

Abstract
RasGRP1 is a guanine nucleotide exchange factor for Ras and a receptor of the second messenger diacylglycerol and its ultrapotent analogs, the phorbol esters. We have recently shown expression of RasGRP1 in the epidermal keratinocytes, where it can mediate Ras activation in response to the phorbol ester 12-O-tetradecanoylphorbol-13-acetate, a well-known mouse skin tumor promoter. To explore the participation of RasGRP1 in skin carcinogenesis, we targeted the overexpression of RasGRP1 to basal epidermal keratinocytes using the keratin 5 promoter. These transgenic mice were viable and indistinguishable from their littermates, with normal differentiation and skin architecture. However, a percentage of the adult transgenic population developed spontaneous skin tumors, mainly squamous cell papillomas. The transgene was detected in the tumors as well as in primary keratinocytes isolated from transgenic mice. The transgenic keratinocytes also displayed elevated levels of active, GTP-loaded Ras compared to the levels observed in keratinocytes derived from wild type littermates. We noticed a correlation between tumor incidence and wounding, which suggests that RasGRP1 overexpression may confer sensitivity to promotional stimuli like wound repair mechanisms. Interestingly, we also found elevated levels of G-CSF in conditioned media derived from transgenic keratinocytes subjected to in vitro wounding. Taken together, these data are the first to provide evidence of a novel role for RasGRP1 in skin carcinogenesis, and suggest that RasGRP1 may participate in tumorigenesis through modulation of Ras and autocrine pathways.
Keywords: RasGRP, skin, carcinogenesis, diacylglycerol, transgenic
INTRODUCTION
The two-stage carcinogenesis protocols have been instrumental in our understanding of the multistep nature of epithelial carcinogenesis. In particular, the chemically induced skin carcinogenesis protocol in the mouse has served to identify cellular alterations that participate in the initiation, promotion and tumor progression steps (1). The typical protocol in the mouse skin carcinogenesis model consists of a single application of the carcinogen 7,12-dimethylbenz[a]anthracene (DMBA) followed by repetitive topical treatment with the tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA). The carcinogen introduces an activating point mutation in the Ha-ras proto-oncogene (2), and the initiated keratinocyte population expands under the effects of TPA. The mechanism of tumor promotion by TPA has begun to be elucidated by elegant studies using transgenic and knockout mice for PKC, which is the major intracellular target for TPA in the skin (3–7). However, the link between Ras, TPA and tumor promotion and progression is still not fully understood.
We have recently shown expression of the novel phorbol ester receptor RasGRP1 in epidermal keratinocytes (8), which are the target cells in the two-stage skin carcinogenesis model. Our initial studies suggested that RasGRP1 is a new link between TPA and Ras activation in the epidermal cells and, potentially, could mediate some of the tumor promoting effects of the phorbol esters in skin. To address the role of RasGRP1 in skin carcinogenesis, we developed a transgenic mouse model for overexpression of RasGRP1 in the epidermis. We found that while the transgenic mice were indistinguishable from their wild type littermates, a percentage of the adult transgenic population developed spontaneous skin tumors. We also noticed a correlation between tumors incidence and wounding and found elevated levels of the cytokine G-CSF in conditioned media from primary transgenic keratinocytes subjected to in vitro wounding, which suggests the participation of an autocrine mechanism in the transgenic susceptibility to tumor development. We propose that RasGRP1 represents a novel target in epidermal carcinogenesis.
MATERIAL AND METHODS
Generation of the K5.RasGRP1 Transgenic Mice
The full-length cDNA of rat RasGRP1 tagged with the HA epitope was cloned into a plasmid cassette containing the regulatory region of the bovine keratin 5 (K5) gene (Figure 1A) (9). The transgene was microinjected into FVB/N mouse embryos according to standard procedures at the University of Michigan Transgenic Animal Core (Ann Arbor, MI). Founder mice were identified by PCR of tail DNA (see Supplementary Data 1) and confirmed by Southern blot analysis. The experiments described in here were carried out on the F2 generation. Mice were maintained at the University of Hawaii Animal Facility.
Figure 1.
