

Abstract
Oxidative stress is central to ischemia-reperfusion injury. The role of the endoplasmic reticulum (ER) in this process is uncertain. In ER signaling, PERK-Nrf2 and Ire-CHOP are two pathways that determine cell fate under stress. PERK-Nrf2 up-regulates antioxidant enzyme expression whereas Ire-CHOP promotes apoptosis. We have identified a novel pathway in ER stress-induced apoptosis after ischemia-reperfusion in vitro involving translational suppression of the survival kinase PKB/Akt (Akt), and elucidated an alternative protective role of antioxidants in the regulation of Akt activity. Using human choriocarcinoma JEG-3 cells, we found that sustained activation of ER stress by tunicamycin or thapsigargin exacerbated apoptosis in oxygen-glucose-deprived cells during reoxygenation. This was mediated via a reduction in phosphorylated Akt secondary to down-regulation of protein translation rather than suppression of phosphorylation. Transient overexpression of wild-type Akt, but not kinase-dead Akt, in JEG-3 cells diminished tunicamycin-OGD reoxygenation-induced apoptosis. The antioxidants Trolox and Edaravone reduced apoptosis, but the protective effect of Trolox was abrogated by the PI3K inhibitor, LY294002. We speculate that sustained ER stress may contribute to the placental dysfunction seen in human pregnancy complications.—Yung, H-w., Korolchuk, S., Tolkovsky, A. M., Charnock-Jones, D. S., Burton, G. J. Endoplasmic reticulum stress exacerbates ischemia-reperfusion-induced apoptosis through attenuation of Akt protein synthesis in human choriocarcinoma cells.
Keywords: unfolded protein response, placenta, oxidative stress, trophoblast, CHOP
Placental oxidative stress has been postulated to be a key factor in the pathogenesis of human pregnancy complications such as intrauterine growth retardation and pre-eclampsia (1, 2). We recently proposed that the cause of the stress is an ischemia-reperfusion type injury (3). Unlike in other organs, where ischemia-reperfusion injury is usually an isolated insult caused by pathological blockage or rupture of blood vessels attenuating oxygen and nutrient supply, in the placenta mild ischemia-reperfusion is likely to be a repetitive process. Maternal blood flow through the intervillous space of the placenta may fluctuate through three principal mechanisms: intrinsic contraction of the spiral arteries supplying the placenta, external compression of the spiral arteries during uterine contractions, and redistribution of maternal blood flow associated with postural changes (2).
Evidence for the first of these comes from angiographic studies of the macaque, during which Martin et al. observed that blood flow from individual spiral arteries into the intervillous space is intermittent (4). Transient periods of placental ischemia therefore are likely to be a feature of normal human pregnancy and will increase in both frequency and severity toward term as uterine contractions intensify. Their incidence is likely to be increased in pathological situations, when deficient trophoblast invasion into the endometrium leads to incomplete conversion of the maternal spiral arteries (5) and to retention of their vasoreactivity. In addition, the impact of ischemia on placental tissues will be greater than that in other organ systems because the placenta continually extracts large quantities of oxygen to meet both its own high metabolic demands and those of its dependent fetus. This dual extraction will exacerbate fluctuations in oxygen and nutrient concentrations during the ischemic period. The result is that a state of chronic oxidative stress is generated within the syncytiotrophoblast of the placenta (2).
There is increasing evidence that ER stress plays a crucial role in ischemia-reperfusion-induced cell dysfunction (6). Most studies have focused on cerebral ischemia, either focal or global, in both in vitro and in vivo models (7-9). By contrast, the role of ER stress in ischemia-reperfusion in the placenta remains unexplored. The ER is the first organelle in the secretory pathway, and serves as a site of secretory protein synthesis and modification prior to transportation to the Golgi bodies for export. Any disturbance of its microenvironment, or disruption of either protein folding or modification within the ER, triggers the unfolded protein response (UPR) and ER-associated protein degradation (ERAD) (10). The UPR coordinates a program of modified gene transcription and protein translation aimed at reducing the burden on the ER, whereas the ERAD pathway up-regulates protein degradation, thereby eliminating accumulated misfolded proteins. Both help to re-establish the cell's homeostatic environment; failure to do so ultimately results in cell death through activation of apoptotic pathways.
The proximal signaling events that are initiated in response to the UPR involve activation of PKR-like endoplasmic reticulum kinase (PERK), inositol requiring 1 (Ire1) protein kinase, and activating transcription factor 6 (ATF6) (11). Activation of PERK in turn phosphorylates eukaryotic translation initiation factor 2α (eIF-2α), which suppresses protein synthesis (12-14). Activated Ire1 protein kinase is required for splicing of the X-box transcription factor (XBP-1) mRNA (15), and the spliced variant is a potent transcriptional activator. A combination of ATF6 and the spliced variant of XBP-1 positively regulates a wide variety of UPR target gene expression, including several ER resident chaperones (15-18). CHOP/GADD153 is a proapoptotic transcription factor that suppresses the transcription of Bcl-2, which can be induced by a combination of the PERK/ATF4 and ATF6 pathways (19). It is expressed at very low levels under physiological conditions but is strongly induced in response to ER stress (20, 21).
PI3K-Akt has been widely reported to be one of the most important survival pathways in a variety of cell types (22). Activation of the PI3K-Akt pathway in response to growth factors promotes cell survival whereas withdrawal of growth factors or a death signal challenge decreases its activity and promotes cell death. A major role of PI3K-Akt is suppression of proapoptotic proteins/enzymes, such as BAD (23) and procaspase-9 (24), via phosphorylation. Srinivasan et al. reported that administration of insulin-like growth factor (IGF-1) reduced thapsigargin-induced apoptosis in insulinoma cells and that this effect is mediated partly by PI3K-Akt (25). A recent study also showed that inactivation of PI3K-Akt is important for inducing CHOP expression in ER-stressed cells (26). Together, these studies suggest that Akt activity plays a crucial role in ER stress-induced apoptosis.
In the present study, we investigated the effects of sustained ER stress on ischemia-reperfusion-induced apoptosis using human choriocarcinoma JEG-3 cells as a model for placental trophoblast. ER stress was induced using tunicamycin, an N-glycosylation linkage inhibitor, or thapsigargin, an inhibitor of the ER Ca2+ pump that depletes ER Ca2+ storage.
MATERIALS AND METHODS
Chemicals and antibodies
Cycloheximide, tunicamycin, thapsigargin, propidium iodide, bisBenzimide H 33342 trihydrochloride (Hoechst 33342), G418, clasto-lactacystin-β-lactone, and 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) were from Sigma-Aldrich (Poole, UK). Boc-Asp(OME)-FMK (BAF) was from Alexis Biochemicals (Bingham, UK). LY294002 and 3-methyl-1-phenyl-2-pyrazolin-5-one (Edaravone) were from Calbiochem (Nottingham, UK). Salubrinal was from Chem-Bridge Corporation (San Diego, CA, USA).
Anti-phospho-Akt (Ser-473), anti-Akt, anti-Akt1 (2H10), anti-phospho-eIF2α (Ser-51, anti-eIF2α, and anti-phospho-Bad (Ser-136) antibodies were from Cell Signaling Technology (Danvers, MA, USA). Anti-Bad antibody was from Transduction Labs (BD Biosciences, Oxford, UK). Anti-hemagglutinin (HA) -tagged antibody was from Santa Cruz Biotechnologies (Autogen Bioclear UK Ltd., Wiltshire, UK). Anti-β-actin antibody was from Sigma-Aldrich. Anti-GADD153/CHOP antibody was from Abcam (Cambridge, UK).
Cell culture
Human choriocarcinoma JEG-3 cells were a gift from Dr. Ashley Moffett and were grown in RPMI 1640 medium (Invitrogen Ltd, Paisley, UK) supplemented with 5% heat-inactivated FBS (HI-FBS) (Invitrogen), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in a 5% CO2 atmosphere.
Oxygen-glucose deprivation
Oxygen-glucose deprivation (OGD was performed as described previously (27). In brief, JEG-3 cells were rinsed once with anoxic glucose-free RPMI 1640 medium, which was purged by degassing glucose-free RPMI 1640 medium with gas containing 5% CO2 in nitrogen for 20 min, followed by incubation in anoxic glucose-free RPMI 1640 medium for the desired time intervals inside an anaerobic chamber (COMPACT-M, Don Whitley Scientific, UK). For reoxygenation, the OGD-treated cells were returned to RPMI 1640 medium containing 2 mg/ml glucose and incubated in a 5% CO2/21% O2 atmosphere.
