

Abstract
The ubiquitously expressed protein glycogen synthase kinase-3 (GSK3) is constitutively active, however its activity is markedly diminished following phosphorylation of Ser21 of GSK3α and Ser9 of GSK3β. Although several kinases are known to phosphorylate Ser21/9 of GSK3, for example Akt, relatively much less is known about the mechanisms that cause the dephosphorylation of GSK3 at Ser21/9. In the present study KCl-induced plasma membrane depolarization of SH-SY5Y cells, which increases intracellular calcium concentrations caused a transient decrease in the phosphorylation of Akt at Thr308 and Ser473, and GSK3 at Ser21/9. Overexpression of the selective protein phosphatase-1 inhibitor protein, inhibitor-2, increased basal GSK3 phosphorylation at Ser21/9 and significantly blocked the KCl-induced dephosphorylation of GSK3β, but not GSK3α. The phosphorylation of Akt was not affected by the overexpression of inhibitor-2. GSK3 activity is known to affect sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) levels. Overexpression of inhibitor-2 or treatment of cells with the GSK3 inhibitors lithium and SB216763 increased the levels of SERCA2. These results indicate that the protein phosphatase-1/inhibitor-2 complex differentially regulates GSK3 dephosphorylation induced by KCl and that GSK3 activity regulates SERCA2 levels.
Keywords: Akt, glycogen synthase kinase-3, inhibitor-2, protein phosphatase-1, sarcoplasmic/endoplasmic reticulum calcium ATPase
Introduction
Glycogen synthase kinase-3 (GSK3) was originally identified as the phosphorylating kinase for glycogen synthase [12, 44]. However it became clearly apparent that GSK3 was much more complex as cumulative evidence showed that GSK3 takes part in numerous signaling pathways and is regulated by multiple mechanisms. The diversity of substrates for GSK3α and GSK3β spans a wide range of proteins from transcription factors, to cell cycle proteins, to metabolic enzymes. Such variation in substrates underscores the involvement of GSK3 in many cellular processes. Considering its widespread actions, alterations in the control of GSK3 activity have been associated with several diseases including Alzheimer’s disease [24], bipolar disorder [26], schizophrenia [11], and non neurological disorders such as cancer [19, 29], heart disease [18], and diabetes [25]. The regulation of GSK3 is intricate, as GSK3 activity is controlled by phosphorylation, protein-protein interactions, and intracellular localization [15]. GSK3 is constitutively active, but its activity can be inhibited by phosphorylation on Ser21 of GSK3α and Ser9 of GSK3β. Several kinases can phosphorylate Ser21/9 of GSK3, including Akt (also called protein kinase B) [6]. Akt is activated by the phosphatidylinositol 3-kinase (PI3K) signaling pathway. Stimulation of the PI3K pathway by growth factors, certain types of stressors [9], and potassium-induced membrane depolarization [33, 8] results in the phosphorylation of Akt at its Thr308 and Ser473 sites. Subsequently, activated Akt phosphorylates Ser21/9 of GSK3, inhibiting GSK3 activity.
In opposition to the manifold signaling pathways that inhibit GSK3 activity are the lesser known mechanisms that activate GSK3 by Ser21/9 dephosphorylation. The regulation of Ser21/9 phosphorylation has been attributed to the actions of protein phosphatase-2A (PP2A) [40, 43, 27, 38] and protein phosphatase-1 (PP1) [46, 34, 41]. The PP1 holoenzyme is comprised of a 37 kDa catalytic subunit which associates with inhibitory subunits and targeting subunits. One such inhibitory subunit, Inhibitor-2 (I-2), binds to PP1 to form a stable PP1/I-2 complex [35]. Unphosphorylated I-2 inhibits PP1 activity [21] and phosphorylation of I-2 at its Thr72 site by GSK3 restores PP1 activity [10, 20]. The phosphorylation of I-2 by GSK3 is well documented; however, relatively little is known about how the PP1/I-2 complex impacts GSK3 phosphorylation. The regulation of phosphatases that contribute to the activation of GSK3 is particularly important, given that the ultimate consequence of increased GSK3β activity can lead to decreased neuronal plasticity.
Calcium is a critical intracellular messenger that triggers the activation of a host of signaling proteins and engages numerous biochemical and cellular processes. Thus, the result of abnormal increases in intracellular calcium concentrations [Ca2+] can result in the dysregulation of signaling circuits and neuronal injury, all of which have been linked to the pathophysiology of neurological disorders such as stroke and Alzheimer’s disease [30]. Therefore, the regulation of the storage of calcium in the endoplasmic reticulum (ER) by the sarcoplasmic/endoplasmic reticulum Ca2+-ATPases (SERCAs) also is important to prevent intracellular [Ca2+] from reaching levels that are toxic to cells. SERCAs, which are encoded by three homologous genes (SERCA1, SERCA2, and SERCA3) [3], undergo alternative splicing in different tissues. SERCA2a and b are expressed in the brain, in skeletal and cardiac muscle as well as in all non-muscle tissues [45]. Interestingly, treatments that cause increases in intracellular [Ca2+] can also increase the activity of GSK3β through the dephosphorylation of its inhibitory Ser9 site [38, 27, 41]. Active GSK3β can in turn regulate the storage of calcium in the ER. Previously, it was shown that overexpression of GSK3β in mouse hearts impaired the re-uptake of calcium into the ER [32]. This was likely due to the regulation of SERCA2a by GSK3β since the expressed constitutively active GSK3β reduced the expression of SERCA2a in mouse hearts. Conversely, inhibition of GSK3β with lithium increased the expression of SERCA2a [32]. In this study, we show that mobilization of intracellular calcium through membrane depolarization caused a transient decrease in the phosphorylation of Akt and GSK3. However, the decrease in the phosphorylation of GSK3 occurred differentially through the activation of PP1. Inhibition of PP1 through I-2 overexpression increased the basal phosphorylation of GSK3α and GSK3β, but only GSK3β dephosphorylation induced by KCl treatment was blocked by I-2 overexpression. Moreover, inhibition of GSK3 through the PP1/I-2 complex increased SERCA2 levels.