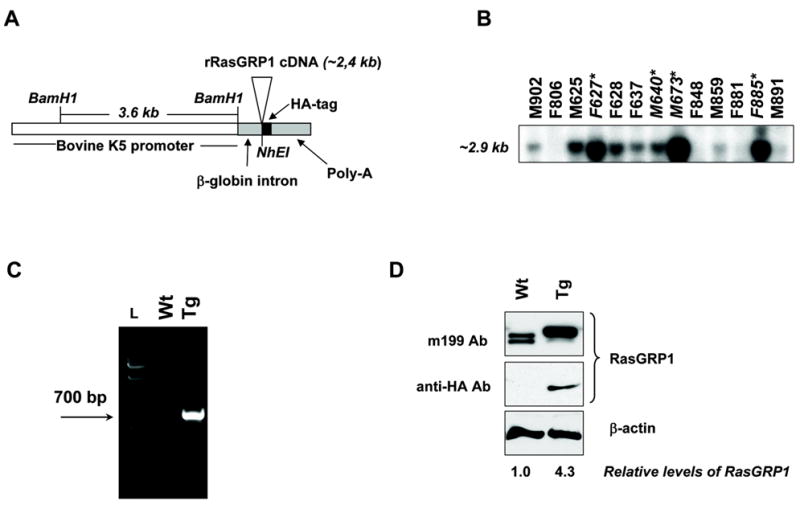
K5.RasGRP1 DNA transgenic construct and transgenic protein expression in mouse epidermal keratinocytes. A, Transgenic cassette. B, Southern blot analysis of NhEI/BamH1 digested tail genomic DNA of founder mice, using a 700-bp specific probe for transgenic RasGRP-HA DNA. Asterisks denoted founders propagated for further analysis. C, Genotyping of wild type (Wt) and transgenic (Tg) mice by PCR of tail DNA using a set of specific primers for the RasGRP1-HA transgene, as described in Materials and Methods. L: DNA ladder. D, RasGRP1 expression in primary keratinocytes isolated from wild type (Wt) and transgenic (Tg) newborn skin. Immunoblots were performed using antibodies against RasGRP1 (m199 Ab) and the HA tag (anti-HA Ab). Transgenic cells expressed approximately 4–5 times higher levels of RasGRP1 than the wild type counterparts, as calculated by densitometry of immunoblots stained with the anti-RasGRP1 antibody and normalized for loading differences using β-actin. Results are representative of six independent experiments.
Immunoblot analysis
Expression of the RasGRP1 transgene was measured by immunoblotting in lysates from primary keratinocytes isolated from newborn mice as previously described (10; see Supplementary Data 2). Cells were harvested in cell lysis buffer containing 25 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 5 mmol/L MagCl2, 1% Igepal, 5% glycerol, and supplemented with Mini Complete Roche Protease Inhibitor Cocktail Tablets (Roche Applied Biosciences, Indianapolis, IN). Samples containing 50 μg of total cell lysate protein were resolved on 8% acrylamide gels and blotted onto nitrocellulose membranes for immunostaining using antibodies against RasGRP1 or the HA probe (m199 and Y-11, respectively, Santa Cruz Biotechnology, Santa Cruz, CA) and β–actin (Sigma-Aldrich, St. Louis, MO).
Histopathology and Immunohistochemistry
Skin tumors and uninvolved sections of dorsal skin were fixed in 10% neutral formalin for 24 h and maintained in 70% ethanol until paraffin-embedded. Hematoxylin & Eosin stained slides were used for descriptive histopathology (IDEXX Veterinary Services, Westbrook, ME; RADIL, University of Missouri, Columbia, MO). For immunohistochemistry, the deparaffinized sections were subjected to heat-induced epitope retrieval using a 2100-Retriever in Buffer A (PickCell Laboratories, Amsterdam, the Netherlands) according to the manufacture’s instructions. Sections were blocked for 1 h at room temperature in TBS-0.5 % Triton X-100 containing 12% normal donkey serum and 1% IgG-free bovine serum albumin (Jackson ImmunoResearch, West Grove, PA). Primary antibodies were diluted in TBS-0.5 % Triton X-100 containing 2% normal donkey serum and 1% IgG-free bovine serum albumin, and added to the slides for 2 h at room temperature in a humidified chamber. Antibodies against keratin 1 (K1) and K5 (Covance, Denver, PA) were used at a 1/500 dilution; anti-HA (Santa Cruz Biotechnology, Santa Cruz, CA) was used at a 1/250 dilution. Normal rabbit IgG (Upstate, Lake Placid, NY) served as negative control. Endogenous peroxidase was blocked after the primary antibody incubation using Peroxidase-Blocking Reagent (Dako, Carpinteria, CA) for 5 min at room temperature. An HRP-conjugated AffinityPure donkey anti-rabbit F(ab')2 fragment specific antibody (Jackson ImmunoResearch, West Grove, PA) was diluted 1/500 in similar diluent as used for the primary antibodies and added to the tissues for 2 h at room temperature, followed by DAB as substrate (Dako, Carpinteria, CA). Tissues were counterstained with Mayer’s Hematoxylin (Inn, San Ramon, CA).