Cell viability assay
The number of dead cells was determined by costaining with two nuclear dyes: Hoechst 33342 (Sigma-Aldrich) and propidium iodide (PI) (Sigma-Aldrich). As PI is a membrane-impermeable dye, it stains the nucleus only in those cells that have lost their membrane integrity. By contrast, Hoechst 33342 is membrane permeable, so its nuclear staining is independent of membrane integrity. Therefore, all cells are stained with Hoechst 33342 (blue), but in dead cells the blue color is overridden by PI (red or pink) staining. After each stress treatment, cells were coincubated with RPMI 1640 medium containing of 5 μg/ml Hoechst 33342 and PI for 10 min at 37°C, and the numbers of Hoechst 33342-positive cells (blue) and PI-positive cells (red/pink) were counted separately under UV fluorescence using a Leitz DM1L microscope. All fragmented or highly condensed nuclei were scored as apoptotic, whereas PI-positive cells with normal nuclear size were scored as necrotic. Six fields were randomly selected and counted in each condition, each field normally containing 150 to 250 cells. When there was severe cell death, the number of fields selected and counted increased to 15 or more to achieve a similar number of cells to be counted for statistical analysis. To obtain the most accurate results, only cells attached on the bottom of the plate were counted, as floating dead cells always concentrated at the center of the plate, possibly due to surface tension effects of the medium.
Transmission electron microscopy (TEM) analysis
Cells were fixed by immersion in 2% glutaraldehyde containing 2 mmol/L CaCl2 in 0.1M PIPES buffer at pH 7.4; 100 μl 33% H2O2 was added to each 10 ml aliquot immediately before use. Cells were fixed in situ in culture plates for 2 h at 4°C. They were rinsed twice in buffer (0.1M PIPES), scraped free of the plates, and transferred to 1.5 ml centrifuge tubes. After buffer washes they were postfixed in 1% osmium ferricyanide for 1 h, rinsed three times in DIW, and bulk stained in 2% uranyl acetate for 1 h. They were rinsed in DIW and dehydrated in an ascending series of ethanol solutions to 100% ethanol, rinsed twice in acetonitrile, and embedded in Quetol epoxy resin. Fifty-nanometer sections were cut on a Leica Ultracut UCT, stained with saturated uranyl acetate in 50% ethanol and lead citrate, and viewed in a FEI Philips CM100 operated at 80 kv.
Western blot analysis
Cells were washed with ice-cold PBS, scraped off in cell lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na3VO4, and complete mini proteases inhibitor cocktail (Roche Diagnostics, East Sussex, UK), and transferred to a microfuge tube. After pipetting up and down ∼30 times, cells were maintained on ice for 20 min with occasional vortexing and centrifuged at 10,000 g for 5 min. The supernatant was kept at −80°C until analysis. Bicinchoninic acid (Sigma-Aldrich) was used to determine protein concentration using 5 μl of cell lysate. Equivalent amounts of protein were resolved by SDS-PAGE, blotted onto nitrocellulose (0.2 μm) and analyzed by enhanced chemiluminescence (ECL) (Amersham Biosciences, Bucks, UK) using Kodak X-OMAT androgen receptor (AR) film (Sigma-Aldrich). Films were scanned using a flat-bed scanner (Cannon 8000F) and intensities of the bands representing phospho- and total kinase forms were determined from two or three different exposures (within the linear detection range) using Image J analysis software (Freeware).
Akt kinase assay
The nonradioactive Akt kinase assay (Cell Signaling Technology) was performed following the manufacturer's instructions. In brief, after harvesting the cell lysate, equal amounts of lysate were mixed with 20 μl immobilized Akt(1G1) mouse monoclonal antibody bead slurry followed by overnight rocking at 4°C. After extensive washing with lysis buffer to remove residual proteins, the beads were mixed with kinase assay reaction buffer containing 1 μM ATP and 1 μg recombinant GSK-3β protein, then incubated at 30°C for 1 h. The reaction was terminated by the addition of protein gel loading buffer and resolved in SDS-PAGE, blotted onto nitrocellulose (0.2 μm), probed with anti-phospho-GSK3β antibody, and analyzed by ECL (Amersham Biosciences) using Kodak X-OMAT AR film (Sigma-Aldrich).
RT-polymerase chain reaction (RT-PCR) analysis of XBP1 mRNA splicing
The assay was adapted from Shang and Lehrman (28). In brief, total RNA was isolated from JEG-3 cells using an RNeasy mini kit (Qiagen, UK) immediately after completion of stress treatments. First-strand cDNA was synthesized using Superscript First-Strand Synthesis System for RT-PCR (Invitrogen). To amplify XBP-1 mRNA, PCR was carried out for 30 cycles using the following profile: 94°C for 30 s; 58°C for 30 s; and 72°C for 1 min. In the final cycle, PCR products were incubated at 72°C for an additional 10 min with XBP-1 forward and reverse primers and TaqDNA polymerase (Invitrogen). Forward and reverse primer sequences were 5′-CTGGAACAGCAAGTGGTAGA-3′ and 5′-CTGGGTCCTTCTGGGTAGAC-3′, respectively. PCR products were resolved by 2% agarose gel electrophoresis with ethidium bromide and documented in a UVP Gel Documentation system (UVP, UK); 398 and 424 bp fragments represented spliced and unspliced XBP1 transcripts, respectively.
Quantitative real-time RT-PCR analysis
The ABI PRISM 7700 Sequence Detection System (TaqMan) was used to perform real-time PCR according to the manufacturer's protocols. “Ct” values for each transcript were compared with those for 18S rRNA. All primers and probes were obtained from Applied Biosystems (ABI, UK) “Assays-on-Demand ” and used a 5′ FAM reporter and 3′ nonfluorescent minor groove binder. The thermocycler parameters were 2 min at 50°C followed by 40 cycles of 15 s 95°C and 1 min at 60°C for PCR amplification.
Expression constructs and transfection
The expression plasmids encoding HA epitope-tagged wild-type Aktα (wt-Akt1), kinase-dead K179A Aktα (kd-Akt1), and green fluorescent protein (GFP) were a gift from Dr. Aviva Tolkovsky (29). To transfect JEG-3 cells, the cells were seeded 1 day in advance to obtain ∼50–70% cell confluence. The transfection was performed using Fugene 6 transfection reagent (Roche, Welwyn Garden City, UK) following the manufacturer's instructions. In brief, a reaction mixture was prepared by mixing 6 μl of Fugene 6 reagent with 89 μl OptiMEM I medium (Invitrogen) and incubated for 5 min at room temperature (RT). One microgram of plasmid DNA (0.2 μg/μl) was added to the reaction mixture and incubated for an additional 30 min at RT. The Fugene/DNA complex mixture was then added dropwise to the JEG-3 cells, supplemented with 5% heat-inactivated FBS and antibiotics. Cells were allowed to express the construct for 48 h before performing tunicamycin-OGD reoxygenation incubation.
Preparation of stable cell line overexpressing wt-Akt1
Due to the lack of a neomycin-resistant gene in the plasmids expressing wt-Akt1, the preparation of stable wt-Akt1-expressed JEG-3 cell preparation was achieved by cotransfection of wt-Akt1 with a GFP plasmid containing a neomycin-resistant gene in a 1:10 ratio. The transfection procedure was the same method used to prepare transient overexpressed wt-Akt1. After transfection, cells were allowed to express the constructs for 48 h before treatment with 500 μg/ml G418 (Sigma) for clone selection followed by culturing in selective medium (RPMI 1640 medium containing 5% FBS and 250 μg/ml G418). After ∼20 passages in the selective medium, immunocytochemical analysis confirmed that all GFP-positive cells also expressed wt-Akt1 (data not shown). Some wt-Akt1-positive cells were GFP negative. The cells that stably expressed wt-Akt1 represented a mixture of different cell clones. Although GFP labeling allowed us to use a FACS cell sorter to sort single cells for homogeneous clones, the cells always died after a few days in culture. JEG-3 cells appear to require a certain cell density to enable survival and proliferation, so it proved impossible to obtain a homogeneous clone of stably expressed wt-Akt1 from this cell type.
Pulse-chase [35S]methionine labeling
Cells were subjected to 8 h ischemia followed by 21 h reoxygenation in the presence of 5 μg/ml tunicamycin, 10 μg/ml cycloheximide, or 75 μM salubrinal. Cells were then washed and incubated with cysteine and methionine-free RPMI 1640 medium (Sigma) for 10 min in the presence of inhibitors twice before being pulsed with [35S]methionine by incubation with cysteine and methionine-free RPMI 1640 medium containing translation grade [35S]methionine (Amersham Biotech) and cysteine in the presence of inhibitors for 1 h. After extensive washing with RPMI 1640 medium, cells were lysed in lysis buffer (Western blot). An equal amount of protein from different samples was mixed with Akt-immobilized (1G1) antibody bead slurry (Cell Signaling Technology) for immunoprecipitation at 4°C for overnight with rocking (according to manufacturer's instructions). After extensive washing with lysis buffer to remove residual proteins, the mixtures were resolved using SDS-PAGE. The gel was stained with 0.25% (w/v) Coomasie Brilliant Blue R 250 to show an equal protein loading between samples and the dried gel was exposed to a PhosphorImager screen (Molecular Dynamics, Sunnyvale, CA, USA). The image was captured and analyzed using ImageQuant software (Molecular Dynamics).