Materials and methods
Cell culture and treatments
Human neuroblastoma SH-SY5Y cells were grown in RPMI medium (Cellgro, Herndon, Virginia, USA) supplemented with 10% horse serum (Invitrogen, Carlsbad, California, USA), 5% fetal clone II (Hyclone, Logan, Utah, USA), 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Experimental agents used were 100 mM KCl, 20 mM LiCl (Sigma, St. Louis, Missouri, USA), and 5 μM SB216763 (Tocris Cookson Inc., Ballwin, Missouri, USA). Cells, with the exception of control cells (time 0), were treated with 100 mM KCl for 5, 10, 15, and 30 min as described previously [27].
Intracellular Calcium Levels
Intracellular calcium levels were measured in cultured SH-SY5Y cells using Fura-2AM as described previously [17]. Briefly, cells were grown on coverslips, and prior to KCl treatment cells were loaded with 5 μM Fura-2AM (TEFLABS, Austin, TX). Coverslips were placed in an imaging chamber (Warner Instrument Co.) and mounted in a heater platform on the stage of a Nikon Diaphot. The cells were maintained at 37°C in a Ringer’s solution for the duration of the experiments. Images were obtained using an Ionoptix ICCD camera (Ionoptix Corp., Milton, MA) and processed with the IonWizard software program. Relative intracellular calcium levels were determined from the ratio of fluorescence using excitation wavelengths of 340 and 380 nm. Background fluorescence was taken from regions not containing cells and subtracted from the cell images at each wavelength. The ratio of fluorescence in the digitized images is a direct indicator of relative intracellular calcium levels. The system was calibrated as previously described [16].
Plasmid preparation and cell transfection
To construct the wt-I-2-myc/HIS vector, a human brain cDNA library was used as a template for RT-PCR of I-2 (Superscript One-Step RT-PCR (Invitrogen)). The primers utilized were: 5’ CCGGTACCATGGCGGCCTCGACGG 3’, forward, and 5’ GGGCTCGAGTGAACTTCGTAATTTGTTTTGCTGTTGGTCACTTGGAGTA 3’, reverse. The PCR product was digested with Kpn 1 and Xho1 and ligated into corresponding sites in a pCDNA3.1myc/HIS vector (Invitrogen). Plasmid integrity was evaluated through DNA sequencing analysis. SH-SY5Y cells were transiently transfected in 35 mm dishes using a RPMI media supplemented with 1% horse serum (Gibco-Invitrogen). After achieving the desired confluency, cells were transfected with 2 μg of pCDNA3.1myc/HIS vector or with wt-I-2 plasmid using Fugene6 (Roche Diagnostics Corporation) according to manufacturer’s instructions. To increase transfection efficiency, plates were spun at 2500 rpm for 30 min immediately following transfection. Cells were subjected to treatments and harvested 24 hours post-transfection.
Immunoblotting
Cells were washed twice with phosphate-buffered saline and lysed in IP lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1 mM sodium orthovanadate, 100 μM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin, 50 mM NaF, 1 nM okadaic acid, and 0.5% NP-40). The lysates were centrifuged at 20,817 x g for 10 min at 4°C and supernatants were collected. Protein concentrations were determined using the bicinchoninic method (Pierce, Rockford, IL). Cell lysates were mixed with Laemmli sample buffer (2% SDS) and placed in a boiling water for 5 min. Proteins (10 μg) were resolved in 7.5% SDS-polyacrylamide gels, and transferred to nitrocellulose. Blots were probed with antibodies to phospho-Ser473-Akt, phospho-Thr308-Akt, total Akt, phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, total GSK3β (Cell Signaling Technology, Beverly, MA), total GSK3α (Southern Biotech, Birmingham, AL), I-2 (BD Transduction Laboratories, Lexington, KY), myc (UAB peptide synthesis core facility), PP1 (Santa Cruz Biotechnologies, Santa Cruz, CA), SERCA2 (Affinity BioReagents, Golden, CO), and β-tubulin (Sigma, St. Louis, MO). The tau antibodies used in this study were Tau5 and 5A6 (Tau5 from Dr. L. Binder), which are phospho-independent tau antibodies [4, 23] and PHF-1 (from Dr. P. Davies), which recognizes tau phosphorylated at Ser-396/404 [36]. Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG (Bio-Rad Laboraories, Hercules, CA), followed by detection with enhanced chemiluminescence.
Immunoprecipitation
To immunoprecipitate myc-tagged I-2 the myc (4 μg) antibody was incubated with 200 μg protein from cell lysate overnight at 4°C. Protein G sepharose (Amersham) beads were washed three times with IP lysis buffer and incubated with antibodies and cell lysate for 3 h at 4°C. The immobilized immune complex was washed three times with IP lysis buffer, mixed with 50 μl 2X Laemmli sample buffer, and placed in a boiling waterbath for 5 min. The proteins were separated in a 7.5% SDS-polyacrylamide gel, transferred to nitrocellulose, and the membrane was incubated with the I-2 or PP1 antibodies. The immunoblot was developed using horseradish peroxidase-conjugated goat anti-mouse heavy chain IgG followed by detection with enhanced chemiluminescence.