Mouse Cytokine Antibody Array and Ras Activation Assay
Conditioned media were harvested from primary wild type or transgenic keratinocytes grown in basal media without serum or growth factors for 24 hours. Mouse Cytokine Antibody Array (Panomics, Fremont, CA) was incubated with 2 mL of conditioned media and processed according to the manufacturer’s instructions. For the in vitro wound assay, the monolayers of keratinocytes were scratched with a 1,000-μl pipette tip along the diameter of the culture dishes 24 h before collecting the conditioned media. Levels of GTP-loaded Ras were determined using the Ras Activation Assay Biochem Kit on 24-h serum-starved cell cultures (Cytoskeleton Inc., Denver, CO).
RESULTS AND DISCUSSION
We have recently shown that the Ras exchange factor and phorbol ester receptor RasGRP1 is expressed in mouse primary keratinocytes, where it mediates Ras activation in response to the tumor promoting phorbol ester TPA (8, 10). To explore in more detail the role of RasGRP1 in the epidermis and in tumor promotion, we generated a transgenic mouse model in which overexpression of RasGRP1 was targeted to the basal layer of epidermal keratinocytes and the outer root sheath of hair follicle using the bovine K5 promoter (Figure 1A) (9). Thirteen founder mice (Figure 1B) were generated from a total offspring of 280 mice (4.6% efficiency), and four founder lines (female 627, male 640, male 673 and female 885) were further expanded. All founders, with the exception of female 885, bred successfully and the transgene transmission to the progeny followed a Mendelian pattern. Line 627 was breed for further analysis and maintained as heterozygous. Routine genotyping was performed by PCR of tail DNA with a primer set specific for the RasGRP1 transgene as described in Materials and Methods (Figure 1C).
The transgenic RasGRP1 protein was detected by immunoblotting in lysates from primary transgenic keratinocytes using a monoclonal antibody against rat RasGRP1 that also recognized mouse endogenous RasGRP1 in the wild type keratinocytes (Figure 1D). The transgenic protein run slightly slower than the endogenous protein. We have noticed similar mobility for RasGRP1 overexpressed in primary keratinocytes upon infection with an adenovirus encoding for an HA-tagged rat RasGRP1 (8). Transgenic RasGRP1 was also detected by immunoblotting using an anti-HA antibody (Figure 1D). β-actin was included as loading control. Keratinocytes derived from adult transgenic skin also expressed the RasGRP1 transgenic protein (data not shown).
The newborn transgenic mice did not show any obvious developmental abnormalities and were not phenotypically different from the wild type littermates. The skin of the K5.RasGRP1 transgenic mice was histologically indistinguishable from that of the wild type mice. They both displayed similar patterns of expression of K1 and K5 and there were no signs of hyperplasia or any defects in the skin architecture in the transgenic animals compared to the wild type mice (Figure 2A).
Figure 2.