Statistical analysis
Differences between means were tested using a two-tailed Student's t test, with a value of P < 0.05 considered significant.
RESULTS
Oxygen-glucose deprivation induces ER stress in JEG-3 cells
Increased phosphorylation of eukaryotic initiation factor-2α (eIF-2α), up-regulation of CHOP protein concentration, and splicing of XBP-1 mRNA are three markers of ER stress. In JEG-3 cells, OGD treatment strongly elevated the phosphorylation level of eIF-2α in a time-dependent manner without changing its total protein level (Fig. 1). CHOP protein concentration also increased after 2 h OGD incubation. There was a gradual increase in the amount of spliced variant of XBP-1 mRNA upon OGD treatment (Fig. 1); after 8 h most XBP-1 mRNA was in this form. Cells recovered completely from the ER stress induced by OGD after 24 h reoxygenation in the presence of 2 mg/ml glucose (Fig. 1, lane 6). Phosphorylation of eIF-2α dropped >80% during the first hour of reoxygenation and returned to control values after 3 h (data not shown). Evidence of ER stress was further confirmed by TEM analysis. In normoxic control cells, a few strands of rough ER with narrow cisternae were observed (Fig. 1B, left panel, arrows), but after 8 h of OGD treatment almost all the cisternae were dilated into circular shapes (Fig. 1B, right panel, arrows). These results demonstrate that there was a reversible increase of ER stress in JEG-3 cells in response to OGD.
Figure 1.
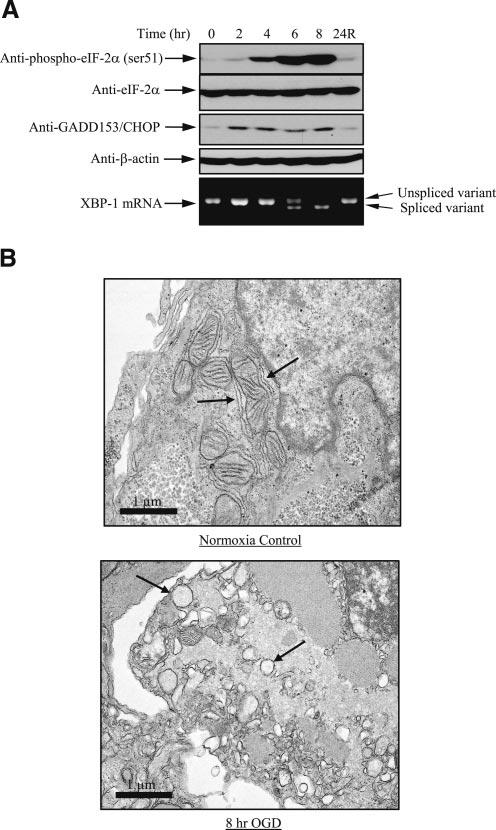
OGD induces ER stress in JEG-3 cells. Cells were subjected to up to 8 h OGD followed by 24 h reoxygenation in the presence of glucose. A) Both protein and RNA were isolated, followed by Western blot analysis for the phosphorylation level of eIF-2α and expression level of GADD153/CHOP, and RT-PCR analysis of the splicing of XBP mRNA. B) TEM analysis showing regular cisternae of rough endoplasmic reticulum (RER) (arrowed) under normoxic conditions and dilated cisternae after 8 h OGD.
Sustained activation of ER stress promotes apoptosis during reoxygenation
Activation of ER stress in cells was not sufficient to induce significant cell death in JEG-3 cells after 8 h OGD (Fig. 2, col. 4). However, an additional 24 h of reoxygenation significantly increased cell death to 24.6% (Fig. 2, col. 5). A pan-caspase inhibitor, Boc-Asp-FMK (BAF) (50 μM), completely inhibited the increase in cell death, suggesting that death occurred by apoptosis.
Figure 2.
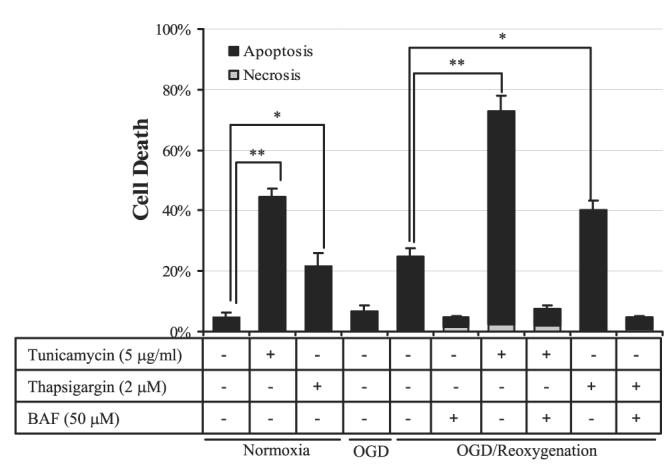
Activation of ER stress pathways exacerbates cell death during OGD reoxygenation in JEG-3 cells. For OGD reoxygenation, cells were subjected to 8 h OGD followed by 24 h rexoygenation in the presence of tunicamycin, thapsigargin, and/or BAF. For tunicamycin and thapsigargin treatments, cells were incubated with either tunicamycin or thapsigargin under normoxic conditions in the presence of glucose for 24 h. Cell viability assay was performed by costaining of Hoechst 33342 and PI. Data are mean ± se from 3 experiments. *P < 0.05; **P < 0.01.
Although there was no obvious ER stress present after 24 h reoxygenation (Fig. 1, lane 6), it is unclear whether sustained activation of ER stress, which is likely to be present in the repetitive placental ischemia-hypoxia reperfusion scenario, promotes cell injury or death. To test this, we performed repetitive hypoxia reoxygenation (HR) in three hourly cycles for 72 h on JEG-3 cells in vitro. Preliminary results showed an elevation of phosphorylated eIF-2α throughout the 72 h incubation whether or not the cells were harvested at the end of the hypoxic or reoxygenation phase of a cycle (unpublished data). However, expression of the proapoptotic ER stress marker, CHOP protein, was not increased and there was no rise in cell death (unpublished data). This is in contrast to the situation in pathological pregnancies, where we observed an increase of CHOP protein concentration in pre-eclamptic placentas (unpublished data); trophoblast apoptosis is elevated in this condition (30). These more severe effects may be the result of chronic mild ER stress throughout several weeks or months of pregnancy. In theory, chronic mild ER stress may be achieved in vitro by increasing the length for repetitive HR challenging, but in practice this is not possible due to the rapid growth rate of JEG-3 cells. This choriocarcinoma cell line requires passaging every 3 days even when the cells are seeded at 10% initial confluence.
Therefore, two different ER stress inducers, tunicamycin and thapsigargin, were introduced into the study. Tunicamycin (5 μg/ml) or thapsigargin (2 μM) alone induced 44.3% and 21.6% cell death, respectively, after 24 h incubation (Fig. 2, cols. 2, 3). Adding tunicamycin or thapsigargin to the OGD-treated cells during the 24 h reoxygenation phase markedly elevated cell death to 70.6% and 39.4%, respectively (Fig. 2, cols. 7, 9). Cell death was suppressed by BAF, suggesting that it was principally apoptotic. To conclude, sustained activation of ER stress exacerbates OGD reoxygenation-induced apoptosis in JEG-3 cells.
Antioxidants Trolox and Edaravone reduce ER stress-induced apoptosis in OGD reoxygenation
There is considerable evidence that reactive oxygen species (ROS) plays an important role in ischemia-reperfusion-induced cell injury or death. Therefore, an antioxidant, Trolox (a water-soluble vitamin E analog), was used to test whether tunicamycin-induced cell death in OGD reoxygenation is mediated by ROS. Application of Trolox (2.5 mM) significantly reduced apoptosis from 67.9% to 28.6% (Fig. 3, cols. 5, 6). The protective effect of Trolox was dose dependent (data not shown) and was restricted to the tunicamycin-mediated apoptosis in ischemically treated cells, as Trolox did not affect cell death in tunicamycin-treated nonischemic cells (Fig. 3, cols. 2, 3).
Figure 3.
