


Abstract
Growing evidence suggests that the Rad9-Rad1-Hus1 complex (the 9-1-1 complex), besides its functions in DNA damage sensing and signaling pathways, plays also a direct role in various DNA repair processes. Recent studies have demonstrated that the 9-1-1 complex physically and functionally interacts with several components of the base excision repair (BER) machinery namely DNA polymerase β (Pol β), flap endonuclease 1 (Fen 1), DNA ligase I (Lig I) and the MutY homologue of Schizosaccharomyces pombe. In this work, we found for the first time that the 9-1-1 complex interacts in vitro and in vivo with the apurinic/apyrimidinic endonuclease 1 (APE 1), an early component of BER, and can stimulate its AP-endonuclease activity. Moreover, we show that the 9-1-1 complex possesses a stimulatory effect on long patch base excision repair (LP-BER) reconstituted in vitro. The enhancement of LP-BER activity is due to the specific stimulation of the two early components of the repair machinery, namely APE 1 and Pol β, suggesting a hierarchy of interactions between the 9-1-1 complex and the BER proteins acting in the repairosome. Overall, our results indicate that the 9-1-1 complex is directly involved in LP-BER, thus providing a possible link between DNA damage checkpoints and BER.
INTRODUCTION
The mammalian genome suffers from many endogenous and exogenous insults that modify DNA leading to base loss or base alterations. It has been estimated that ∼100 000 modifications occur daily in the DNA of a single cell (1). In order to remove damage and to maintain the integrity of the genome, different DNA repair pathways have evolved. The major repair pathway protecting cells against a single-base DNA damage is base excision repair (BER) (2), although other pathways such as nucleotide excision repair and methyltransferase-mediated error-free repair are also involved (3). BER is often initiated by a damage type specific DNA glycosylase that cleaves the N-glycosydic bond of the damaged base, leaving an apurinic/apyrimidinic site, also referred to as an abasic site or apurinic/apyrimidinic (AP) site (4). Subsequently, AP endonuclease 1 (APE 1) cleaves the sugar-phosphate backbone at the 5′-side of the AP site resulting in 3′-hydroxyl and 5′-deoxyribose phosphate (dRP) groups flanking a one-nucleotide gap (5). DNA polymerase β (Pol β) inserts a first nucleotide into the gap, leaving nicked DNA with a 5′-dRP flap (6). At this point, the repair can be accomplished via two different BER sub-pathways depending on the nature of the AP site (2). In case of regular AP sites, Pol β removes the 5′-dRP group through its associated dRP-lyase activity (7), and the resulting nick is sealed by the DNA ligase III/XRCC1 complex. This sub-pathway is referred to as short-patch BER (SP-BER). However, if the sugar group is oxidized or reduced, Pol β cannot remove the 5′-dRP moiety and repair proceeds through the alternate long-patch BER (LP-BER) sub-pathway involving removal and replacement of 2–10 nucleotides (3,8). It has been found that, in the LP-BER pathway, the collaboration of Pol 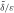 , proliferating cell nuclear antigen (PCNA), replication factor C (RF-C), and flap endonuclease 1 (Fen 1) can displace the strand 3′ to the nick and synthesize up to 10 nucleotides. The resulting flap is cut by Fen 1 and the final nick is sealed by Lig I (9–13). However, growing evidence indicates that Pol β is a key enzyme in BER, that not only initiates both sub-pathways (14) but can also perform efficient strand displacement via the ‘hit and run’ mechanism (4). In this model Pol β and Fen 1 act successively, followed by the action of Lig I.
, proliferating cell nuclear antigen (PCNA), replication factor C (RF-C), and flap endonuclease 1 (Fen 1) can displace the strand 3′ to the nick and synthesize up to 10 nucleotides. The resulting flap is cut by Fen 1 and the final nick is sealed by Lig I (9–13). However, growing evidence indicates that Pol β is a key enzyme in BER, that not only initiates both sub-pathways (14) but can also perform efficient strand displacement via the ‘hit and run’ mechanism (4). In this model Pol β and Fen 1 act successively, followed by the action of Lig I.
DNA repair is coordinated with cell-cycle progression (1,15) and with DNA-damage checkpoints (16). DNA-damage checkpoint pathways are activated at specific points during the cell cycle when the integrity of DNA is examined before progression to the next cell-cycle phase is allowed. The checkpoint response is a signal transduction cascade, where sensor proteins detect a lesion in the double helix and stimulate several effectors through the activity of different protein kinases. This may lead to temporary cell cycle arrest, slowing down of DNA replication, changes in the cellular transcriptional program, chromatin remodeling, induction of DNA repair genes, and occasionally to apoptosis (17,18). Some of the major checkpoint proteins namely ataxia telangectasia mutated protein (ATM), ATM-related protein (ATR), ATR interacting protein (ATRIP), Rad17, Rad9, Rad1, and Hus1, are thought to be involved in sensing and triggering DNA repair processes. Among those, the three human proteins Rad9, Hus1, and Rad1 form a heterotrimeric complex (the 9-1-1 complex) which exhibits structural similarity with the homotrimeric clamp formed by PCNA (19–21). The 9-1-1 complex has been characterized as a sensor of DNA damage and is targeted to damage sites following genotoxic stress (17,22–27). In addition, Rad17 associates with the four small subunits of the heteropentameric RF-C complex in a manner similar to the RF-C complex (20,21,28–30). Several studies showed that the 9-1-1 complex and Rad17-RF-C2-5 function as a clamp/clamp–loader pair, similarly to PCNA and RF-C (22,23,27,31,32). Moreover, the 9-1-1 complex, Rad17-RF-C2−5 and PCNA co-localize in foci formed upon DNA damage (22,33,34). These data suggested a mechanism in which Rad17- RF-C2−5 would recognize DNA lesions, allowing the recruitment of the 9-1-1 complex to those sites. ATM and ATR kinases are recruited simultaneously to the same sites of DNA damage but in a 9-1-1 complex and Rad17-RFC2−5 independent manner (35,36). The current model suggests that the 9-1-1 complex may facilitate the recruitment of the checkpoint effector kinase Chk1 (24,26) that is subsequently phosphorylated by the ATR/ATM kinases (17). Additionally, it has been recently proposed by two different groups, that the 9-1-1 complex and the Rad17–RFC2−5 alternative clamp loader could stabilize stalled replication forks (23,27).