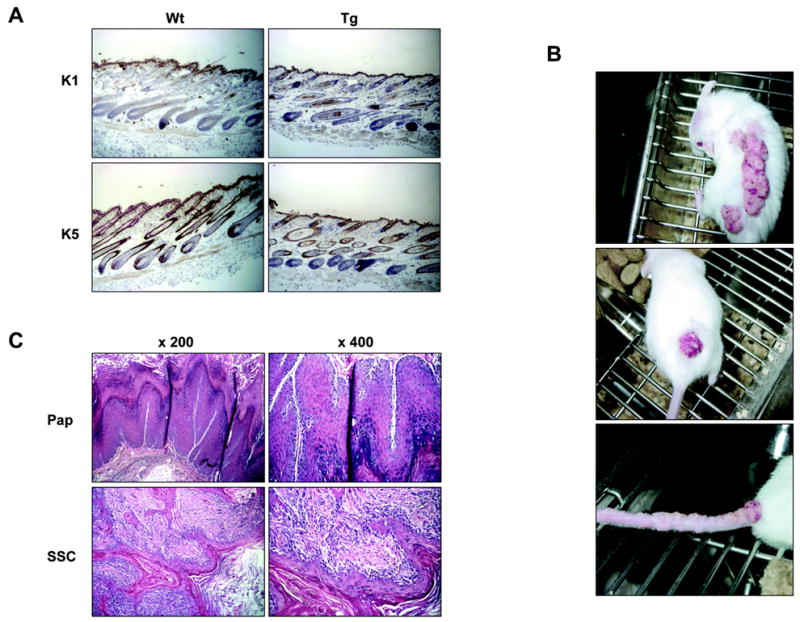
Analysis of the K5.RasGRP1 transgenic skin and spontaneous tumors. A, Expression of markers of differentiation K1 and K5 in wild type (Wt) and transgenic (Tg) skin. B, Representative photographs of spontaneous tumors in the transgenic mice. Note the presence of multiple tumors along the tail (lower panel). C, Hematoxylin & Eosin staining of tumor specimens from the K5-RasGRP1 mice. Pap, papilloma; SSC, squamous cell carcinoma. Results are representative of eleven tumors and are shown at two magnifications (x 200 and x 400).
Interesting, a percentage of the transgenic mice developed spontaneous tumors in the skin as the colony aged, with a higher frequency in mice housed in pairs or groups than in single-caged animals. To evaluate tumor incidence, we observed a group of 47 mice, 23 of which were K5.RasGRP1 transgenic animals at the F2 generation and the rest wild type mice. At 3 months old, a small number of the transgenic population showed wart-like skin tumors on the dorsal skin. Seven months later, eleven of the twenty-three (48 %) transgenic mice had developed spontaneous tumors, while none of the wild type counterparts showed any signs of tumor development or lesions on the skin (p< 0.0001, Fisher’s exact test). The spontaneous tumors arose in the dorsal skin, along the tail or in the base of the tail (Figure 2B). Some tumors were also observed at the tail tip. Eighty percent of the tumors were diagnosed histologically as squamous cell papillomas (Figure 2C, Pap). These papillomas were usually sessile and multifocal, with multiple arborizing cords. The dermis was usually infiltrated with a mixed inflammatory cell population. Twenty percent of the dorsal tumors were malignant, either intermediate squamous cell carcinomas or squamous cell carcinoma with spindle cell differentiation (Figure 2C, SSC). Two transgenic mice developed nodular growths in the ventral areas, next to the genitalia. One of the lesions could no be analyzed due to the premature death of the animal; the second one was diagnosed histologically as a ruptured infundibular cyst. It should be noted that some of the transgenic mice had to be euthanized before they reached 10 months old, due to the large tumor size (> 10 mm).
To evaluate if the tumors expressed the transgenic protein, we performed immunohistochemistry using an anti-HA rabbit polyclonal antibody. The exogenous protein was detected in the basal layer of the papillary projection of the tumor (Figure 3A, middle panels), with a distribution pattern similar to that of K5 (Figure 3A, right panels). Normal rabbit IgG (Figure 3A, left panels) was used as negative control in place of the primary antibody. Carcinomas also tested positive for the expression of the RasGRP1 transgenic protein (Figure 3B). Closer examination of the keratinocytes showed cytoplasmic and perinuclear localization of RasGRP1 in the cells (Figure 3C), a localization that matched the subcellular distribution of RasGRP1 in keratinocytes in culture (8, 10).
Figure 3.
