required for the sustained growth of Eμ-myc lymphomas that have not arisen as a consequence of Bcl-2 overexpression indicates that targeting Bcl-2 specifically might not be effective for this class of tumor. Targeting several of the Bcl-2 homologs (eg, using ABT-737) may provide a more efficacious approach. Eventually, however, we surmise that antagonizing a critical individual prosurvival Bcl-2 family member, such as Bcl-xL, offers the best prospect for selective killing of certain types of tumor cells while minimizing damage to normal tissues. Moreover, when the inherent ability of myc overexpression to sensitize cells to apoptotic stimuli is better understood, that may also provide an attractive adjunct strategy for targeted cancer therapy.
Supplementary Material
Acknowledgments
We are grateful to the late Dr A. Harris, and Drs S. Cory, P. Bouillet, L. Coultas, M. Bath, V. Marsden, S. Nutt, C. Scott, and A. Wiegmans and L. Lee for scientific advice and assistance. We thank Drs A. Harris and S. Cory for Eμ-myc transgenic mice and Drs D. Loh and P. Bouillet for bcl-2+/− mice; K. Vella, G. Siciliano, A. Naughton, K. Pioch, N. Iannarella, and J. Allen for expert animal care; B. Helbert and M. Robati for genotyping; Dr F. Battye, V. Milovac, C. Tarlinton, C. Young, and J. Garbe for cell sorting; J. Corbin for automated blood analysis; Dr S. Mihajlovic, E. Tsui, and A. Hasanein for histology; and T. Nikolaou, D. Quilici, G. Thomas, S. Kwok, and D. Baum for radiation.
Supported by fellowships and grants from the National Health and Medical Research Council (Australia; program no. 257502), the Leukemia and Lymphoma Society (New York; SCOR grant no. 7015) and the National Cancer Institute (National Institutes of Health; CA 80188 and CA 43540).
Footnotes
The online version of this manuscript contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Author contributions: P.N.K. performed experiments and contributed to experimental design, data interpretation, and manuscript writing; H.P. contributed to some experiments and assisted with interpretation; J.M.A. contributed to experimental design and manuscript writing; and A.S. conceived the study, designed experiments, and contributed to writing of the manuscript.
Conflict-of-interest statement: The authors declare no competing financial interests.
Correspondence: Andreas Strasser, The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, 3050 Victoria, Australia; e-mail: strasser@wehi.edu.au.
References
- 1.Prendergast GC. Mechanisms of apoptosis by c-Myc. Oncogene. 1999;18:2967–2987. doi: 10.1038/sj.onc.1202727. [DOI] [PubMed] [Google Scholar]
- 2.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 3.Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Ann Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 4.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 5.Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM. Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science. 1984;224:1403–1406. doi: 10.1126/science.6610211. [DOI] [PubMed] [Google Scholar]
- 6.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 7.Adams JM, Harris AW, Pinkert CA, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 8.Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in Eμ-myc transgenic mice. Cell. 1986;47:11–18. doi: 10.1016/0092-8674(86)90361-2. [DOI] [PubMed] [Google Scholar]
- 9.Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Eμ-myc transgenic mouse: a model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ, Lowe SW. Suppression of tumorigenesis by the p53 target PUMA. Proc Natl Acad Sci U S A. 2004;101:9333–9338. doi: 10.1073/pnas.0403286101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 14.Swanson PJ, Kuslak SL, Fang W, et al. Fatal acute lymphoblastic leukemia in mice transgenic for B cell-restricted bcl-xL and c-myc. J Immunol. 2004;172:6684–6691. doi: 10.4049/jimmunol.172.11.6684. [DOI] [PubMed] [Google Scholar]
- 15.Eischen CM, Roussel MF, Korsmeyer SJ, Cleveland JL. Bax loss impairs Myc-induced apoptosis and circumvents the selection of p53 mutations during Myc-mediated lymphomagenesis. Mol Cell Biol. 2001;21:7653–7662. doi: 10.1128/MCB.21.22.7653-7662.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonnell TJ, Deane N, Platt FM, et al. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 17.Strasser A, Whittingham S, Vaux DL, et al. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A. 1991;88:8661–8665. doi: 10.1073/pnas.88.19.8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strasser A, Harris AW, Cory S. Eμ-bcl-2 transgene facilitates spontaneous transformation of early pre-B and immunoglobulin-secreting cells but not T cells. Oncogene. 1993;8:1–9. [PubMed] [Google Scholar]
- 19.Yunis JJ, Mayer MG, Arnesen MA, Aeppli DP, Oken MM, Frizzera G. bcl-2 and other genomic alterations in the prognosis of large-cell lymphoma. N Engl J Med. 1989;320:1047–1054. doi: 10.1056/NEJM198904203201605. [DOI] [PubMed] [Google Scholar]