Recently several new links between DNA damage checkpoints and various DNA repair processes have been discovered (34,37,38). Moreover, recent investigations showed several possible connections between the human 9-1-1 complex and the BER pathway (Table 1). First, a physical and functional interaction of the 9-1-1 complex with Pol β revealed that the 9-1-1 complex has a specific stimulatory effect on Pol β activity (39). Pol β stimulation resulted in an increase in its affinity for the primer-template. Interaction with the 9-1-1 complex stimulated DNA strand displacement synthesis raising the possibility that the 9-1-1 complex might attract Pol β to DNA-damage sites, thus connecting directly checkpoints and DNA repair. Very recently, we showed that the 9-1-1 complex interacts with and stimulates Lig I (40,41). In addition, similar physical and functional interactions with the 9-1-1 complex were identified for Fen 1 (42,43) and for the MutY DNA glycosylase homolog (44,45). In summary, these results suggested that the 9-1-1 complex could act as a recruiting platform for different factors involved in LP-BER.
Table 1.
Functional consequences of the 9-1-1 complex interactions with components of the LP-BER machinery
| LP-BER component | Functional consequence | References |
|---|---|---|
| MutY DNA glycosylase | Stimulation of glycosylase activity | (44,45) |
| APE 1 | Stimulation of AP endonuclease activity | Present study |
| Pol β | Stimulation of polymerase and strand displacement activities | (39) |
| Fen 1 | Stimulation of endo- and exonuclease activities | (42,43) |
| Lig I | Stimulation of ligase activity | (40,41) |
In order to get more insight into the functional consequences of these interactions, we have reconstituted LP-BER in vitro by using a duplex oligonucleotide substrate containing a lesion (a tetrahydrofuran (THF) moiety) that mimics a reduced AP site and the four human proteins APE 1, Pol β, Fen 1, and Lig I. Our results suggest for the first time that the 9-1-1 complex specifically stimulates the endonuclease activity of APE 1 in a single-enzyme assay as well as in the reconstituted LP-BER in vitro. Further analysis revealed that the 9-1-1 complex physically interacts with APE 1 in vitro as well as in vivo. We also present evidence that the 9-1-1 complex specifically stimulates LP-BER in vitro through the stimulation of strand displacement activity of Pol β. Importantly, under conditions applied in the reconstituted LP-BER assay, we could not detect any effect of the 9-1-1 complex on the enzymatic activities of Fen 1 and Lig I. Our data suggest that in a reconstituted LP-BER in vitro system, a hierarchy of interactions between the 9-1-1 complex and the components of the LP-BER repairosome exists.
MATERIALS AND METHODS
Chemicals
[γ-32P]ATP (3000 Ci/mmol), [α-32P]dTTP (3000 Ci/mmol) and Sephadex G-25 spin column were from Amersham Biosciences; unlabeled dNTPs were from Roche Molecular Biochemicals. Oligonucleotides were from Microsynth (Switzerland). The THF (dSpacer) was from Glen Research. All other reagents were of analytical grade and were purchased from Merck or Fluka.
Nucleic acid substrates
The 100 mer oligonucleotide containing the synthetic (THF) AP site as well as the complementary strand and the corresponding primers were chemically synthesized and purified on denaturing polyacrylamide gel. The sequence of 100 mer with THF moiety is as follows: 5′ATCCTGATTGCTATCTGAATATGGTGGTGGTGGGCGCCGGCG(X/G)TGTGAATTCGGCACTGGCCGTCATCGTGATCTATCTTACAGTATGCTCTTGGTTGTA3′
In brackets, the position of the lesion located 43 from the 5′ end or the corresponding G residue in the undamaged template is indicated in bold letters (X, THF moiety). This double stranded substrate was used for the single enzyme assays as well as for the reconstituted LP-BER in vitro. Alternatively the ends of the substrate were blocked with biotin by annealing a THF moiety-containing strand to the complementary one, containing biotin at the 3′ and 5′ end respectively. For the pol β assay the substrate contained an additional 1 nucleotide gap; for the Fen 1 assay a 10 nt flap (5′-ATCTGATCGC) and for Lig I a nick. The structures of these substrates are depicted in Figure 1B.
Figure 1.

Proteins and substrates used in reconstitution of long patch base excision repair in vitro. (A) Recombinant proteins were purified as described in ‘Materials and Methods’ and separated on a 8–20% gradient SDS-PAGE gel, and stained with Coomassie Blue. Lane 1: molecular weight markers; lane 2: APE 1 (2 μg); lane 3: Pol β (2 μg); lane 4: Fen 1 (2 μg); lane 5: Lig I (2 μg); lane 6: 9-1-1 complex (6 μg). (B) Schematic representation of the 32P-5′-labeled oligonucleotide substrates used in the study: a 100 bp duplex oligonucleotide containing a THF moiety at the position 43 was used for repair reactions, the ends of the substrate were either free (unblocked substrate) or blocked with a biotin at each end (blocked substrate); a 100 bp duplex oligonucleotide with a 1 nucleotide gap at the same position was used for the Pol β assay; with a nick for the Lig I assay and with a 10 nucleotide flap for the Fen 1 reaction
Proteins and antibodies
Human AP endonuclease 1 was obtained from Enzymax LLC. Bovine serum albumin (BSA) was purchased from New England BioLabs. The 9-1-1 complex was isolated by co-expressing in Sf21 cells the three baculoviruses encoding the recombinant hRad1, his-hRad9, and hHus1. The complex was subsequently purified as previously described (39). Human PCNA was produced in Escherichia coli using the plasmid pT7/hPCNA and purified to homogeneity as described (46). Human recombinant Pol β, Fen 1, and Lig I were expressed in E. coli and purified as previously described (47–49). The anti-Hus1 and anti-Rad9 antibodies described in Toueille et al. (39) were a gift of R. Freire (Teneriffe, Spain). The goat anti-Rad1 antibody (N18), as well as the rabbit anti-APE 1 (Anti-Ref1, H300) antibody were from Santa Cruz biotechnology.