Protein Phosphatase-1 Assay
A fluorometric assay was used to measure PP1 activity. The ProFluor™ Ser/Thr Phosphatase Assay kit (Promega, Madison, WI) was used to measure the activity of PP1 in cell lysates. Briefly, cells were harvested in lysis buffer (10 mM Tris, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.05% NP-40, and 1mM EGTA, 50 mM NaF, 100 μM Na3VO4, 200 μM leupeptin, 40 μg/ml aprotinin, and 2 mM phenylmethylsulfonyl fluoride) and protein concentrations were determined using the bicinchoninic method (Pierce, Rockford, IL). Cell lysates (50 μg) were mixed with a 1X phosphatase inhibitor cocktail (100 mM NaF, 5 μM okadaic acid, 100 μM Na3VO4, and 5 mM EGTA) and substrate/ATP mix (10 μM phosphorylated bisamide rhodamine-110 peptide substrate, 10 μM AMC substrate, 100 μM ATP, 20 μM cAMP, 40 mM Tris pH7.5, 0.1% bovine serum albumin) and incubated in a 96-well plate for 20 min at room temperature. Subsequently, a protease reagent (1 mU/μl of a proprietary protease provide with the kit, 40 mM Tris pH 8.0, 10 mM EDTA, 100 mM EGTA, 0.1% bovine serum albumin) was mixed with the cell lysates and incubated for 30 min at room temperature. PP1 activity was determined using a fluorescent plate reader set at wavelengths 485/527 nm to measure the fluorescence of the rhodamine-110 peptide substrate. Thus the decrease in fluorescence intensity is correlated with decreased PP1 activity and is standardized to total protein concentrations in the samples.
Statistical analysis
All statistical significance was analyzed using ANOVA.
Results and discussion
The first goal of the study was to test the effects of intracellular calcium mobilization on the phosphorylation state of Akt and its downstream substrate GSK3. SH-SY5Y cells were treated with 100 mM KCl to induce membrane depolarization. Intracellular calcium measurements showed that 100 mM KCl treatment causes a rapid and transient increase in intracellular calcium levels (Fig 1A), confirming KCl-induced membrane depolarization. Intracellular calcium levels reached maximum within 30 sec of KCl treatment, and then decreased to 32% of maximum levels after 1 min of KCl treatment and remained stable at this level for 5 min post KCl treatment to 30 min post KCl treatment, data not shown. Treatment of SH-SY5Y cells with KCl also rapidly and transiently decreased the phosphorylation of Akt at Ser473, although the decrease in Akt Ser473 phosphorylation was delayed relative to the increase in intracellular calcium levels. Decreased Akt Ser473 phosphorylation was evident beginning at 2.5 min with maximum dephosphorylation occurring by 5 min (60% that of control) following treatment (Fig. 1B). The Akt dephosphorylation immediately followed the rapid transient increase in intracellular calcium levels. This rapid Akt dephosphorylation was followed by a near return to basal phosphorylation levels between 10 min and 30 min. The magnitude of dephosphorylation of Akt at Thr308 (58% that of control) was similar to that of Ser473, but it occurred within 2.5 min followed by an increase in phosphorylation between 5 min and 30 min (Fig. 1B). Effects that closely paralleled Akt dephosphorylation were observed in the phosphorylation states of both isoforms of GSK3 following KCl treatment. Treatment with KCl caused a transient decrease in the phosphorylation of GSK3α beginning at 2.5 min with maximum dephosphorylation occurring at 5 min (67% that of control) (Fig. 2). A similar trend in phosphorylation was seen with the GSK3β isoform. The phosphorylation of GSK3β decreased transiently with maximum dephosphorylation occurring at 5 min (54% that of control) following KCl treatment (Fig. 2).
Fig. 1.
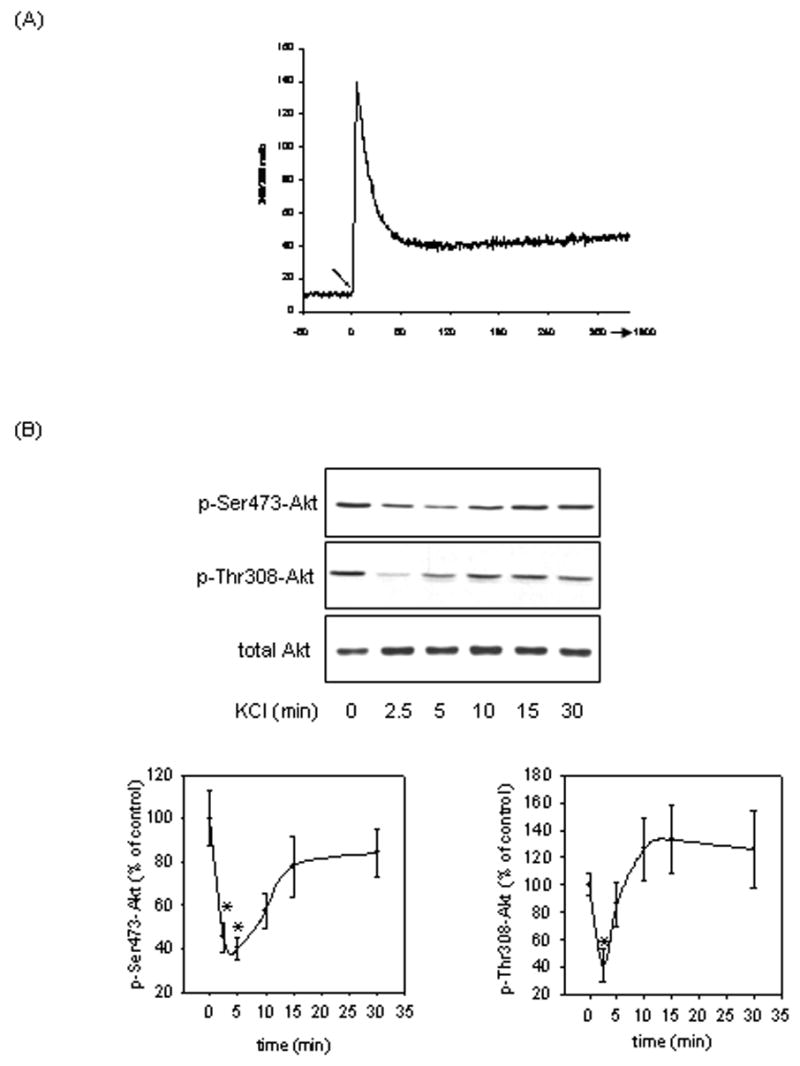
KCl treatment transiently increased intracellular calcium levels and decreased the phosphorylation of Akt. (A) Intracellular calcium levels were measured in cultured SH-SY5Y cells as described in “Materials and methods”. KCl (100 mM) was added to the cell culture at time 0, indicated by the arrow. Image analysis was conducted for 30 min post KCl treatment, although the graph is truncated at approximately 5 min post KCl treatment. (B) SH-SY5Y cells maintained in serum-containing media were treated with 100 mM KCl for 2.5, 5, 10, 15, or 30 min, and whole-cell lysates were immunoblotted for p-Ser473-Akt, p-Thr308-Akt, and total Akt. Means ± SE, n=3 experiments; p*<0.05 (ANOVA) compared to values from untreated cells.