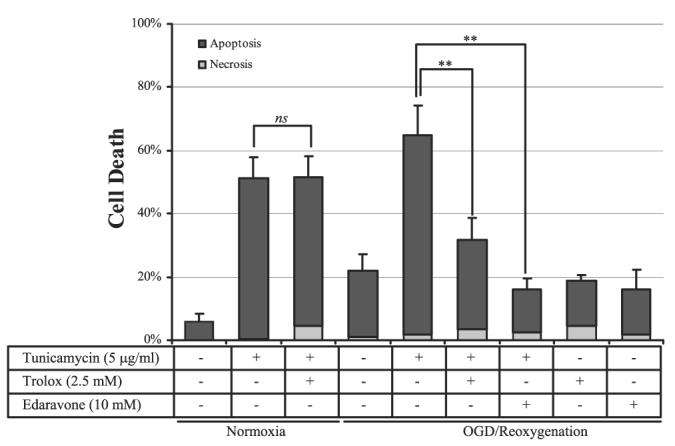
Antioxidants Trolox and Edaravone protect against ER stress-induced apoptosis in OGD reoxygenation. Cells were subjected to 8 h OGD followed by 24 h reoxygenation incubation with or without tunicamycin, Trolox, or Edaravone. Normoxic controls were incubated with the same concentrations of tunicamycin and Trolox for 24 h. Cell viability assay was performed as previously. Data are mean ± sd from 3 to 6 experiments. **P < 0.01.
To confirm these findings, Edaravone (3-methyl-1–1phenyl-2-pyrazolin-5-one), a lipophilic free radical scavenger that has been shown to be neuroprotective even after the onset of cerebral ischemia (31), was used. Application of Edaravone (10 mM) markedly suppressed tunicamycin-induced apoptosis in OGD-treated cells after 24 h reoxygenation (Fig. 3, col. 7).
These results demonstrate that sustained ER stress during reoxygenation enhances ROS damage, thereby promoting oxidative injury to the cells. To elucidate the protective mechanisms of Trolox, the PKB/Akt (Akt) survival pathway was investigated.
Sustained activation of ER stress reduces Akt activity by down-regulation of Akt total protein, but not phosphorylation in tunicamycin-OGD reoxygenation-induced apoptosis
After 8 h of OGD treatment, there was no detectable activity of Akt in the Akt kinase assay (Fig. 4A, lane 2), associated with a marked reduction in the concentration of immunoprecipitated Akt protein (Fig. 4A, lane 2). After reoxygenation, Akt activity and protein concentration returned to control levels (Fig. 4A, lane 3). Addition of tunicamycin during OGD reoxygenation not only prevented resumption of Akt activity to control levels, but also induced a further reduction in the amount of immunoprecipitated Akt compared with 8 h OGD treatment (Fig. 4A, lane 4).
Figure 4.
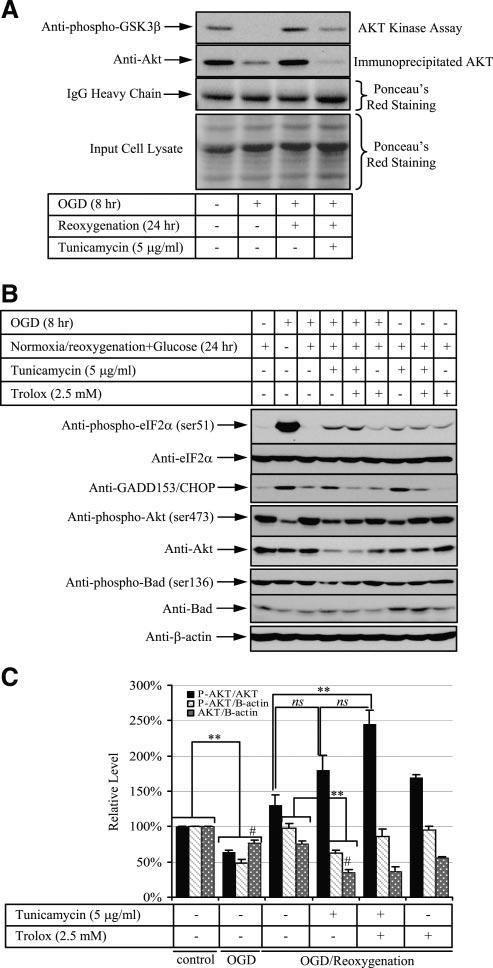
Sustained activation of ER stress inhibits Akt activity, but Trolox reverses this process by elevation of its phosphorylation. Cells were subjected to 8 h OGD followed by 24 h reoxygenation with either tunicamycin, Trolox, or both. A) Akt activity was reflected by phosphorylation of the substrate GSK3-β level after probing with phosphor-GSK3-β antibody. IgG heavy chain and input cell lysate showed equal loading of Akt antibody and the cell lysate used for immunoprecipitation. B) Western blot analysis of phosphorylated eIF-2α (Ser-51) and its total form, CHOP, phosphorylated Akt (Ser-473) and its total form, and phosphorylated BAD (Ser-136) and its total protein. β-Actin was used to normalize protein loading. C) Densitometric quantification of band intensity expressed relative to the control (untreated). Phosphorylation status and its protein level are presented as a ratio in a combination between phosphorylated Akt (P-AKT), total Akt (AKT) and β-actin. Data are mean ± se from 3 experiments. **P < 0.01. #P < 0.01 of Akt/β-actin ratio between OGD and tunicamycin-OGD reoxygenation.
Akt activity is dependent on both its phosphorylation status and its total protein concentration. The loss of Akt activity may thus be the result of suppression of protein phosphorylation, reduction of protein expression, or a combination of the two. Because the immobilized Akt (1G1) antibody used in the immunoprecipitation binds preferentially to Akt phosphorylated at serine 473 but also to nonphosphorylated Akt (manufacturer's data sheet), we could not distinguish between these effects. Therefore, the phosphorylation status and protein concentration of Akt during tunicamycin-OGD reoxygenation were further investigated using anti-phospho-Akt (Ser-473) and anti-Akt-specific antibodies. Anti-Akt antibody recognizes all three isoforms of Akt protein (manufacturer's data sheet). OGD suppressed 40% of Akt (Ser-473) phosphorylation and 20% total protein expression. Loss of Akt activity following tunicamycin-OGD reoxygenation was associated with significant reductions in both serine 473 phosphorylation and total Akt protein level compared with OGD reoxygenation alone (Fig. 4B, lanes 3, 4, Fig. 4C, cols. 3, 4). When normalization was carried out between phosphorylated and total Akt, there was a trend of increasing Akt phosphorylation in tunicamycin-OGD reoxygenation compared with OGD reoxygenation alone (Fig. 4C, cols. 3, 4), though the difference did not reach statistical significance.
Phosphorylation of the proapoptotic protein BAD at serine 136 by Akt has been reported to be crucial for the inhibition of mitochondrial-mediated apoptosis (32). Reduction of Akt phosphorylation in tunicamycin-OGD reoxygenation-treated cells was accompanied by a decrease of BAD serine 136 phosphorylation while the total BAD protein concentration remained relatively constant (Fig. 4B, lane 4).
To conclude, these observations suggest that tunicamycin induces cell death during OGD reoxygenation not only through an increase in oxidative damage, but also by suppressing Akt activity through lowering its protein expression level, thereby releasing BAD, which in turn promotes apoptosis via the mitochondrial pathway.
Trolox suppression of sustained ER stress-induced apoptosis is mediated by Akt
In addition to acting as an antioxidant, vitamin E/Trolox has been reported to modulate signaling pathways (33). Treating JEG-3 cells with Trolox alone did not alter Akt phosphorylation at serine 473 (Fig. 4B, lane 9). However, treating ischemic cells with tunicamycin and Trolox together elevated the relative phosphorylation level of Akt (245.4%±19.4%, mean±se, n=3) significantly compared with OGD reoxygenation (130.7%±13.6%, mean±se, n=3) (Fig. 4C, cols. 3, 5). There was also a trend of increasing Akt phosphorylation compared with tunicamycin-OGD reoxygenation (179.6%± 20.8%, mean±se, n=3) (Fig. 4C, cols. 4, 5). However, the total amount of Akt protein was almost constant between tunicamycin-OGD reoxygention in the presence or absence of Trolox (Fig. 4B, lanes 4, 5; Fig. 4C, cols. 4, 5).
To investigate the role of Akt in the protective effect of Trolox, a PI3K inhibitor, LY294002, was used to suppress Akt activity. Addition of LY294002 (75 μM) reduced Akt phosphorylation by >90% (data not shown). To observe the effect of LY294002, reoxygenation time was shortened to 16 h. Addition of LY294002 to OGD-treated cells during reoxygenation induced a similar amount of cell death (34±5.3%, mean±se, n=3) as did tunicamycin-induced cell death (46.2±11.2%, mean±se, n=3), demonstrating the importance of Akt activity in maintaining cell survival during reoxygenation (Fig. 5A, cols. 4, 5). Addition of Trolox significantly reduced tunicamycin-induced cell death in ischemic cells (Fig. 5A, col. 6), but suppression of Akt activity by LY294002 abolished this protective effect (Fig. 5A, col. 7).