Enzymatic assays
APE 1 incision assay. A 100 bp duplex oligonucleotide substrate was prepared as follows: a 100- mer oligonucleotide containing THF moiety at the position 43 was annealed to the complementary strand (Figure 1B). Prior to annealing a lesion containing strand was 5′-end labeled with T4 polynucleotide kinase and [γ-32P]ATP. Unincorporated labeled nucleotides were removed on a Sephadex G-25 spin column. APE 1 incision reactions were carried out in a reaction mixture (10 μl) containing 45 mM Hepes/KOH (pH 7.8), 70 mM KCl, 7.5 mM MgCl2, 2 mM dithiothreitol, 0.5mM EDTA, 0.1mg/ml BSA, 2 mM ATP, 50 fmol of oligonucleotide substrate, and the indicated amounts of APE 1. The ends of the oligonucleotide substrate were either free or blocked with biotin as indicated in the Figure legends. Reactions were incubated at 37°C for 20 min and stopped by adding an equal volume of gel loading buffer (95% formamide, 20 mM EDTA, 0.02% bromophenol blue, and 0.02% xylene cyanol). Following incubation at 100°C for 5 min, the reaction products were separated by electrophoresis on a 10% denaturing polyacrylamide gel and visualized by autoradiography. To determine the effect of the 9-1-1 complex on APE 1 activity, the reactions were performed as described above, with the exception that constant amounts of APE 1 were included in the reaction mixture and increasing amount of the 9-1-1 complex were added as indicated in Figure 4A.
Figure 4.

The 9-1-1 complex specifically stimulates the endonuclease activity of APE 1 in vitro. The APE 1 incision assay was performed as described in ‘Materials and Methods’. Reactions were stopped by adding an equal volume of formamide-dye solution and products were analyzed on a 10% denaturing polyacrylamide gel. (A) An APE 1 reaction mixture (10 μl) contained (besides all components described in ‘Materials and Methods’) 32P-5′-labeled 100 bp duplex oligonucleotide (50 fmol see Figure 1B), APE 1 (2 fmol). Reactions were incubated for 20 min at 37°C with the indicated amounts of the 9-1-1 complex. (B) As A but with the blocked substrate (50 fmol). (C) Quantification of the stimulation of APE 1 endonuclease cleavage by the 9-1-1 complex on the substrate with free ends (closed circles) and with the ends blocked with biotin (open circles). The values represent the mean of three independent experiments. The error bars correspond to the standard error of the mean.
Pol β assay
A 42 mer oligonucleotide was radiolabeled at 5′ end and annealed with a 57 mer downstream oligonucleotide to the complementary 100 mer strand. The resulting 32P-labeled duplex oligonucleotide has a 1-nt gap at the position 43 (Figure 1B). Pol β DNA synthesis was determined by measuring nucleotide insertion into the DNA substrate described above. Reactions were carried out in a final volume of 10 μl containing 45 mM Hepes/KOH (pH 7.8), 70 mM KCl, 7.5 mM MgCl2, 2mM dithiothreitol, 0.5mM EDTA, 0.1mg/ml BSA, 2 mM ATP, 20 μM each of dATP, dCTP, dGTP, dTTP, 50 fmol of gapped oligonucleotide substrate, and the indicated amounts of Pol β. Reactions were incubated at 37°C for 20 min and stopped by adding an equal volume of gel loading buffer (95% formamide, 20 mM EDTA, 0.02% bromophenol blue,and 0.02% xylene cyanol). Following incubation at 100°C for 5 min, the reaction products were separated by electrophoresis on a 10% denaturing polyacrylamide gel and visualized by autoradiography.
Pol β activity assay on the lesion-containing substrate
Pol β activity on the 32P-5′-labeled, duplex oligonucleotide substrate containing THF moiety at the position 43 was assayed in the presence of a constant amount of APE 1. Reactions were incubated for 20 min at 37°C in the reaction buffer (10 μl) that contained 45 mM Hepes/KOH (pH 7.8), 70 mM KCl, 7.5 mM MgCl2, 2 mM dithiothreitol, 0.5 mM EDTA, 0.1mg/ml BSA, 2 mM ATP, 20 μM each of dATP, dCTP, dGTP, dTTP, 50 fmol of THF containing oligonucleotide substrate, 55.5 fmol of APE 1, and the indicated amounts of Pol β. Following incubation, reactions were stopped by adding an equal volume of gel loading buffer (95% formamide, 20 mM EDTA, 0.02% bromophenol blue, and 0.02% xylene cyanol), heated at 100°C for 5 min, and the products were separated by electrophoresis on a 10% denaturing polyacrylamide gel and visualized by autoradiography.
Fen 1 Endonuclease Assay
The Fen 1 assay was performed as described with some modifications (42). To prepare a flap substrate, 42 and 68 mer oligonucleotides were annealed to the complementary 100 mer strand. Prior to annealing a 68 mer oligonucleotide was radiolabeled at the 3′ end with terminal deoxynucleotidyltransferase and [α-32P]ddATP. A 32P-Labeled duplex oligonucleotide, thus prepared, had a 10-nt flap at the position 43 (Figure 1B). The Fen 1 cleavage assay was carried out in the reaction mixture (10 μl) containing 45 mM Hepes/KOH (pH 7.8), 70 mM KCl, 7.5 mM MgCl2, 2 mM dithiothreitol, 0.5 mM EDTA, 0.1 mg/ml BSA, 2 mM ATP, 50 fmol of flap oligonucleotide substrate and, the indicated amounts of Fen 1. Reactions were incubated at 37°C for 20 min and stopped by adding an equal volume of gel loading buffer (95% formamide, 20 mM EDTA, 0.02% bromophenol blue, and 0.02% xylene cyanol). Following incubation at 100°C for 5 min, the reaction products were separated by electrophoresis on a 10% denaturing polyacrylamide gel and visualized by autoradiography. In order to measure the stimulatory effect of the 9-1-1 complex on Fen 1 endonuclease activity the reactions were performed under the same conditions, except that constant amounts of Fen 1 were present in the reaction buffer (25 fmol) and the indicated amounts of the 9-1-1 complex were added as indicated in Figure 8C.
Figure 8.