Fig. 2.
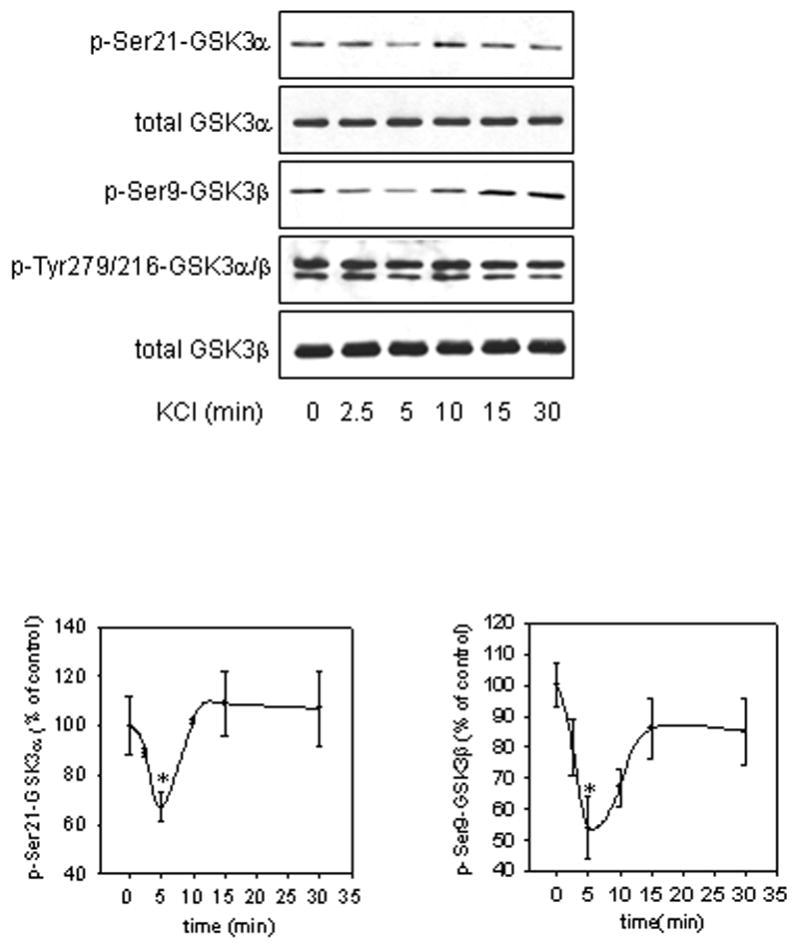
KCl treatment transiently decreased the phosphorylation of GSK3. Cells were treated with 100 mM KCl for 2.5, 5, 10, 15, or 30 min, and cell lysates were immunoblotted for p-Ser21-GSK3α, p-Ser9-GSK3β, p-Tyr279/216-GSK3α/β, total GSK3α, and total GSK3β. Means ± SE, n=3 experiments; p*<0.05 (ANOVA) compared to values from untreated cells.
Next, we examined the contribution of PP1 to the dephosphorylation of GSK3 following KCl treatment. Cells overexpressing I-2 and vector-transfected cells were subjected to KCl treatment, and the phosphorylation of GSK3 was analyzed. Firstly, confirmation of I-2 overexpression and association with PP1 were determined by immunoprecipitating cell lysates with the anti-myc antibody and immunoblotting the samples with the I-2 or PP1 antibodies (Fig. 3A). Then PP1 activity, as well as GSK3α Ser21 and GSK3β Ser9 phosphorylations were measured in control and I-2 overexpressing cells. I-2 overexpression significantly inhibited the activity of PP1 by 50% compared to that in vector-transfected cells (Fig. 3A). There were significant increases in the phosphorylation of GSK3α at Ser21 (799% that of control) and GSK3β at Ser9 (161% that of control) in the I-2-overexpressing cells compared to vector-transfected cells (Fig. 3A). Treatment of vector-transfected cells with KCl decreased the phosphorylation of GSK3α and GSK3β by 5 min (Fig. 3B). However, I-2 overexpression markedly blocked the KCl-induced dephosphorylation of GSK3β (60% that of KCl-treated cells) but only slightly blocked dephosphorylation of GSK3α. Neither the basal phosphorylation nor the KCl-induced dephosphorylation of Akt at Thr308 was affected by I-2 overexpression (Fig. 3C). These results indicate that PP1 differentially contributes to the KCl-mediated dephosphorylation of GSK3 but does not mediate the inactivation of Akt signaling.
Fig. 3.