Figure 5.
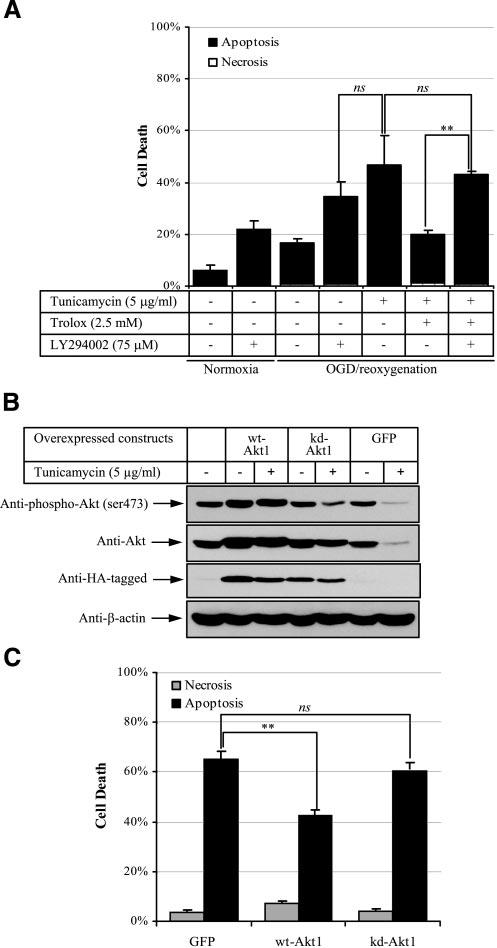
Akt activity is crucial for suppression of apoptosis induced by sustained ER stress in ischemic JEG-3 cells. A) Inhibition of Akt activity abolishes the protective effect of Trolox in OGD reoxygenation-tunicamycin-induced cell death. Cells were subjected to 8 h OGD followed by 16 h reoxygenation with a combination of LY294002, tunicamycin, and/or Trolox. Cell viability assay was performed as previously. Data are mean ± se from 3 experiments. **P < 0.01. B, C) Overexpression of Akt suppresses tunicamycin-induced cell death in OGD reoxygenation. Cells were transfected with GPF, wt-Akt1, or kd-Akt1 plasmids and allowed to express proteins for 2 days before being subjected to 8 h OGD followed by 24 h reoxygenation in the presence of 5 μg/ml tunicamycin. A, B) Western blot: proteins were probed by specific antibodies against phospho-Akt (Ser-473), Akt, HA-tagged, and β-actin. B, C) Cell viability assay was performed as previously. Data are mean ± se from 3 experiments. **P < 0.01.
To confirm these results, transient overexpression of plasmid cDNA encoding containing wild-type Aktα (wt-Akt1), kinase-dead Aktα (kd-Akt1), or GFP in JEG-3 cells was performed. Minimizing Fugene 6 toxicity while maximizing transfection efficiency, we obtained ∼30%-positive cells with ∼5% dead cells 48 h after transfection. The cultures were then subjected to the tunicamycin-OGD reoxygenation challenge for 24 h. Western blot analysis revealed that the amount of phosphorylated Akt was reduced in the presence of tunicamycin in both kd-Akt1- and GFP-transfected cells; however, the total concentration of Akt remained constant in the kd-Akt1-transfected cells but was reduced in the GFP-transfected cells. In contrast, the amounts of phosphorylated and total Akt remained about the same in wt-Akt1 tunicamycin-treated cells (Fig. 5B). Tunicamycin treatment induced 65.3% and 59.8% cell death in GFP- and kd-Akt1-transfected cells, respectively (Fig. 5C, cols. 1, 3); there was a statistically significant reduction of cell death, to 42.5%, in wt-Akt1-transfected cells (Fig. 5C, col. 2).
To conclude, sustained ER stress during ischemia-reperfusion has a dual action. It not only enhances ROS-mediated cell injury; it also attenuates Akt activity by lowering the amount of total Akt. Both effects will weaken survival pathways and exacerbate apoptotic cell death. Trolox reduces apoptosis via activation of Akt phosphorylation. The question remains as to how sustained ER stress down-regulates the amount of Akt protein.
Sustainable ER stress during OGD reoxygenation inhibits Akt protein synthesis
The cellular protein concentration is the result of an equilibrium between three processes: transcription, translation, and degradation. Inhibition of either transcription, translation, or activation of degradation will cause a reduction in the amount of total protein level. To elucidate which process or processes are involved in the loss of Akt protein in cells treated with tunicamycin and OGD reoxygenation, all three processes were investigated. Quantitative real-time RT-PCR was used to measure the relative level of akt transcripts. Akt protein has three common isoforms: Akt1, Akt2, and Akt3 (22). After 8 h OGD, akt1, akt2, and akt3 transcript levels were down-regulated significantly (Fig. 6A, cols. 1, 4), and 24 h reoxygenation in the presence or absence of tunicamycin neither increased nor decreased mRNA levels (Fig. 6A, cols. 5, 6). Addition of Trolox in ischemic cells during reoxygenation significantly elevated expression of akt1 transcripts but not akt2 or akt3 transcripts (Fig. 6A, col. 7).
Figure 6.
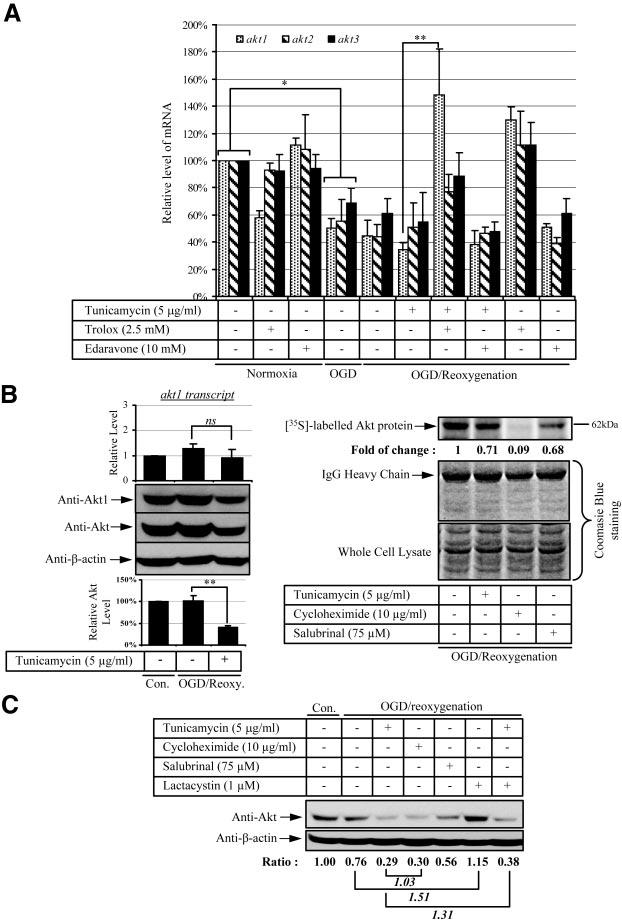
Sustained ER stress attenuates Akt protein translation in ischemic JEG-3 cells. A, C) Cells were subjected to 8 h OGD followed by 24 h reoxygenation with a combination of tunicamycin, Edaravone, Trolox, cycloheximide, salubrinal, and/or clasto-lactacystin-β-lactone. B) Stable overexpressing Akt1 cells were subjected to 8 h OGD followed by 21 h reoxygenation in the presence of tunicamycin, cycloheximide, or salubrinal. A) Expression of akt transcripts: quantitative real-time PCR was used to measure the relative level of transcripts of the three Akt isoforms between samples. B) Translation of Akt protein: Left panel shows that in Akt1-overex-pressing cells, akt1 transcripts do not change but that both Akt1 and total Akt protein concentrations are reduced after tunicamycin-OGD reoxygenation. In all cases, the result was compared with control, which is expressed as 1. Data are mean ± se from 3 experiments. *P < 0.05; **P < 0.01. In right panel, Akt protein translation is halted under sustained ER stress. Cells were exposed for 1 h to [35S]methionine before protein extraction and immunoprecipitation. The IgG heavy chain and whole cell lysate were used to show an equal input of antibody and protein used for immunoprecipitation, respectively. Fold of change is the ratio between the absence and presence of drugs and is expressed as 1. C) Degradation of Akt protein: after treatment, proteins were isolated and resolved in SDS-PAGE. Anti-Akt antibody was used to reveal the total Akt protein level. All results are compared with control, which is expressed as 1.