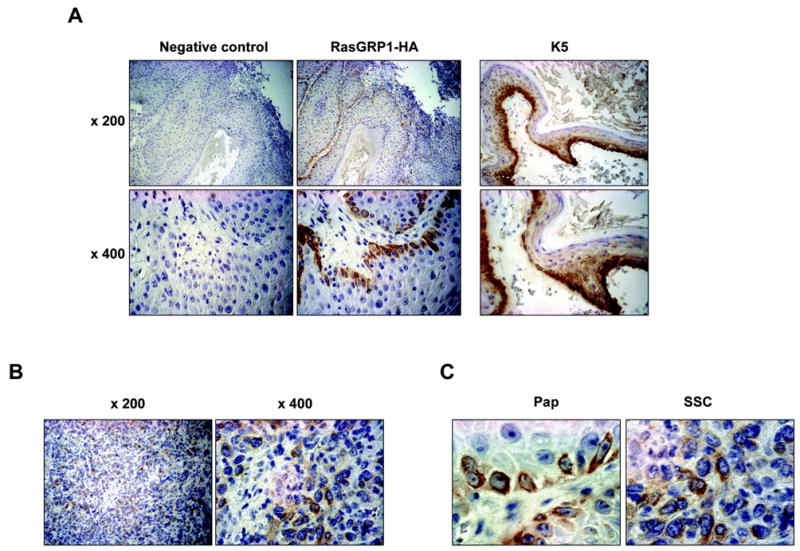
Expression of RasGRP1 in the K5.RasGRP1 transgenic skin and tumors. A, Immunohistochemistry of spontaneous papillomas using an anti-HA antibody to detect the transgenic RasGRP1-HA protein (brown staining). Normal rabbit IgG was used as negative control (left panels). Note the expression of the transgenic RasGRP1 in the basal keratinocytes of the papilloma (middle panels), which coincided with K5 distribution (right panels). Photographs were taken at two magnifications (x 200 and x 400). B, Expression of transgenic RasGRP1 in squamous cell carcinomas. Photographs were taken at two magnifications (x 200 and x 400). C, Photographs of papillomas (Pap) and carcinomas (SSC) at a higher magnification (x 630), showing cytoplasmic and perinuclear localization of RasGRP1.
Activation of the Ha-ras proto-oncogene by specific point mutations is frequently associated with skin tumors in the mice and is the initiation event in the two-stage chemically induced mouse skin carcinogenesis model (2). One of the most frequent mutations is a transversion mutation of an A to T in codon 61 of Ha-ras. To evaluate whether the spontaneous tumors originated in the K5.RasGRP1 transgenic mice carried that mutation, we utilized a PCR approach developed by Nelson et al. (11), based on the specific amplification of mutant Ha-Ras. From a sample of seven tumors, none of them tested positive for the presence of the mutant gene, whereas wild type Ha-ras was readily amplified (Figure 4A). Control mouse genomic DNA (Promega, Madison, WI) and DNA from papilloma cells carrying a A–T mutation in codon 61 of Ha-ras were used as negative and positive controls, respectively (data not shown). Other potential mutations in Ha-ras were also investigated using PCR-based amplification followed by sequencing. No other ras activating mutations were found in the spontaneous tumors (data not shown).
Figure 4.
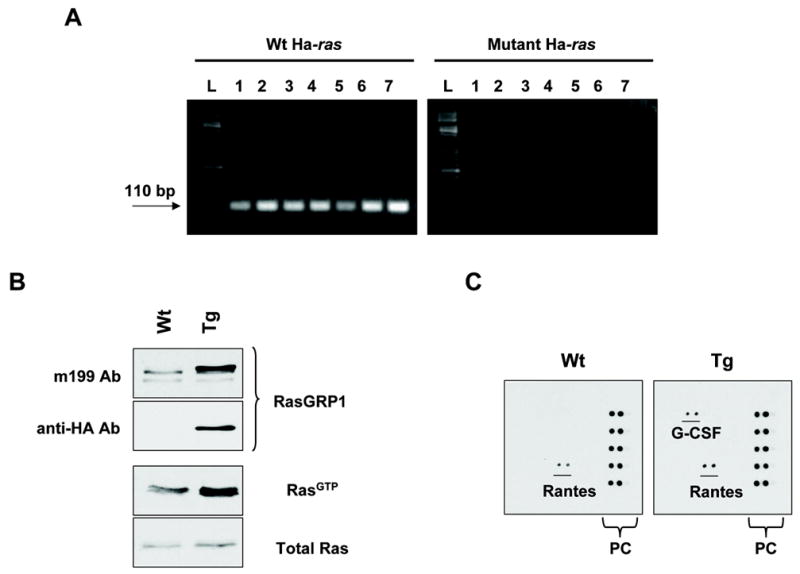
Ras activation status and cytokine profile in K5.RasGRP1 transgenic mice. A, Mutations in codon 61 of the Ha-ras proto-oncogene were evaluated by a PCR approach as described in Materials and Methods, using genomic DNA isolated from tumor samples from seven transgenic mice (L: DNA ladder; tumor samples: 1 to 7). Amplification of wild type Ha-ras was included for comparison. B, Levels of GTP-loaded, active Ras in primary keratinocytes isolated from newborn skin from K5-RasGRP1 transgenic mice (Tg) and their wild type littermates (Wt). Expression of the RasGRP1 transgene was confirmed by immunoblotting using antibodies against RasGRP1 (m199 Ab) and the HA tag (anti-HA Ab). Ras activation status (level of RasGTP) was determined as described in Materials and Methods. C, Profile of cytokine expression in primary keratinocytes from K5-RasGRP1 transgenic mice and their wild type littermates. A mouse cytokine antibody array was probed with serum-deprived conditioned media obtained from wild type (Wt) or transgenic (Tg) keratinocytes grown for 24 hours after in vitro wounding. PC: array positive controls. Results are representative of three independent experiments.