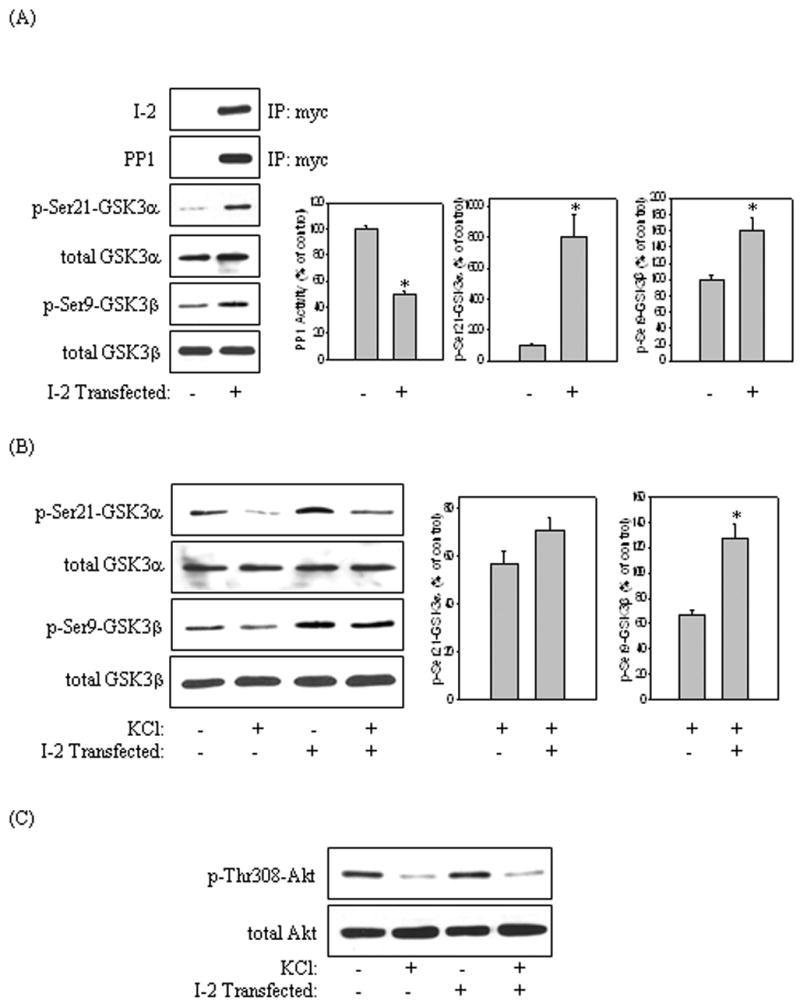
Inhibition of PP1 by I-2 differentially blocked KCl-induced dephosphorylation of GSK3. I-2 was overexpressed in SH-SY5Y cells. (A) Cell lysates from I-2-transfected and vector-transfected cells were immunoprecipitated as described in “Materials and methods” using the myc antibody and were immunoblotted for I-2 and PP1. Cell lysates also were immunoblotted for p-Ser21-GSK3α, p-Ser9-GSK3β, total GSK3α, and total GSK3β. PP1 activity was measured as described in “Materials and methods”. Quantification is based on the ratio of phospho-protein levels to total protein levels. Means ± SE, n=3 experiments; p*<0.05 (ANOVA) compared to values from vector-transfected cells. (B) I-2-transfected cells and vector-transfected cells were treated with 100 mM KCl for 5 min. Cell lysates were immunoblotted for p-Ser21-GSK3α, p-Ser9-GSK3β, total GSK3α, and total GSK3β. Means ± SE, n=3 experiments; p*<0.05 (ANOVA) compared to values from vector-transfected cells. (C) Cell lysates were immunoblotted for p-Thr308-Akt, and total Akt.
Previously, both GSK3β [32] and PP1 [5] were shown to regulate the expression and activity of SERCA2, respectively. Therefore, we tested if the inhibition of GSK3 pharmacologically or through the inhibition of PP1 with I-2 would affect SERCA2 levels in SH-SY5Y cells. Treatment of cells with 20 mM lithium, which inhibits GSK3 activity [26, 39] increased SERCA2 levels by 235% compared to control cells by 4 h following treatment (Fig. 4A). The levels of SERCA2 also increased 216% compared to control cells by 6 h following treatment with 5 μM SB216763, a specific GSK3 inhibitor [7] (Fig. 4B). To confirm that GSK3 was inhibited by both inhibitors, the phosphorylation of tau was examined. Tau, a GSK3 substrate, is known to be phosphorylated by GSK3 at its paired helical filament (PHF-1) region (Ser-396/404) [28]. Treatment with lithium (Fig. 4A) or SB216763 (Fig. 4B) resulted in a time-dependent decrease in PHF-1 immunoreactivity, thus indicating that both lithium and SB216763 inhibited GSK3 activity. Interestingly, overexpression of I-2 also increased SERCA2 levels 198% compared to vector-transfected cells (Fig. 5). These results suggest that inhibition of GSK3 pharmacologically or via the PP1/I-2 complex can boost SERCA2 levels in cells.
Fig. 4.
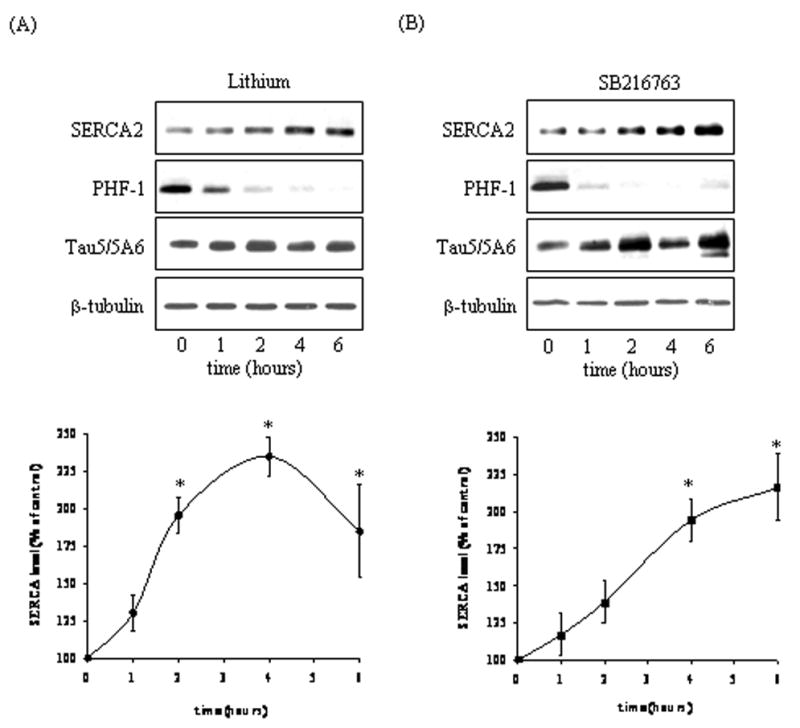
GSK3 inhibitors increased SERCA2 levels. Cells in serum-free media were treated with (A) 20 mM lithium or (B) 5 μM SB216763 for 1, 2, 4, or 6 h and cell lysates were immunoblotted for SERCA2, PHF-1, Tau5/5A6, and β-tubulin. Means ± SE, n=3 experiments; p<0.05 (ANOVA) compared to values from untreated cells.