The 9-1-1 complex has no effect on the activities of Fen 1 and Lig I in long patch base excision repair in vitro. The LP-BER in vitro reaction was performed as described in ‘Materials and Methods’. Reactions were stopped by adding an equal volume of formamide-dye solution and products were analyzed on a 10% denaturing polyacrylamide gel. (A) The reaction mixtures (10 μl) contained (besides all components described in Materials and Methods) 32P-5′-labeled 100 bp duplex oligonucleotide (50 fmol), APE 1 (55 fmol), Pol β (64 fmol), Fen 1 (9 fmol), and of Lig I (245 fmol). Reactions were incubated for 20 min at 37°C with the indicated amounts of the 9-1-1 complex. (B) The reaction mixtures (10 μl) contained (besides all components described in Materials and Methods) 32P-5′-labeled 100 bp duplex oligonucleotide (50 fmol), APE 1 (55 fmol), Pol β (64 fmol), Fen 1 (93 fmol), and Lig I (25 fmol). Reactions were incubated for 20 min at 37°C with the indicated amounts of the 9-1-1 complex. (C) A Fen 1 reaction mixture (10 μl) contained (besides all components described in Materials and Methods) a 10 nucleotide flap substrate (50 fmol, see Figure 1B) and Fen 1 (25 fmol). Reactions were incubated for 20 min at 37°C with the indicated amounts of the 9-1-1 complex. (D) A Lig I reaction mixture (10 μl) contained (besides all components described in Materials and Methods) a nicked oligonucleotide (50 fmol, see Figure 1B) and Lig I (0.5 fmol). Reactions were incubated for 20 min at 37°C with the indicated amounts of the 9-1-1 complex.
Lig I Assay
The Lig I assay was performed as described with modifications (40) by using a 42 mer and 32P-5′-labeled 58 mer oligonucleotides annealed to the complementary 100 mer strand. Reaction mixtures (10 μl) contained 45 mM Hepes/KOH (pH 7.8), 70 mM KCl, 7.5 mM MgCl2, 2 mM dithiothreitol, 0.5 mM EDTA, 0.1 mg/ml BSA, 2 mM ATP, 50 fmol of a nicked oligonucleotide substrate, and the indicated amounts of Lig I. Reactions were incubated at 37°C for 20 min and stopped by adding an equal volume of gel loading buffer (95% formamide, 20 mM EDTA, 0.02% bromophenol blue, and 0.02% xylene cyanol). Following incubation at 100°C for 5 min, the reaction products were separated by electrophoresis on a 10% denaturing polyacrylamide gel and visualized by autoradiography. To measure the stimulatory effect of the 9-1-1 complex on Lig I activity the reactions were performed under the same conditions except that constant amounts of Lig I were present in the reaction buffer (0.5 fmol) and the indicated amounts of the 9-1-1 complex were added as indicated in Figure 8D.
Reconstituted in vitro LP-BER
The LP-BER assay was performed as described previously with modifications (14,50). The complete repair reactions were carried out in the reaction buffer (10 μl) that included: 45 mM Hepes/KOH (pH 7.8), 70 mM KCl, 7.5 mM MgCl2, 2 mM dithiothreitol, 0.5 mM EDTA, 0.1 mg/ml BSA, 2 mM ATP, 20 μM each o dATP, dCTP, dGTP, dTTP, and 50 fmol of 32P-5′-labeled 100 bp duplex oligonucleotide substrate containing a THF moiety at the position 43. The ends of the oligonucleotide substrate were either free or blocked with biotin as indicated in the Figure legends. For the reconstitution of LP-BER using unlabeled substrate the same conditions were used with the exception that the concentration of dTTP was reduced to 8 μM and [α-32P] dTTP (2.5 μCi) was added to the reaction mixtures. The reactions were initiated by the addition of purified APE 1 (55 fmol), Pol β (64 fmol), Fen 1 (93 fmol), and Lig I (245 fmol). Increasing amounts of the 9-1-1 complex were added to the reconstituted LP-BER in vitro reaction under limiting conditions for each of the four enzymes APE 1, Pol β, Fen 1, and Lig I (see ‘Results’). After incubation at 37°C for 20 min, the reactions were stopped by adding an equal volume of gel loading buffer (95% formamide, 20 mM EDTA, 0.02% bromophenol blue, and 0.02% xylene cyanol), heated at 100°C for 5 min, and the final products were separated by electrophoresis on a 10% denaturing polyacrylamide gel and visualized by autoradiography.
Pulldown assays
For the His-pulldowns, 6 μg of the his-tagged 9-1-1 complex or the his-tagged subunits Rad9, Rad1 or Hus1 were incubated with 1 μg APE 1 for 2 h at 4°C in pulldown buffer [50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 0.05% (v/v) NP-40]. Ni2+ beads, previously coated by overnight incubation in pulldown buffer containing 5 mg/ml BSA, followed by three washes in pulldown buffer, were then added to the proteins. The samples were subsequently incubated at 4°C for 1 h. After washing five times in pulldown buffer containing 5 mM imidazole, the beads were heated for 5 min at 95°C in Laemmli buffer and the co-precipitated proteins were analyzed by western blot using the corresponding antibodies according to established methods.
For the APE 1-sepharose pulldowns, purified APE 1 or BSA, used as a negative control, was covalently coupled to CNBr-activated Sepharose (Amersham Pharmacia Biotech) as described by the supplier, at a final ratio of 1 μg of protein/μl of beads. APE 1-sepharose or BSA-sepharose (10 μl) were subsequently washed once in pulldown buffer containing 10 mg/ml BSA and three times in pulldown buffer. The beads were subsequently incubated with 300 ng of his9-1-1 complex for 2 h in pulldown buffer at 4°C. After washing five times in pulldown buffer, the beads were heated for 5 min at 95°C in Laemmli buffer and the co-precipitated proteins were analyzed by western blot using the corresponding antibodies according to established methods.
Whole cell extracts
For preparing total cell extracts, 7×106 293T cells were harvested by trypsinisation followed by centrifugation. The cell pellet was subsequently lysed in cell lysis buffer [50 mM Hepes-KOH, pH 7.5, 400 mM NaCl, 1 mM DTT, 2.5 mM MgCl2, 20% glycerol, 0.5% (v/v) NP40, 2 μg/ml leupetpin, 1 μg/ml bestatin, 1 μg/ml pepstatin, 2 mM PMSF, 10 mM glycerophosphate, 1 mM NaF, 10 mM Na4P2O7] for 15 min at 4°C. The cell lysate was subsequently centrifuged for 15 min at 10 000 rpm, the supernatant was collected and kept as a total cell extract. The protein concentration was determined by using the Bradford assay.