Our finding of an ∼40% reduction in Akt protein concentration after tunicamycin-OGD reoxygenation compared with OGD reoxygenation without any change in the amount of akt transcripts suggests that loss of Akt protein is not due to a change at the transcriptional level. This conclusion is further supported by the finding that addition of Trolox to tunicamycin-OGD reoxygenation elevated akt1 transcript to control levels whereas the amount of Akt protein remained unchanged (Fig. 4B, lane 5; Fig. 4C, col. 5; Fig. 6A, col. 7).
Numerous studies have revealed that phosphorylation of eIF-2α is able to inhibit protein translation (34), and there was an increase of eIF-2α phosphorylation upon tunicamycin challenge during OGD reoxygenation (Fig. 4B, lane 4). To further confirm the involvement of translation inhibition in the reduction of Akt protein, a stable cell line overexpressing Akt1 was established from transiently overexpressing wt-Akt1 cells by G418 antibiotic selection. It was hypothesized that cells expressing wt-Akt1 from the plasmid would drive akt1 mRNA transcription from the cytomegalovirus (CMV) promoter, and thus should bypass any endogenous transcriptional regulation in akt1 mRNA expression. In Akt1-overexpressing cells, the level of total Akt protein level was ∼4-fold higher than in parental cells (data not shown). Unlike parental cells in which OGD reoxygenation induced a 50% reduction in akt1 transcripts, the stable cell line exhibited a relatively constant expression of akt1 transcripts irrespective of either OGD reoxygenation or tunicamycin-OGD reoxygenation challenge (Fig. 6B, left upper panel, cols. 1–3); Western blot analysis revealed a 55% decrease in Akt protein (Fig. 6B, left middle panel and lower graph). A similar result was obtained using an anti-Akt1 antibody (Fig. 6B, left middle panel). Transcript levels of akt2 and akt3 were down-regulated to an extent similar to that observed in parental cells upon treatment (data not shown). This observation suggested there was indeed an inhibition of protein translation of Akt upon tunicamycin treatment in ischemic cells irrespective of the mRNA level. However, it does not exclude the possibility of activation of Akt protein degradation under these conditions.
To confirm the above finding, a pulse-chase experiment using [35S]methionine was conducted in the stable wt-Akt1-overexpressing cells. A brief exposure (1 h) of the cells to [35S]methionine allowed newly synthesized radiolabeled Akt to be detected prior to its targeting for degradation. Addition of tunicamycin to the OGD reoxygenation-treated cells induced an ∼29% loss of Akt protein (Fig. 6B, right panel, lane 2). The protein synthesis inhibitor cycloheximide used as a positive control caused >90% inhibition of Akt protein translation (Fig. 6B, right panel, lane 3). Salubrinal is a newly identified inhibitor for eIF-2α dephosphorylation (35). It acts as a phosphatase inhibitor and specifically targets the phosphatases involved in dephosphorylation of phospho-eIF-2α protein. Treating cells with salubrinal induced eIF-2α phosphorylation at serine 51 (data not shown), and therefore should yield a result similar to that of tunicamycin. Indeed, addition of salubrinal to the OGD reoxygenation cells reduced Akt protein concentrations by 32% (Fig. 6B, right panel, lane 4), similar to the reduction caused by tunicamycin. The results are consistent with the idea that an increase in eIF-2α phosphorylation selectively attenuates protein synthesis.
Protein degradation of newly synthesized proteins is carried out mainly inside proteosomes. Application of the proteosomal inhibitor clasto-lactacystin-β-lactone (1 μM) increased the amount of Akt protein by ∼1.5-fold compared with OGD reoxygenation alone (Fig. 6C, lanes 2, 6). A similar result was obtained after tunicamycin-OGD reoxygenation in which addition of clastolactacystin-β-lactone elevated Akt protein levels by ∼ 1.3-fold (Fig. 6C, lanes 3, 7). There was no difference in the amount of Akt protein in ischemic cells treated with cycloheximide or tunicamycin (Fig. 6C, lanes 3, 4), further excluding a role for activation of protein degradation in regulating Akt concentrations under ER stress.
To conclude, loss of Akt activity in response to sustained ER stress in ischemic cells is primarily the result of persistent inhibition of Akt protein translation.
DISCUSSION
In this study we used an in vitro model of ischemia-reperfusion to generate oxidative stress in human choriocarcinoma JEG-3 cells. This cell line was used in preference to primary cultures of trophoblast cells due to the increased variability and lack of proliferative activity in the latter. OGD induced ER stress, which resolved upon 24 h reoxygenation, with phosphorylation of eIF-2α falling to control values at ∼4 h of rexoygenation. In brain tissues, phosphorylation of eIF-2α similarly fell to control values by 4–6 h of reperfusion after focal and global ischemia (36, 37). Application of tunicamycin or thapsigargin to ischemic cells sustained ER stress during reoxygenation, exacerbating OGD-induced apoptosis. This was associated with elevation of CHOP protein, similar to our observations in pre-eclamptic placentas (unpublished data). The increase in apoptotic cell death is likely to occur through reduction in Akt activity, secondary to suppression of its protein translation. The antioxidants Trolox and Edaravone exerted a protective role by enhancing Akt phosphorylation. These findings reveal a novel role for ER stress in mediating apoptosis during ischemia-reperfusion (Fig. 7).
Figure 7.
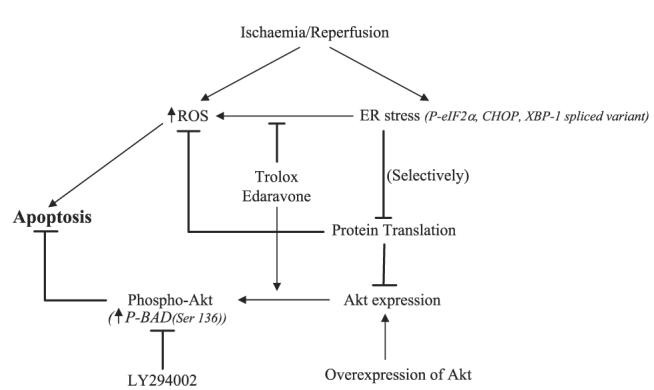
Summary diagram showing the roles of ER stress in oxidative stress-induced apoptotic cell death.
Severe cellular energy deficiency may provide one explanation for activation of the UPR during OGD. Around 90% of ATP was depleted in astrocytes upon 6 h OGD incubation (27). Under normal conditions, calcium ions in the cytosol are actively loaded into the ER by an ATP-dependent process through the sarcoendoplasmic reticulum Ca2+ ATPase (SERCA). Depletion of intracellular ATP suppresses SERCA activity, perturbing ionic homeostasis inside the ER lumen and thereby promoting protein misfolding and activation of the UPR. Resumption of ATP supply after reoxygenation re-establishes the ER homeostatic environment, limiting ER stress.
Addition of either tunicamycin or thapsigargin exacerbated apoptotic cell death in OGD-treated JEG-3 cells during reoxygenation. This effect could be ameliorated by antioxidants, suggesting there is increased oxidative damage upon sustained ER stress. ROS can be generated within the ER during disulfide bond formation (38, 39). Alleviation of ER stress through the UPR can cause further accumulation of ROS from the UPR-regulated oxidative machinery in the ER and from the mitochondria (40). Weakening of the antioxidant defense systems by suppression of translation of the principal antioxidant enzymes, such as manganese and copper/zinc superoxide dismutase (SOD), catalase, and glutathione peroxidase, as a result of protein synthesis inhibition (PSI) after ischemic insult or sustained ER stress, may also sensitize cells to oxidative injury/death (13, 34, 41). However, our immunoblotting results did not reveal any change in the protein concentrations of these enzymes under sustained activation of ER stress (data not shown). However, we cannot exclude the possibility that ER stress induced protein misfolding in these enzymes, thereby reducing their activities; further work is required to investigate this possibility.
Both Trolox and Edaravone were effective in reducing apoptosis after sustained ER stress in ischemic JEG-3 cells. However, the protective effect of Trolox, but not Edaravone, was limited to ischemic cells, as it failed to reduce apoptosis induced by tunicamycin in nonischemic JEG-3 cells. The protective effect of Trolox acts through activation of Akt phosphorylation rather than by increasing the amount of Akt protein level (Fig. 4C, Fig. 5A). Vitamin E/α-tocopherol has been linked to activation of protein phosphatase 2A (PP2A), which in turn dephosphorylates PKC, thereby inhibiting its activity (42, 43). However, our data do not fit these findings. Logically, if PP2A is being activated, Akt should be dephosphorylated (27). The mechanism by which Trolox activates Akt phosphorylation is currently under investigation.