- 20.Pegoraro L, Palumbo A, Erikson J, et al. A 14;18 and an 8;14 chromosome translocation in a cell line derived from an acute B-cell leukemia. Proc Natl Acad Sci U S A. 1984;81:7166–7170. doi: 10.1073/pnas.81.22.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–249. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 22.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 23.Li Y-S, Hayakawa K, Hardy RR. The regulated expression of B lineage associated genes during B cell differentiation in bone marrow and fetal liver. J Exp Med. 1993;178:951–960. doi: 10.1084/jem.178.3.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merino R, Ding L, Veis DJ, Korsmeyer SJ, Nuñez G. Developmental regulation of the Bcl-2 protein and susceptibility to cell death in B lymphocytes. EMBO J. 1994;13:683–691. doi: 10.1002/j.1460-2075.1994.tb06307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strasser A, Harris AW, Vaux DL, et al. Abnormalities of the immune system induced by dysregulated bcl-2 expression in transgenic mice. Curr Top Microbiol Immunol. 1990;166:175–181. doi: 10.1007/978-3-642-75889-8_22. [DOI] [PubMed] [Google Scholar]
- 26.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 27.Nakayama K, Nakayama K-I, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of bcl-2αβ in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci U S A. 1994;91:3700–3704. doi: 10.1073/pnas.91.9.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamada S, Shimono A, Shinto Y, et al. bcl-2 deficiency in mice leads to pleiotropic abnormalities: accelerated lymphoid cell death in thymus and spleen, polycystic kidney, hair hypopigmentation, and distorted small intestine. Cancer Res. 1995;55:354–359. [PubMed] [Google Scholar]
- 29.Bouillet P, Cory S, Zhang L-C, Strasser A, Adams JM. Degenerative disorders caused by Bcl-2 deficiency are prevented by loss of its BH3-only antagonist Bim. Dev Cell. 2001;1:645–653. doi: 10.1016/s1534-5807(01)00083-1. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama K-i, Nakayama K, Izumi N, et al. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science. 1993;261:1584–1588. doi: 10.1126/science.8372353. [DOI] [PubMed] [Google Scholar]
- 31.Evan GI, Wyllie AH, Gilbert CS, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 32.Strasser A, Elefanty AG, Harris AW, Cory S. Progenitor tumours from Eμ-bcl-2–myc transgenic mice have lymphomyeloid differentiation potential and reveal developmental differences in cell survival. EMBO J. 1996;15:3823–3834. [PMC free article] [PubMed] [Google Scholar]
- 33.O'Reilly LA, Huang DCS, Strasser A. The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 1996;15:6979–6990. [PMC free article] [PubMed] [Google Scholar]
- 34.Mazel S, Burtrum D, Petrie HT. Regulation of cell division cycle progression by bcl-2 expression: a potential mechanism for inhibition of programmed cell death. J Exp Med. 1996;183:2219–2226. doi: 10.1084/jem.183.5.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linette GP, Li Y, Roth K, Korsmeyer SJ. Cross talk between cell death and cell cycle progression: BCL-2 regulates NFAT-mediated activation. Proc Natl Acad Sci U S A. 1996;93:9545–9552. doi: 10.1073/pnas.93.18.9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott CL, Schuler M, Marsden VS, et al. Apaf-1 and caspase-9 do not act as tumor suppressors in myc-induced lymphomagenesis or mouse embryo fibroblast transformation. J Cell Biol. 2004;164:89–96. doi: 10.1083/jcb.200310041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell. 2002;1:289–298. doi: 10.1016/s1535-6108(02)00047-8. [DOI] [PubMed] [Google Scholar]
- 38.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 39.Grillot DAM, Merino R, Pena JC, et al. bcl-x exhibits regulated expression during B cell development and activation and modulates lymphocyte survival in transgenic mice. J Exp Med. 1996;183:381–391. doi: 10.1084/jem.183.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eischen CM, Woo D, Roussel MF, Cleveland JL. Apoptosis triggered by myc-induced suppression of Bcl-XL or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol. 2001;21:5063–5070. doi: 10.1128/MCB.21.15.5063-5070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Motoyama N, Wang FP, Roth KA, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 42.Lindeman GJ, Adams JM, Cory S, Harris AW. B-lymphoid to granulocytic switch during hematopoiesis in a transgenic mouse strain. Immunity. 1994;1:517–527. doi: 10.1016/1074-7613(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 43.Ngan BY, Chen Levy Z, Weiss LM, Warnke RA, Cleary ML. Expression in non-Hodgkin's lymphoma of the bcl-2 protein associated with the t(14;18) chromosomal translocation. N Engl J Med. 1988;318:1638–1644. doi: 10.1056/NEJM198806233182502. [DOI] [PubMed] [Google Scholar]
- 44.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–885. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}