We next assessed the activation status of Ras in primary keratinocytes isolated from transgenic and wild type skin. Under serum-starved condition, the levels of active, GTP-loaded Ras were at least 2 times higher in the transgenic keratinocytes compared to the wild type counterparts (Figure 4B). It is tempting to speculate that in the same way that activating mutations in the ras proto-oncogenes initiate cells in the skin, the elevated levels of active Ras driven by RasGRP1 overexpression could confer an initiation status to the transgenic keratinocytes.
The incidence of spontaneous tumors in the transgenic mice was higher in animals housed in pairs or groups than in single-housed mice. Additionally, there was a positive correlation between tumor incidence and wounding sites in the mice. This somehow resembled the response of the v-Ha-ras Tg.AC mouse, which develops spontaneous dorsal skin tumors upon wounding or treatment with the tumor promoter TPA (12, 13). The potential sensitivity of the K5.RasGRP1 transgenic mice to wounding as a promotional stimulus lead us to investigate the profile of cytokines secreted by the transgenic keratinocytes, as some of the cytokines released during the inflammatory events associated to wounding are believed to contribute to tumor development and progression in a susceptible microenvironment. In response to in vitro wounding (scratch wound assay), levels of G-CSF were significantly elevated in conditioned media from transgenic keratinocytes compared with the wild type group (Figure 4C). G-CSF, and the related cytokine GM-CSF, can promote keratinocyte growth and angiogenesis, and they have been associated with tumor growth and progression in human skin cancer models (14, 15). In mice, a transgenic model for overexpression of PKCε in the epidermis that is susceptible to the formation of metastatic squamous cell carcinomas, is highly sensitive to induction of specific cytokines -among them G-CSF- in response to the tumor promoter TPA and UV radiation (16). Taken together, this evidence strongly implicates G-CSF as one of the cytokines involved in skin carcinogenesis. In the context of our study, one could argue that overexpression of RasGRP1 in the epidermis causes an aberrant response to wound repair mechanisms, leading to keratinocyte secretion of G-CSF that could participate in tumorigenesis. The mechanisms involved in the increased production of this cytokine by the K5.RasGRP1 transgenic keratinocytes remain to be elucidated. Further studies using a full-thickness incisional wound model as well as tumor promotion stimuli like TPA will help to delineate the events involved in the effects of RasGRP1 in the epidermis and will contribute to our understanding of the mechanisms of carcinogenesis in the skin.
Acknowledgments
We thank José Luis Jorcano for providing the bovine K5 cassette (CIEMAT, Spain) and Thomas Saunders and his team (Transgenic Animal Model Core, University of Michigan) for the transgenic mouse production.