Immunoprecipitations
For immunoprecipitations, 25 μl of protein G sepharose were coated for 3 hours at 4°C with 100 μg BSA in IP buffer (40 mM Hepes-KOH, pH 7.5, 100 mM NaCl, 8 mM MgCl2, 2 μg/ml leupetpin, 1 μg/ml bestatin, 1 μg/ml pepstatin, 2 mM PMSF, 10 mM glycerophosphate, 1mM NaF, 10 mM Na4P2O7) and subsequently incubated overnight with 3 μg of anti-Rad9 antibody or the unimmunized rabbit IgG. After three washes in IP buffer, the beads were incubated with 1mg of 293 T total cell extract for 3 h at 4°C. After incubation, the beads were washed three times in IP buffer containing 0.05% NP40 and subsequently heated for 5 min at 95°C in Laemmli buffer. The co-precipitated proteins were analyzed by western blot using the corresponding antibodies according to established methods.
RESULTS
Proteins and substrates used in this study and their requirements in long patch base excision repair
The 9-1-1 complex has been shown to interact and stimulate the activity of several LP-BER factors in vitro (Table 1). Therefore, our aim was to determine if the 9-1-1 complex could also stimulate the complete repair of an abasic site through the stimulation of different components of LP-BER. To address this question, we first reconstituted LP-BER in vitro using a set of the four different protein components APE 1, Polβ, Fen 1, and Lig I, all of which were highly purified from recombinant sources (Figure 1A). First they were tested for their enzymatic activities as described in ‘Material and Methods’. As a substrate for all repair reactions we used a 100 bp oligonucleotide duplex containing a THF moiety at the position 43, which resembles a reduced abasic site (AP site) (Figure 1B). The requirement of the different components of LP-BER was tested as shown in Figure 2B. In this experiment we used a THF-containing substrate labeled at its 5′ end. For this reason the substrate (Figure 2 lane 8) and the product of the reaction (Figure 2 lane 7) showed the same migration. However, when deoxyribonucleotides or Pol β were absent from the reaction (lane 2 and 4, respectively) the product corresponded to the DNA substrate cleaved by APE 1. Subsequently, the reaction performed in the absence of Fen 1 showed an efficient strand displacement synthesis performed by Pol β (Figure 2 lane 5). However, the resulting flap-structure intermediates could not be ligated by Lig I to generate the fully repaired product. When Lig I was excluded from the reaction mixture, Pol β synthesized up to 6 nucleotides, which led to the generation of the flap structure, subsequently removed by Fen 1 cleavage (Figure 2B, lane 6). This limited DNA synthesis (compare lanes 5 and 6) reflects the effect of Fen 1 on Pol β strand displacement synthesis, illustrating the previously described so-called ‘hit and run’ mechanism (4). Surprisingly, incubation of the substrate with all four protein components but without addition of ATP resulted in the complete repair of the damaged strand even though ATP is known to be required for Lig I activity (Figure 2B, lane 1). The explanation for this observation is that a fraction of Lig I used in this assay might have been purified in its pre-adenylated form, thus allowing ligation to occur in the absence of ATP. As expected, omission of APE 1 resulted in the absence of cleavage and of the subsequent LP-BER reaction. This results show that the reconstituted LP-BER reaction is dependent on the presence and proper activity of all required enzymatic components, hence demonstrating that all the steps of the reconstituted reaction are functional. Incubation of the oligonucleotide substrate in the reaction buffer alone (Figure 2B, lane 8) or with addition of the maximal amount of the 9-1-1 complex used in the study, did not show any enzymatic contamination of the purified 9-1-1 checkpoint clamp.
Figure 2.

Protein requirements for the long patch base excision repair in vitro system. (A) Schematic representation of the 32P-5′-labeled oligonucleotide substrates used. (B) The LP-BER in vitro reaction was performed as described in ‘Materials and Methods’. The complete reaction contained a 100 bp duplex oligonucleotide (50 fmol) with a THF moiety at the position 43, APE 1 (55 fmol), Pol β (64 fmol), Fen 1 (93 fmol) and Lig I (245 fmol). Lane 1: ATP omitted; Lane 2: dNTPs omitted; Lane 3: APE 1 omitted; lane 4: Pol β omitted; lane 5: Fen 1 omitted; lane 6: Lig I omitted; lane 7: complete reaction; lane 8: the 100 bp duplex oligonucleotide (50 fmol) was incubated in the reaction buffer alone and (lane 9) with 1 pmol of the 9-1-1 complex. After incubation reactions were stopped by adding an equal volume of formamide-dye solution and products were analyzed on a 10% denaturing polyacrylamide gel.
Fine-tuning of the different steps of long patch base excision repair in vitro
Next, in order to study the effect of the 9-1-1 complex on the LP-BER machinery, each protein component was titrated against all the others (Figure 3). This allowed us to determine the amount of enzyme that significantly reduces repair at any given step, therefore serving as a limiting factor of the total reaction. First, the repair was performed by adding increasing amounts of APE 1 to the reaction mixture, which contained saturating amounts of Pol β, Fen 1 and Lig I (Figure 3A). In order to avoid the problem of the substrate and the BER reaction product running at the same position on the gel, the substrate remained unlabeled in this experiment, and the repair was visualized by the incorporation of [α-32P] dTTP, the first nucleotide incorporated opposite A after the lesion. The minimal amount of APE 1 that incised the damaged strand and allowed repair to take place although at a low level, was 14-fold lower than the amount of enzyme required for the complete repair (Figure 3A, compare 5 and 55 fmols). Incubation of 5′-32P labeled substrate with saturating amounts of APE 1, Fen 1, and Lig I and increasing amounts of Pol β then allowed determining the amount of enzyme performing a limited strand displacement DNA synthesis (15 fmol, Figure 3B). That was approximately 4-fold less as compared with the amount of Pol β used to obtain complete repair (compare to Figure 3B, 50 fmols). To determine the fine-tuning of Fen 1 and Lig1 activities in LP-BER, both enzymes were titrated in the presence of 5′-32P labeled substrate. All the proteins were present in saturating amounts in the reaction mixture except of Fen 1 (Figure 3C) and Lig I (Figure 3D), respectively. For further experiments we chose amounts of Fen 1 (Figure 3C, compare 20 and 100 fmols) and Lig I (Figure 3D, compare 25 and 250 fmols) that were 10-fold decreased as compared with the level required for the complete LP-BER.