By contrast, Trolox failed to suppress apoptosis induced by OGD reoxygenation alone (Fig. 3, col. 8). This finding, combined with the different efficacy of Trolox in suppressing tunicamycin-OGD reoxygenation- and tunicamycin-induced apoptosis, strongly suggests that apoptosis induced by tunicamycin, OGD reoxygenation, and tunicamycin-OGD reoxygenation occurs through three distinct pathways. It also excludes the possibility that the increase in apoptosis observed after tunicamycin-OGD reoxygenation is due simply to the additive effects of OGD reoxygenation and tunicamycin. Indeed, tunicamycin induces apoptosis via Trolox-sensitive and -insensitive pathways in ischemic or nonischemic cells, respectively.
CHOP has been reported to sensitize cells to ER stress-induced apoptosis by down-regulating expression of the antiapoptotic protein Bcl-2 (44). Trolox suppresses expression of CHOP and so attenuates cell death, but also maintains eIF-2α at a high phosphorylation status. This will lead to persistent PSI, thereby reducing the UPR and aiding recovery from the injury. Edaravone similarly increases Akt phosphorylation (data not shown) and suppresses apoptosis in sustained ER stress-treated cells. Addition of LY294002 did not eliminate its protective role (data not shown). Edaravone also inhibits apoptosis induced by tunicamycin in nonischemic JEG-3 cells (data not shown). Qi et al. showed that Edaravone protects against hypoxia-ischemia-induced ER dysfunction (45). Although the exact molecular mechanisms involved in the Edaravone-mediated protective effect under ER stress are uncertain, from our results we speculate that it operates as an antiapoptotic molecule downstream of Akt pathway. This hypothesis is supported by the study of Rajesh et al., in which Edaravone inhibited the mitochondrial permeability transition pore and up-regulated Bcl-2 expression (46).
The PI3K-Akt pathway has been shown to be a positive regulator of ER stress-induced apoptosis (25, 26). In OGD, reduction of Akt activity was associated with lowering both protein phosphorylation and expression. Severe ATP depletion and activation of phosphatases are two major factors that regulate kinase dephosphorylation during ischemia (27). However, it is unlikely that ATP depletion was the cause here, as eIF-2α was still strongly phosphorylated after 8 h OGD. Application of the PP1 and PP2A inhibitors, okadaic acid and calyculin A, respectively, inhibits dephosphorylation of Akt under OGD in cultured astrocytes, supporting the role for phosphatase activation (27).
Sustained activation of the UPR during reoxygenation induced a reduction in cellular Akt protein concentration of 60% compared with controls and of 40% compared with reoxygenation alone. The results in Fig. 6 excluded significant contributions to these reductions from Akt transcriptional inhibition and activation of protein degradation. By contrast, our data support the notion of PSI being the principal effect. This result is not consistent with the finding of Hyoda's study in which ER stress reduced Akt phosphorylation without affecting its total protein concentration (26). However, our result is consistent with the principle of ER stress-induced PSI, which is due to inhibition of translation initiation (6, 41). Phosphorylation of eIF-2α leads to a reduction in the rate of Met-tRNAiMet delivery by acting as a competitive inhibitor of eIF-2β, hence slowing down protein synthesis (34). Addition of tunicamycin to the ischemic cells prolongs eIF-2α phosphorylation (Fig. 4B, lane 4), thereby prolonging the effect of PSI. Recent studies have revealed that Akt also regulates the protein translation process by direct phosphorylation of the mammalian target of rapamycin (mTOR) (47), which in turn regulates protein synthesis through phosphorylating 4E binding protein 1 (4E-BP1) and p70 ribosomal protein S6 kinase (S6K1) (48-50). Therefore, down-regulation of Akt activity upon ER stress provides a negative feedback loop, preventing further translation of nascent protein into the ER and thereby reducing the UPR.
Unlike in tunicamycin-OGD reoxygenation, the 20% reduction in Akt protein observed in OGD may result from a combination of transcriptional and translational inhibition, and activation of protein degradation. Quantitative real-time RT-PCR showed a 50% reduction of all akt transcripts. Using cycloheximide, the half-life of Akt protein in JEG-3 was estimated to be ∼36 h (data not shown). There was no change in the amount of Akt protein after 8 h of cycloheximide treatment compared with control (data not shown). Hence, the loss of 20% of Akt protein after 8 h of OGD must be the result of activation of protein degradation. After 24 h reoxygenation, both Akt transcripts and protein levels were at the same level as those found after 8 h of OGD. The inability of reoxygenated JEG-3 cells to increase Akt protein concentration back to control levels may be the result of persistent transcriptional and/or translational inhibition, although phosphorylated eIF-2α had already dropped to an undetectable level. This observation is consistent with the findings in cerebral reperfusion, where PSI is persistent although eIF-2α is completely dephosphorylated (51, 52). Persistent inhibition of the protein synthesis of transcription factor(s) promoting Akt transcription may also provide an explanation as to why akt transcripts failed to return to control level.
eIF-2α phosphorylation is postulated to attenuate global protein translation. However, the concentration of CHOP and ATF4 protein paradoxically increased in response to high phosphorylation of eIF-2α (19, 53). In addition, the protein concentration of an early response gene, c-Fos, was up-regulated in response to tunicamycin-OGD reoxygenation (data not shown), further challenging this assertion. It has been reported that the mRNAs containing small upstream open reading frames within their promoter regions (uORF) or internal ribosome entry sites (IRES sequences) are selectively increased independent of eIF-2α regulation (19, 54, 55). Therefore, PSI is likely to be a selective process in sustained ER stress in ischemic JEG-3 cells.
Activated Akt phosphorylates its target substrates, thereby inhibiting apoptosis (23, 24, 56). Phosphorylation of BAD at serine 136 by Akt facilitates 14–3-3 protein binding and sequestration of BAD in the cytosol, thereby inhibiting the mitochondrial-dependent apoptotic pathway (23, 56). Our data also show a decrease in BAD serine 136 phosphorylation in response to sustained activation of ER stress. In addition, inhibition of Akt activity by LY294002 promoted apoptosis in JEG-3 cells, highlighting the importance of Akt activity in JEG-3 survival. This may also explain why JEG-3 cells maintain a high basal activity of Akt independent of serum withdrawal. Overexpression of a BAD construct induced apoptosis in JEG-3 (unpublished data), further suggesting the essential role Akt-BAD pathways in determining the fate of JEG-3 cells. This finding also provides an additional mechanism by which ER stress may induce mitochondrial-mediated apoptosis. Our unpublished data further support this explanation by demonstrating that cytochrome c is released from mitochondria upon tunicamycin challenge in JEG-3 cells undergoing sustained ER stress after 24 h reoxygenation.
In conclusion, our findings provide a novel role for ER stress in the induction of apoptosis via down-regulation of Akt protein translation.
Acknowledgments
We are grateful to Dr. A. Moffett, Department of Pathology, University of Cambridge, for the gift of the JEG-3 choriocarcinoma cells. We thank Dr. J. N. Skepper of the Multiple Imaging Centre, School of Biological Sciences, for TEM and for taking the photomicrographs. Supported by The Wellcome Trust (069027/Z/02/Z).