Footnotes
Grant Support: NCI grant 1R01 CA096841 (P. S. Lorenzo)
References
- 1.Hennings H, Glick AB, Greenhalgh DA, et al. Critical aspects of initiation, promotion, and progression in multistage epidermal carcinogenesis. Proc Soc Exp Biol Med. 1993;202:1–8. doi: 10.3181/00379727-202-43511a. [DOI] [PubMed] [Google Scholar]
- 2.Quintanilla M, Brown K, Ramsden M, Balmain A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature. 1986;322:78–80. doi: 10.1038/322078a0. [DOI] [PubMed] [Google Scholar]
- 3.Wang HQ, Smart RC. Overexpression of Protein Kinase Cα in the epidermis of transgenic mice results in striking alterations in phorbol ester-induced inflammation and COX-2, MIP-2 and TNF-α expression but not tumor promotion. J Cell Sci. 1999;112:3497–506. doi: 10.1242/jcs.112.20.3497. [DOI] [PubMed] [Google Scholar]
- 4.Reddig PJ, Dreckschimdt NE, Ahrens H, et al. Transgenic mice overexpressing Protein Kinase Cδ in the epidermis are resistant to skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1999;59:5710–8. [PubMed] [Google Scholar]
- 5.Reddig PJ, Dreckschmidt NE, Zou J, Bourguignon SE, Oberley TD, Verma AK. Transgenic mice overexpressing Protein Kinase Cε in their epidermis exhibit reduced papilloma burden but enhanced carcinoma formation after tumor promotion. Cancer Res. 2000;60:595–602. [PubMed] [Google Scholar]
- 6.Hara T, Saito Y, Hirai T, et al. Deficiency of Protein Kinase Cα in mice results in impairment of epidermal hyperplasia and enhancement of tumor formation in two-stage skin carcinogenesis. Cancer Res. 2005;65:7356–62. doi: 10.1158/0008-5472.CAN-04-4241. [DOI] [PubMed] [Google Scholar]
- 7.Cataisson C, Joseloff E, Murillas R, et al. Activation of cutaneous Protein Kinase Cα induces keratinocyte apoptosis and intraepidermal inflammation by independent signaling pathways. J Immunol. 2003;171:2703–13. doi: 10.4049/jimmunol.171.5.2703. [DOI] [PubMed] [Google Scholar]
- 8.Rambaratsingh RA, Stone JC, Blumberg PM, Lorenzo PS. RasGRP1 represents a novel non-Protein Kinase C phorbol ester signaling pathway in mouse epidermal keratinocytes. J Biol Chem. 2003;278:52792–801. doi: 10.1074/jbc.M308240200. [DOI] [PubMed] [Google Scholar]
- 9.Ramirez A, Bravo A, Jorcano JL, Vidal M. Sequences 5' of the bovine keratin 5 gene direct tissue- and cell-type-specific expression of a lacz gene in the adult and during development. Differentiation. 1994;58:53–64. doi: 10.1046/j.1432-0436.1994.5810053.x. [DOI] [PubMed] [Google Scholar]
- 10.Tuthill MC, Oki CE, Lorenzo PS. Differential effects of bryostatin 1 and 12-O-tetradecanoylphorbol-13-acetate on the regulation and activation of RasGRP1 in mouse epidermal keratinocytes. Mol Cancer Ther. 2006;5:602–10. doi: 10.1158/1535-7163.MCT-05-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson MA, Futscher BW, Kinsella T, Wymer J, Bowden GT. Detection of mutant Ha-ras genes in chemically initiated mouse skin epidermis before the development of benign tumors. Proc Natl Acad Sci U S A. 1992;89:6398–402. doi: 10.1073/pnas.89.14.6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leder A, Kuo A, Cardiff RD, Sinn E, Leder P. v-Ha-ras transgene abrogates the initiation step in mouse skin tumorigenesis: effects of phorbol esters and retinoic acid. Proc Natl Acad Sci U S A. 1990;87:9178–82. doi: 10.1073/pnas.87.23.9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spalding JW, Momma J, Elwell MR, Tennant RW. Chemically induced skin carcinogenesis in a transgenic mouse line (Tg. Ac) carrying a v-Ha-ras gene. Carcinogenesis. 1993;14:1335–41. doi: 10.1093/carcin/14.7.1335. [DOI] [PubMed] [Google Scholar]
- 14.Mueller MM, Fusenig NE. Tumor-stroma interactions directing phenotype and progression of epithelial skin tumor cells. Differentiation. 2002;70:486–97. doi: 10.1046/j.1432-0436.2002.700903.x. [DOI] [PubMed] [Google Scholar]
- 15.Mueller MM, Peter W, Mappes M, et al. Tumor progression of skin carcinoma cells in vivo promoted by clonal selection, mutagenesis, and autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor. Am J Pathol. 2001;159:1567–79. doi: 10.1016/S0002-9440(10)62541-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wheeler DL, Li Y, Verma AK. Protein Kinase Cε signals ultraviolet light-induced cutaneous damage and development of squamous cell carcinoma possibly through induction of specific cytokines in a paracrine mechanism. Photochem Photobiol. 2005;81:9–18. doi: 10.1562/2004-08-12-RA-271. [DOI] [PubMed] [Google Scholar]