Figure 3.

Fine-tuning of the different enzymes in the long patch base excision repair in vitro. The LP-BER in vitro reaction was performed as described in ‘Materials and Methods’. All reactions were stopped by adding an equal volume of formamide-dye solution and products were analyzed on a 10% denaturing polyacrylamide gel. (A) The reaction mixtures (10 μl) contained (besides all components described in ‘Materials and Methods’) unlabeled 100 bp duplex oligonuleotide (50 fmol), [α-32P] dTTP (2.5 μCi), Pol β (64 fmol), Fen 1 (93 fmol), Lig I (245 fmol) and the indicated amounts of APE 1. Reactions were incubated for 20 min at 37°C. (B) The reaction mixtures (10 μl) contained (besides all components described in ‘Materials and Methods’) 32P-5′-labeled 100 bp duplex oligonucleotide (50 fmol), APE 1 (55 fmol), Fen 1 (93 fmol) and Lig I (245 fmol). Reactions were incubated for 20 min at 37°C with the indicated amounts of Pol β. (C) The reaction mixtures (10 μl) contained (besides all components described in ‘Materials and Methods’) 32P-5′-labeled 100 bp duplex oligonucleotide (50 fmol), APE 1 (55 fmol), Pol β (64 fmol), Lig I (245 fmol) and increasing amounts of Fen 1. Reactions were incubated for 20 min at 37°C. (D) The reaction mixtures (1 μl) contained (besides all components described in ‘Materials and Methods’) 32P-5′-labeled 100 bp duplex oligonucleotide (50 fmol), of APE 1 (55 fmol) of Pol β (64 fmol), Fen 1 (93 fmol) and indicated amounts of Lig I. Reactions were incubated for 20 min at 37°C.
The Rad9-Rad1-Hus1 complex specifically stimulates the endonuclease activity of APE 1 without encircling the DNA substrate
Previous studies have shown that the 9-1-1 complex stimulates the activities of Pol β, Fen 1, and Lig I on the substrates that mimic transient intermediates of the respective steps of LP-BER. We sought to determine whether the 9-1-1 complex exerts the same effect on the activity of APE 1. Incubation of 5′-32P labeled, THF-containing substrate with APE 1 and increasing amounts of the 9-1-1 complex led to a 6-fold stimulation of the APE 1 endonuclease activity, as shown by the accumulation of APE 1 cleavage product (42 nucleotides) (Figure 4A, 45 and 450 fmols and Figure 4C). Moreover, in control experiments this stimulation was not observed when the substrate and APE 1 were incubated with increasing amounts of either PCNA or BSA, thus confirming the specificity of this stimulation (data not shown). Next we tested whether the loading of the 9-1-1 complex onto DNA is necessary for the observed stimulatory effect. It has been shown that 9-1-1 complex can be loaded onto DNA in vitro in a Rad17-RFC(2−5) dependent manner (22,23,27,31,32). In order to determine if the 9-1-1 complex has to encircle DNA to stimulate APE 1 activity, we used a THF-containing substrate with both ends blocked with biotin. Efficient blockage of the ends was checked in a pol δ stimulation assay in the presence of PCNA, and resulted in a complete abolishment of PCNA dependent pol δ activity (data not shown). As shown in Figure 4B, incubation of 5′-32P labeled, blocked substrate with APE 1 and increasing amounts of the 9-1-1 complex (Figure 4B, 45 and 900 fmols) led to a similar extent of APE 1 stimulation as compared with the reactions performed in the presence of unblocked substrate (Figure 4C). Thus, we concluded that the loading of the checkpoint clamp onto DNA is not required to stimulate APE 1 endonuclease cleavage in vitro.
The 9-1-1 complex physically interacts with APE 1 in vitro and in vivo
Next, in order to assess the relevance of the observed stimulation of APE 1 by the 9-1-1 complex, we investigated the physical interaction of the two proteins. We first performed pulldown experiments in order to determine whether the two proteins interact specifically and directly in vitro (Figure 5A). By incubation of APE 1-bound sepharose beads with purified 9-1-1 complex, we were able to show that the 9-1-1 complex was interacting with APE 1 as Rad9, Rad1 and Hus1 were detected in the pulled-down fraction (Figure 5A, left panel). Next, to confirm these data, his-9-1-1 complex was incubated together with APE 1 followed by binding of the 9-1-1 complex to Ni2+ beads via the his-Rad9 subunit (Figure 5A, right panel). We were first able to show that the three subunits hisRad9, Rad1, and Hus1, indeed form a complex under those conditions, as the three subunits were co-precipitated with the Ni2+ beads in the presence or absence of APE 1 (Figure 5A, right panel and data not shown), although only Rad9 possessed a his-tag. In this pulldown, APE 1 was also co-precipitated with the 9-1-1 complex. Hence, these results demonstrate for the first time a direct physical interaction of the 9-1-1 complex with APE 1.
Figure 5.

The 9-1-1 complex physically interacts with APE 1 in vitro and in vivo. (A) Direct interaction of the 9-1-1 complex with APE 1 in vitro. APE 1-sepharose pulldowns (right panel) were performed by incubating APE 1-sepharose beads, or BSA-sepharose as a negative control, together with purified his9-1-1 complex as described in ‘Materials and Methods’. His9-1-1 pulldowns (left panel) were performed by incubating purified his9-1-1 complex with purified APE 1 and subsequent binding to Ni2+ beads. Five percent of the pulldown were used to check the presence of all the subunits of the 9-1-1 complex (his-Rad9, Rad1 and Hus1), and the remaining sample was used to detect co-precipitated APE 1 by SDS-PAGE followed by western blot analysis. Input represents 2% of the total amount of interacting protein used in the pulldown experiments. (B) Physical interaction of APE 1 with the 9-1-1 complex subunits. His-pulldowns were performed by using the individual subunits his-Rad1, his-Rad9, and his-Hus1 as described in A. (C) The 9-1-1 complex interacts with APE1 in vivo. Immunoprecipitation experiments were performed as described in ‘Material and Methods’ by incubating 293T total cell extracts with anti-human Rad9 antibody. Presence of immuno-precipitated Rad9 and co-precipitated APE1 were analyzed by SDS-PAGE followed by western blot analysis. The lane IP IgG contains the control immunoprecipitation performed in the presence of un-immunized rabbit IgG. Input represents 5% of the amount of extract used for immunoprecipitation. Arrows indicate the positions of endogenous APE1 and Rad9. The bands indicated by asterisks correspond to the antibody heavy and light chain respectively.