REFERENCES
- 1.Roberts JM, Hubel CA. Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet. 1999;354:788–789. doi: 10.1016/S0140-6736(99)80002-6. [DOI] [PubMed] [Google Scholar]
- 2.Burton GJ, Jauniaux E. Placental oxidative stress: from miscarriage to preeclampsia. J. Soc. Gynecol. Investig. 2004;11:342–352. doi: 10.1016/j.jsgi.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Hung TH, Skepper JN, Charnock-Jones DS, Burton GJ. Hypoxia reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ. Res. 2002;90:1274–1281. doi: 10.1161/01.res.0000024411.22110.aa. [DOI] [PubMed] [Google Scholar]
- 4.Martin CB, Jr., McGaughey HS, Jr., Kaiser IH, Donner MW, Ramsey EM. intermittent functioning of the uteroplacental arteries. Am. J. Obstet. Gynecol. 1964;90:819–823. doi: 10.1016/0002-9378(64)90948-2. [DOI] [PubMed] [Google Scholar]
- 5.Brosens JJ, Pijnenborg R, Brosens IA. The myometrial junctional zone spiral arteries in normal and abnormal pregnancies: a review of the literature. Am. J. Obstet. Gynecol. 2002;187:1416–1423. doi: 10.1067/mob.2002.127305. [DOI] [PubMed] [Google Scholar]
- 6.DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J. Neurochem. 2004;91:1–8. doi: 10.1111/j.1471-4159.2004.02703.x. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Oxidative injury to the endoplasmic reticulum in mouse brains after transient focal ischemia. Neurobiol. Dis. 2004;15:229–239. doi: 10.1016/j.nbd.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Benavides A, Pastor D, Santos P, Tranque P, Calvo S. CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia. 2005;52:261–275. doi: 10.1002/glia.20242. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J. Cereb. Blood Flow. Metab. 2005;25:41–53. doi: 10.1038/sj.jcbfm.9600005. [DOI] [PubMed] [Google Scholar]
- 10.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat. Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 11.Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006;38:317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 12.Kimball SR, Jefferson LS. Regulation of protein synthesis by modulation of intracellular calcium in rat liver. Am. J. Physiol. 1992;263:E958–E964. doi: 10.1152/ajpendo.1992.263.5.E958. [DOI] [PubMed] [Google Scholar]
- 13.Wong WL, Brostrom MA, Kuznetsov G, Gmitter-Yellen D, Brostrom CO. Inhibition of protein synthesis and early protein processing by thapsigargin in cultured cells. Biochem. J. 1993;289:71–79. doi: 10.1042/bj2890071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sood R, Porter AC, Ma K, Quilliam LA, Wek RC. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. Biochem. J. 2000;346:281–293. [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002;366:585–594. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ubeda M, Habener JF. CHOP gene expression in response to endoplasmic-reticular stress requires NFY interaction with different domains of a conserved DNA-binding element. Nucleic Acids Res. 2000;28:4987–4997. doi: 10.1093/nar/28.24.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 22.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 24.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 25.Srinivasan S, Ohsugi M, Liu Z, Fatrai S, Bernal-Mizrachi E, Permutt MA. Endoplasmic reticulum stress-induced apoptosis is partly mediated by reduced insulin signaling through phosphatidylinositol 3-kinase/Akt and increased glycogen synthase kinase-3beta in mouse insulinoma cells. Diabetes. 2005;54:968–975. doi: 10.2337/diabetes.54.4.968. [DOI] [PubMed] [Google Scholar]
- 26.Hyoda K, Hosoi T, Horie N, Okuma Y, Ozawa K, Nomura Y. PI3K-Akt inactivation induced CHOP expression in endoplasmic reticulum-stressed cells. Biochem. Biophys. Res. Commun. 2006;340:286–290. doi: 10.1016/j.bbrc.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 27.Yung HW, Tolkovsky AM. Erasure of kinase phosphorylation in astrocytes during oxygen-glucose deprivation is controlled by ATP levels and activation of phosphatases. J. Neurochem. 2003;86:1281–1288. doi: 10.1046/j.1471-4159.2003.01946.x. [DOI] [PubMed] [Google Scholar]
- 28.Shang J, Lehrman MA. Discordance of UPR signaling by ATF6 and Ire1p-XBP1 with levels of target transcripts. Biochem. Biophys. Res. Commun. 2004;317:390–396. doi: 10.1016/j.bbrc.2004.03.058. [DOI] [PubMed] [Google Scholar]
- 29.Virdee K, Xue L, Hemmings BA, Goemans C, Heumann R, Tolkovsky AM. Nerve growth factor-induced PKB/Akt activity is sustained by phosphoinositide 3-kinase dependent and independent signals in sympathetic neurons. Brain Res. 1999;837:127–142. doi: 10.1016/s0006-8993(99)01643-1. [DOI] [PubMed] [Google Scholar]
- 30.Ishihara N, Matsuo H, Murakoshi H, Laoag-Fernandez JB, Samoto T, Maruo T. Increased apoptosis in the syncytiotrophoblast in human term placentas complicated by either preeclampsia or intrauterine growth retardation. Am. J. Obstet. Gynecol. 2002;186:158–166. doi: 10.1067/mob.2002.119176. [DOI] [PubMed] [Google Scholar]
- 31.Yoneda Y, Uehara T, Yamasaki H, Kita Y, Tabuchi M, Mori E. Hospital-based study of the care and cost of acute ischemic stroke in Japan. Stroke. 2003;34:718–724. doi: 10.1161/01.STR.0000056171.55342.FF. [DOI] [PubMed] [Google Scholar]
- 32.Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- 33.Rimbach G, Minihane AM, Majewicz J, Fischer A, Pallauf J, Virgli F, Weinberg PD. Regulation of cell signalling by vitamin E. Proc. Nutr. Soc. 2002;61:415–425. doi: 10.1079/pns2002183. [DOI] [PubMed] [Google Scholar]
- 34.DeGracia DJ. Acute and persistent protein synthesis inhibition following cerebral reperfusion. J. Neurosci. Res. 2004;77:771–776. doi: 10.1002/jnr.20225. [DOI] [PubMed] [Google Scholar]
- 35.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 36.Althausen S, Mengesdorf T, Mies G, Olah L, Nairn AC, Proud CG, Paschen W. Changes in the phosphorylation of initiation factor eIF-2alpha, elongation factor eEF-2 and p70 S6 kinase after transient focal cerebral ischaemia in mice. J. Neurochem. 2001;78:779–787. doi: 10.1046/j.1471-4159.2001.00462.x. [DOI] [PubMed] [Google Scholar]
- 37.Martin de la Vega C, Burda J, Nemethova M, Quevedo C, Alcazar A, Martin ME, Danielisova V, Fando JL, Salinas M. Possible mechanisms involved in the down-regulation of translation during transient global ischaemia in the rat brain. Biochem. J. 2001;357:819–826. doi: 10.1042/0264-6021:3570819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bader M, Muse W, Ballou DP, Gassner C, Bardwell JC. Oxidative protein folding is driven by the electron transport system. Cell. 1999;98:217–227. doi: 10.1016/s0092-8674(00)81016-8. [DOI] [PubMed] [Google Scholar]
- 39.Tu BP, Weissman JS. The FAD- and O2-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell. 2002;10:983–994. doi: 10.1016/s1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- 40.Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell. 2004;15:767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 41.Hossmann KA. Disturbances of cerebral protein synthesis and ischemic cell death. Prog. Brain Res. 1993;96:161–177. doi: 10.1016/s0079-6123(08)63265-3. [DOI] [PubMed] [Google Scholar]
- 42.Clement S, Tasinato A, Boscoboinik D, Azzi A. The effect of alpha-tocopherol on the synthesis, phosphorylation and activity of protein kinase C in smooth muscle cells after phorbol 12-myristate 13-acetate down-regulation. Eur. J. Biochem. 1997;246:745–749. doi: 10.1111/j.1432-1033.1997.t01-2-00745.x. [DOI] [PubMed] [Google Scholar]
- 43.Ricciarelli R, Tasinato A, Clement S, Ozer NK, Boscoboinik D, Azzi A. alpha-Tocopherol specifically inactivates cellular protein kinase C alpha by changing its phosphorylation state. Biochem. J. 1998;334:243–249. doi: 10.1042/bj3340243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi X, Okuma Y, Hosoi T, Nomura Y. Edaravone protects against hypoxia/ischemia-induced endoplasmic reticulum dysfunction. J. Pharmacol. Exp. Ther. 2004;311:388–393. doi: 10.1124/jpet.104.069088. [DOI] [PubMed] [Google Scholar]
- 46.Rajesh KG, Sasaguri S, Suzuki R, Maeda H. Antioxidant MCI-186 inhibits mitochondrial permeability transition pore and upregulates Bcl-2 expression. Am. J. Physiol. 2003;285:H2171–H2178. doi: 10.1152/ajpheart.00143.2003. [DOI] [PubMed] [Google Scholar]
- 47.Ruggero D, Sonenberg N. The Akt of translational control. Oncogene. 2005;24:7426–7434. doi: 10.1038/sj.onc.1209098. [DOI] [PubMed] [Google Scholar]
- 48.Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–446. doi: 10.1038/377441a0. [DOI] [PubMed] [Google Scholar]
- 49.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. U. S. A. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 51.DeGracia DJ, Sullivan JM, Neumar RW, Alousi SS, Hikade KR, Pittman JE, White BC, Rafols JA, Krause GS. Effect of brain ischemia and reperfusion on the localization of phosphorylated eukaryotic initiation factor 2 alpha. J. Cereb. Blood Flow. Metab. 1997;17:1291–1302. doi: 10.1097/00004647-199712000-00004. [DOI] [PubMed] [Google Scholar]
- 52.Kumar R, Krause GS, Yoshida H, Mori K, DeGracia DJ. Dysfunction of the unfolded protein response during global brain ischemia and reperfusion. J. Cereb. Blood Flow. Metab. 2003;23:462–471. doi: 10.1097/01.WCB.0000056064.25434.CA. [DOI] [PubMed] [Google Scholar]
- 53.Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005;24:3470–3481. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 55.Jan E, Thompson SR, Wilson JE, Pestova TV, Hellen CU, Sarnow P. Initiator Met-tRNA-independent translation mediated by an internal ribosome entry site element in cricket paralysis virus-like insect viruses. Cold Spring Harb. Symp. Quant. Biol. 2001;66:285–292. doi: 10.1101/sqb.2001.66.285. [DOI] [PubMed] [Google Scholar]
- 56.Del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]