Fig. 5.
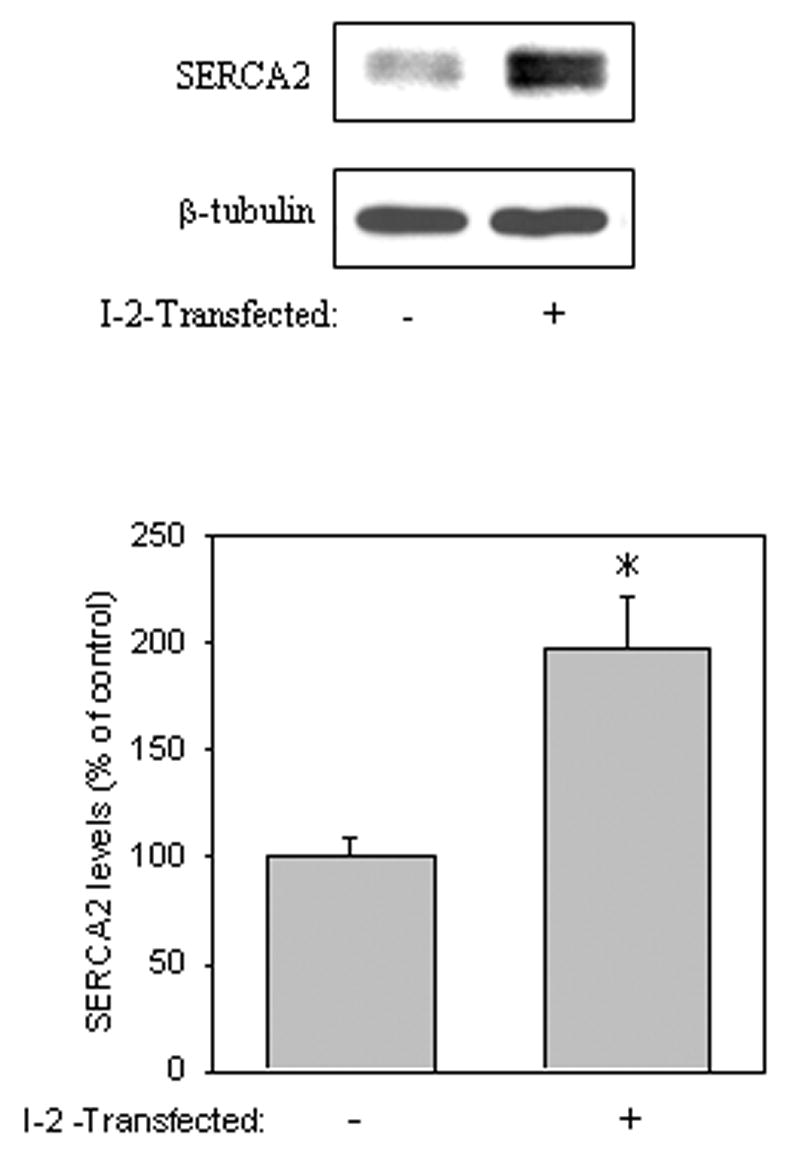
I-2 overexpression increased SERCA2 levels. Cells were transfected with vector or I-2 and incubated at 37°C for 24 h prior to harvesting, and cell lysates were immunoblotted for SERCA2. Equal protein loading was verified by blotting for β-tubulin. Means ± SE, n=3 experiments; p<0.05 (ANOVA) compared to values from untreated cells.
The regulation of GSK3 is multi-tiered and intricate. While much is known about the upstream signaling proteins, such as the PI3K/Akt signaling pathway, which leads to the inhibition of GSK3 activity, much less is known about signaling systems that result in the dephosphorylation of GSK3 at Ser21/9. This is puzzling given that the dephosphorylation of GSK3 at Ser21/9 and the resulting increased GSK3 activity can affect numerous critical cellular functions and can mitigate the ability of cells to survive under adverse conditions. Much of the GSK3-directed phosphatase activity has been linked to the actions of PP2A and PP2B [40, 43, 27, 38]. The present study shows that the PP1/I-2 complex also contributes to the regulation of GSK3 phosphorylation. Furthermore, the present study shows the close involvement of GSK3 in calcium signaling as it was found that GSK3 regulates SERCA2 levels in cells, which is mediated, in part, by the actions of the PP1/I-2 complex on GSK3 phosphorylation activity.
The transient flux of ions across the plasma membrane is a fundamental aspect of signal propagation in neurons. KCl-induced plasma membrane depolarization causes a rapid influx of calcium into cells. In addition, potassium depolarization of excitable cells activates PI3K [33] which results in the phosphorylation and activation of Akt [8]. It is widely accepted that PP2A dephosphorylates Akt [1, 31]. Indeed, our results show that expression of I-2 did not increase the basal phosphorylation of Akt compared to vector-transfected cells nor did it block the dephosphorylation of Akt following KCl treatment.
Recently, it was reported that treatment of SH-SY5Y cells with KCl resulted in the undulating phosphorylation of GSK3β at Ser9 [27]. In the present study transient dephosphorylation of GSK3β, was found to be dependent on the PP1/I-2 complex. The overexpression of I-2, which forms a stable complex with PP1 and inhibits PP1 activity [35], increased the basal phosphorylation of GSK3 at Ser21/9 and markedly blocked the KCl-induced dephosphorylation of GSK3β at Ser9, but only slightly blocked dephosphorylation of GSK3α at Ser21. These findings are interesting because they suggest that under basal conditions, PP1 dephosphorylates both isoforms of GSK3; however, following KCl-induced membrane depolarization, PP1 selectively dephosphorylates GSK3β. The finding that I-2 expression failed to block KCl-induced GSK3α Ser21 dephosphorylation was intriguing given that overexpression of I-2 increased basal phosphorylation of both GSK3α and GSK3β isoforms.