We next investigated whether APE 1 was able to interact with all three subunits of the 9-1-1 complex, as previously shown for the other LP-BER proteins (39–43). To address this question we performed pulldown experiments using his-tagged Rad1, Rad9, or Hus1 incubated with APE 1 and subsequently bound to Ni2+ beads (Figure 5B). As revealed by western-blot against APE 1, APE 1 was able to interact with the three separated subunits of the 9-1-1 complex, although with a weaker intensity for Rad1.
Finally we tested whether the interaction of the 9-1-1 complex with APE 1 could also be detected in cell extracts, in order to confirm its in vivo relevance. Indeed by performing co-immunoprecipitation with an anti-human Rad9 antibody we were able to detect APE 1 in the immuno-precipitated fraction, whereas it was not detectable in the negative control performed with un-immunized rabbit IgG (Figure 5C, bottom). The immuno-precipitation of Rad9 was checked by detecting of one of its phosphorylated forms, known to be present in untreated cells (19), that migrated above the heavy IgG chains, whereas the main band corresponding to the non-phosphorylated form was not visible due to its migration at the same level than the heavy IgG chains (Figure 5C, top). In addition, the same experiment performed with HeLa cell extracts gave similar results (data not shown). We therefore concluded that the interaction of the 9-1-1 complex with APE 1 also occurs in human cells, thus supporting an in vivo role for this interaction.
The Rad9-Rad1-Hus1 complex specifically stimulates the endonuclease activity of APE 1 in LP-BER in vitro
Having established that APE 1 can be stimulated by the 9-1-1 complex via a direct interaction, an important question arose; namely if the 9-1-1 complex is able to enhance the activity of APE 1 directly in the reconstituted LP-BER reaction. To address this question we performed a LP-BER assay where APE 1 was the limiting factor of the reaction (Figure 6B, compare 4 and 70 fmols of APE 1). Indeed, addition of increasing amounts of the 9-1-1 complex to the reaction mixture enhanced the yield of repair up to 4-fold (Figure 6B, 45–450 fmols). In contrast, PCNA and BSA had no significant effect on the repair efficiency (Figure 4C, D and E, respectively). In addition, we tested if the blockage of the substrate ends influenced the reported stimulation. We observed that the 9-1-1 complex was able to stimulate APE 1 activity without being loaded onto DNA in the reconstituted BER reaction, as well as in the single enzyme assay (Figure 6F). Thus, our results demonstrate for the first time that the 9-1-1 complex specifically stimulates LP-BER in vitro via stimulation of the endonucleolytic cleavage by APE 1.
Figure 6.

The 9-1-1 complex specifically stimulates the endonuclease activity of APE 1 in LP-BER in vitro. (A) Schematic representation of the substrate used in the LP-BER reaction. (B) LP-BER assay was performed as described in ‘Materials and Methods’ The reaction mixtures (10 μl) contained (besides all components described in ‘Materials and Methods’) unlabeled 100 bp duplex oligonucleotide (50 fmol), [α-32P] dTTP (2.5 μCi), APE 1 (4 fmol), Pol β (64 fmol), Fen 1 (93 fmol), and Lig I (245 fmol). Reactions were incubated for 20 min at 37°C with the indicated amounts of the 9-1-1 complex, (C) as B but with the indicated amounts of PCNA (D) as B but with the indicated amounts of BSA. (E) Quantification of the stimulation of APE 1 activity in LP-BER in vitro by the 9-1-1 complex (circles); PCNA (rectangles) and BSA (triangles). The values represent the mean of three independent experiments. The error bars correspond to the standard error of the mean. (F) Quantification of the stimulation of APE 1 activity in LP-BER in vitro by the 9-1-1 complex, on the substrate with free ends (closed circles) and with the ends blocked with biotin (open circles). The values represent the mean of three independent experiments. The error bars correspond to the standard error of the mean.
The Rad9-Rad1-Hus1 complex enhances the strand displacement activity of Pol β in long patch base excision repair in vitro
Our next aim was to examine whether the 9-1-1 complex can influence LP-BER in vitro via stimulation of Pol β activity. We have previously shown that the 9-1-1 complex increases the affinity of Pol β for the primer-template and stimulates its DNA strand displacement activity (39). To test if the same effect could be observed in LP-BER, we performed a repair reaction using a 5′-32P labeled, THF-containing substrate in the presence of limiting amounts of Pol β (Figure 7, compare 64 and 15 fmols of pol β). Addition of the 9-1-1 complex to the reaction resulted in approximately 3-fold stimulation of the Pol β strand displacement activity and therefore increased efficiency of the total repair. In contrast no effect was observed when PCNA and BSA were tested under the same conditions, which confirms the specificity of the reported stimulation (Figure 7B and C).
Figure 7.

The 9-1-1 complex specifically stimulates the activity of Pol β in long patch base excision repair in vitro. The LP-BER in vitro reaction was performed as described in ‘Materials and Methods’. Reactions were stopped by adding an equal volume of formamide-dye solution and products were analyzed on a 10% denaturing polyacrylamide gel. (A) The reaction mixtures (10 μl) contained (besides all components described in ‘Materials and Methods’) 32P-5′-labeled 100 bp duplex oligonucleotide (50 fmol), APE 1(55 fmol), Pol β (15 fmol), Fen 1 (93 fmol), and Lig I (245 fmol). Reactions were incubated for 20 min at 37°C with increasing amounts of the 9-1-1 complex. (B) as A but with the indicated amounts of PCNA or BSA. (C) Quantification of the stimulation of Pol β activity in LP-BER in vitro by the 9-1-1 complex (rectangles); PCNA (rhomboids) and BSA (triangles). The error bars correspond to the standard error of the mean.