There is previous evidence which shows that GSK3 is the target of the PP1/I-2 complex. Initially, Zhang et al [46] reported the existence of an autoregulatory loop whereby GSK3 regulates its own phosphorylation at Ser21/9. In this case, the phosphorylation and inhibition of I-2 by GSK3 results in increased PP1 activity and decreased Ser21/9 phosphorylation of GSK3. Szatmari et al [41] reported that NMDA treatment of cortical and hippocampal neurons caused the dephosphorylation of Ser9 on GSK3β via the activation of PP1 [41] which was blocked by lithium treatment. Furthermore, they reported that inhibition of GSK3β by lithium reduced I-2 phosphorylation and increased GSK3β Ser9 phosphorylation. Lee et al [27] reported that the KCl-induced dephosphorylation of GSK3β in SH-SY5Y cells could be blocked by the PP1/PP2A inhibitor okadaic acid and the PP2B inhibitor cyclosporine A [27]. However, in that study, 30 nM okadaic acid was used, which is well above the IC50 values of okadaic acid for PP1 (10 nM) and PP2 (0.1 nM) [34], thus making okadaic acid a broadly-specific phosphatase inhibitor. Therefore, it is likely that PP1 also is involved in dephosphorylating GSK3β following KCl treatment.
Lee et al [27] also reported that GSK3β and PP2A form a complex, and this complex formation changed in parallel with the phosphorylation level of GSK3β. This indicates that the interaction of PP2A and GSK3β is highly dynamic. In the present study the differential effects of I-2-mediated inhibition of PP1 on GSK3α and GSK3β dephosphorylation following KCl treatment could potentially be explained by a redistribution of GSK3-PP complexes. For instance, under basal conditions GSK3α may be primarily complexed with PP1. Thus I-2-mediated inhibition of PP1 greatly increases GSK3α Ser21 phosphorylation, as is shown in Fig 3A. During membrane depolarization, PP1 complexed to GSK3α may be displaced by another phosphatase, in which case the blockade of PP1 activity by I-2 has little or no effect on GSK3α dephosphorylation. By comparison, under basal conditions GSK3β is complexed to PP2A and possibly to PP1 also. Therefore inhibition of PP1 only modestly increases basal GSK3β Ser9 phosphorylation compared to GSK3α Ser21 phosphorylation. Membrane depolarization could increase the association of PP1 and GSK3β, such that inhibition of PP1 blocks GSK3β Ser9 dephosphorylation. This could partly explain the differential effects of KCl-induced GSK3α and GSK3β dephosphorylation, but this mechanism will have to be tested in future studies.
Activation of calcium signaling triggers a host of signaling cascades including the activation of both phosphatases and kinases. Within this framework, it has been reported previously that GSK3β is dephosphorylated on its Ser9 site following thapsigargin treatment [38], which inhibits SERCA2 activity [42]. SERCA2 is an important regulator of calcium homeostasis and was previously shown to regulate diastolic [Ca2+] in the mouse heart [2]. Moreover, diastolic function in the mouse heart is adversely affected as a result of the downregulation of SERCA2a expression by GSK3β [32]. Our study showed that SERCA2 levels increased following the pharmacological inhibition of GSK3 with lithium and SB216763. Inhibition of GSK3 through the PP1/I-2 complex also increased SERCA2 levels. Overall, this study shows that the PP1/I-2 complex differentially regulates the phosphorylation of GSK3α and GSK3β which downstream modulates SERCA2 levels.
Acknowledgments
We wish to thank Dr. Richard S. Jope for helpful suggestions during the course of this study and Abigail Rountree for her excellent technical assistance. This research was supported by grants from The National Alliance for Research in Schizophrenia and Depression Young Investigator Award to GNB, and from the National Institutes of Health grant NS044853.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andjelkovic M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci U S A. 1996;93:5699–5704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 3.Burk SE, Lytton J, MacLennan DH, Shull GE. cDNA cloning, functional expression, and mRNA tissue distribution of a third organellar Ca2+ pump. J Biol Chem. 1989;264:18561–18568. [PubMed] [Google Scholar]
- 4.Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- 5.Ceulemans H, Bollen M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev. 2004;84:1–39. doi: 10.1152/physrev.00013.2003. [DOI] [PubMed] [Google Scholar]
- 6.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 7.Cross DA, Culbert AA, Chalmers KA, Facci L, Skaper SD, Reith AD. Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurons from death. J Neurochem. 2001;77:94–102. doi: 10.1046/j.1471-4159.2001.t01-1-00251.x. [DOI] [PubMed] [Google Scholar]
- 8.Crowder RJ, Freeman RS. The survival of sympathetic neurons promoted by potassium depolarization, but not by cyclic AMP, requires phosphatidylinositol 3-kinase and Akt. J Neurochem. 1999;73:466–475. [PubMed] [Google Scholar]
- 9.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;12:3499–3511. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 10.DePaoli-Roach AA. Synergistic phosphorylation and activation of ATP-Mg-dependent phosphoprotein phosphatase by F A/GSK-3 and casein kinase II. J Biol Chem. 1984;259:12144–12152. [PubMed] [Google Scholar]
- 11.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 12.Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3: an emerging therapeutic target. Trends Mol Med. 1980;8:9660–0664. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- 13.Fang X, Yu SX, Lu Y, Bast RC, Jr, Woodgett JR, Mills GB. Phosphorylation and inactivation of glycogen synthase kinase-3 by protein kinase A. Proc Natl Acad Sci U S A. 2000;97:11960–11965. doi: 10.1073/pnas.220413597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulation of glycogen synthase kinase-3β by protein kinase C isotypes. J Biol Chem. 1992;267:16878–16882. [PubMed] [Google Scholar]
- 15.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase-3β in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 16.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 17.Hartigan JA, Johnson GV. Transient increases in intracellular calcium result in prolonged site-selective increases in Tau phosphorylation through a glycogen synthase kinase 3beta-dependent pathway. J Biol Chem. 1999;274:21395–21401. doi: 10.1074/jbc.274.30.21395. [DOI] [PubMed] [Google Scholar]
- 18.Hardt SE, Sadoshima J. Glycogen synthase kinase-3β: a novel regulator of cardiac hypertrophy and development. Circ Res. 2002;90:1055–1063. doi: 10.1161/01.res.0000018952.70505.f1. [DOI] [PubMed] [Google Scholar]
- 19.Hill MM, Hemmings BA. Inhibition of protein kinase B/Akt. implications for cancer therapy. Pharmacol Ther. 2002;93:243–251. doi: 10.1016/s0163-7258(02)00193-6. [DOI] [PubMed] [Google Scholar]
- 20.Holmes CF, Kuret J, Chisholm AA, Cohen P. Identification of the sites on rabbit skeletal muscle protein phosphatase inhibitor-2 phosphorylated by casein kinase-II. Biochim Biophys Acta. 1986;870:408–416. doi: 10.1016/0167-4838(86)90248-7. [DOI] [PubMed] [Google Scholar]
- 21.Huang FL, Glinsmann WH. Separation and characterization of two phosphorylase phosphatase inhibitors from rabbit skeletal muscle. Eur J Biochem. 1976;70:419–426. doi: 10.1111/j.1432-1033.1976.tb11032.x. [DOI] [PubMed] [Google Scholar]
- 22.Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993;12:803–808. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D. The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J Neurochem. 1997;68:430–433. doi: 10.1046/j.1471-4159.1997.68010430.x. [DOI] [PubMed] [Google Scholar]
- 24.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Kaidanovich O, Eldar-Finkelman H. The role of glycogen synthase kinase-3 in insulin resistance and Type 2 diabetes. Expert Opin Ther Targets. 2002;6:555–561. doi: 10.1517/14728222.6.5.555. [DOI] [PubMed] [Google Scholar]
- 26.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YI, Seo M, Kim Y, Kim SY, Kang UG, Kim YS, Juhnn YS. Membrane depolarization induces the undulating phosphorylation/dephosphorylation of glycogen synthase kinase-3β, and this dephosphorylation involves protein phosphatases 2A and 2B in SH-SY5Y human neuroblastoma cells. J Biol Chem. 2005;280:22044–22052. doi: 10.1074/jbc.M413987200. [DOI] [PubMed] [Google Scholar]
- 28.Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- 29.Manoukian AS, Woodgett JR. Role of glycogen synthase kinase-3 in cancer: regulation by Wnts and other signaling pathways. Adv Cancer Res. 2002;84:203–229. doi: 10.1016/s0065-230x(02)84007-6. [DOI] [PubMed] [Google Scholar]
- 30.Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD. Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000;23:222–229. doi: 10.1016/s0166-2236(00)01548-4. [DOI] [PubMed] [Google Scholar]
- 31.Meier R, Thelen M, Hemmings BA. Inactivation and dephosphorylation of protein kinase Bα (PKBα) promoted by hyperosmotic stress. EMBO J. 1998;24:7294–7303. doi: 10.1093/emboj/17.24.7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michael A, Haq S, Chen X, Hsich E, Cui L, Walters B, Shao Z, Bhattacharya K, Kilter H, Huggins G, Andreucci M, Periasamy M, Solomon RN, Liao R, Patten R, Molkentin JD, Force T. Glycogen synthase kinase-3β regulates growth, calcium homeostasis, and diastolic function in the heart. J Biol Chem. 2004;279:21383–21393. doi: 10.1074/jbc.M401413200. [DOI] [PubMed] [Google Scholar]
- 33.Miller TM, Tansey MG, Johnson EM, Jr, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 34.Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliver CJ, Shenolikar S. Physiologic importance of protein phosphatase inhibitors. Front Biosci. 1998;3:961–972. doi: 10.2741/a336. [DOI] [PubMed] [Google Scholar]
- 36.Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- 37.Park IK, DePaoli-Roach AA. Domains of phosphatase inhibitor-2 involved in the control of the ATP-Mg-dependent protein phosphatase. J Biol Chem. 1994;269:28919–28928. [PubMed] [Google Scholar]
- 38.Song L, De Sarno P, Jope RS. Central role of glycogen synthase kinase-3β in endoplasmic reticulum stress-induced caspase-3 activation. J Biol Chem. 2002;277:44701–44708. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- 39.Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 40.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3β by phosphorylation: new kinase connections in insulin and growth-factor signaling. Biochem J. 1993;296:15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szatmari E, Habas A, Yang P, Zheng J, Hagg T, Hetman M. A positive feedback loop between glycogen synthase kinase-3β and protein phosphatase-1 after stimulation of NR2B NMDA receptors in forebrain neurons. J Biol Chem. 2005;280:37526–37535. doi: 10.1074/jbc.M502699200. [DOI] [PubMed] [Google Scholar]
- 42.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welsh GI, Proud CG. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J. 1993;294:625–629. doi: 10.1042/bj2940625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woodgett JR, Jr, Cohen P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase-II (glycogen synthase kinase-5) Biochem Biophys Acta. 1984;14:339–47. doi: 10.1016/0167-4838(84)90047-5. [DOI] [PubMed] [Google Scholar]
- 45.Wu KD, Lee WS, Wey J, Bungard D, Lytton J. Localization and quantification of endoplasmic reticulum Ca2+-ATPase isoform transcripts. Am J Physiol. 1995;269:775–784. doi: 10.1152/ajpcell.1995.269.3.C775. [DOI] [PubMed] [Google Scholar]
- 46.Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]