The Rad9-Rad1-Hus1 complex has no effect of the activities of Fen 1 and Lig I in long patch base excision repair in vitro
To further elucidate the involvement of the 9-1-1 complex in LP-BER we studied the effect of the checkpoint clamp on the activities of Fen 1 and Lig I in LP-BER in vitro. Similar to the previous experiments, we performed the LP-BER assay in the presence of limiting amounts of Fen 1 (Figure 8A) or Lig I (Figure 8B). Surprisingly, under those conditions, the 9-1-1 complex was unable to stimulate the enzymatic activities of Fen 1 and Lig I in LP-BER in vitro. However, the same 9-1-1 complex used in a single enzyme assay using DNA substrates representing the corresponding LP-BER intermediates was, as previously reported (40,42), able to stimulate enzymatic activities of Fen 1 and Lig I (Figure 8C and D). Thus, it appears that in the reconstituted LP-BER pathway, the 9-1-1 complex predominantly stimulates the enzymatic activities of those components of BER that act in the early steps of repair, such as APE 1 and Pol β, whereas the repair enzyme activities occurring during later steps, i.e. Fen 1 removal of the displaced strand or Lig I ligation, are not influenced directly by the checkpoint clamp in the presence of all the LP-BER proteins.
DISCUSSION
A growing amount of evidence indicates that the 9-1-1 complex, initially shown to be important for the DNA damage sensing and signaling pathways (17), plays also a more direct role in various DNA repair processes such as nucleotide excision repair (37), double strand break repair (34), translesion synthesis (34,37,38), and BER (Table 1). In previous reports we and others have demonstrated that the 9-1-1 complex physically and functionally interacts with several important components of the BER machinery namely Pol β (39), Fen 1 (42,43), Lig I (40,41), and the MutY DNA glycosylase homolog (MYH) (44,45). In this work we provide evidence that the 9-1-1 complex is directly involved in LP-BER. The evidence is derived from our demonstration that the 9-1-1 complex markedly stimulates a reconstituted LP-BER system in vitro and that this stimulation is due to the effect the 9-1-1 complex exerts on the enzymatic activities of the two early components of LP-BER namely APE 1 and Pol β.
Interestingly, we showed for the first time that the 9-1-1 complex directly interacts with APE 1 in vitro and in vivo, and that the 9-1-1 complex specifically stimulates the APE 1 endonuclease activity on a THF-containing substrate independently of its loading onto DNA. However, it is well known that in vivo, APE 1 is a very abundant protein possessing multiple cellular functions. In addition to its role in BER, it is involved in oxidative DNA damage repair and stimulates the DNA binding activity of AP-1 (Fos, Jun) proteins as well as nuclear factor-κB (NF-κB), the polyoma virus enhancer-binding protein 2 (PEBP2), the early growth response-1 (Egr-1), Myb, members of the ATF/CREB family, the hypoxia inducible factor-1α(HIF-1α), the HIF-like factor (HLF), Pax-5, and Pax-8 [reviewed in (51)]. APE 1 possesses also the major AP-1 redox activity in cells and represents a novel redox component of signal transduction cascades that regulates eukaryotic gene expression. In addition APE 1 has been implicated in the control of p53 activity through redox dependent and independent mechanisms. It has been also closely linked to apoptosis and altered levels of APE 1 have been found in some cancers. Therefore, APE 1 appears to form a link between the BER pathways, cancer, and regulation of transcription factors, oxidative signaling and cell cycle control. Hence, although APE 1 is known to be very abundant, some regulatory mechanisms must exist, that target this multifunctional enzyme in a timely and spatially regulated manner towards its required functions in specific cellular conditions. For this reason and considering our data, we hypothesize that the 9-1-1 complex may not simply stimulate APE 1 but it may also attract it to the sites of damage and target its functions towards the BER pathway. On the other hand, our efforts to determine the mechanism of stimulation of APE 1 by the 9-1-1 complex by biochemical ways remained still unsolved. First, the use of kinetic studies to address this point proved difficult, since the stimulatory effect of the 9-1-1 complex on APE1 appears to be of a complex nature and does not follow the classical rules of enzymology (e.g. reduction of the Km and/or increase of the Vmax). Second, in electro-mobility shift assays (EMSA) performed in the absence of Mg2+, addition of the 9-1-1 complex did not lead to an increase in the binding of APE 1 to a double stranded THF-containing substrate (data not shown). However, the absence of divalent cation in this experiment might create a context in which the 9-1-1 complex can not exert its stimulatory effect on APE 1, thus making it impossible to conclude whether the 9-1-1 complex does not affect the binding of APE 1 to DNA at all, or only in the system we used. More importantly we showed in this manuscript that the 9-1-1 complex interacts in vivo with APE 1 in human cells, as it has been shown previously for other LP-BER enzymes (41, 43, 45), thus supporting a functional role for the interaction of the 9-1-1 complex with APE 1. In addition preliminary experiments performed in our group indicated that APE 1 and the 9-1-1 complex co-localize in vivo and that this effect is enhanced upon H2O2 and IR treatment (Gembka and Hübscher, unpublished data).
Moreover, we showed that in our in vitro system the 9-1-1 complex significantly stimulates Pol β strand displacement activity, which is in agreement with previously reported functional and physical interactions between these two proteins (39). However, we could not observe any effect of the 9-1-1 complex on the activities of Fen 1 and Lig I in the reconstituted LP-BER in vitro. At first sight, this suggests a strong discrepancy between our findings and previous reports indicating that the 9-1-1 complex stimulates the enzymatic activities of Fen 1 (42,43) and Lig I (40,41). However, we suggest that in our in vitro system a hierarchy of stimulation and protein–protein interactions between the 9-1-1 complex and the BER components exists. Thus, it seems to be reasonable to speculate that the 9-1-1 complex, which functions as a sensor of DNA damage, once localized to the damage site recruits other repair proteins where their functions are necessary. This is in agreement with the reported in vivo interaction studies showing that the 9-1-1 co-immunoprecipitates with nearly all components of LP-BER (this work, 41,43,45). However, at the site of lesion, the 9-1-1 complex may exert its stimulatory effect only on the enzymatic activities of the early components of the repair machinery e.g. APE 1 and Pol β. This effect seems to be no longer necessary at the latter steps of repair involving the action of Fen 1 and Lig I. However, those enzymes, acting late in the BER process, are known to be stimulated by PCNA whereas this has not been shown for Pol β and APE 1 so far. Hence, one might speculate that after a first step of recruitment of the early repair enzymes to the lesion by the 9-1-1 complex, PCNA might be responsible for completion of the pathway through its action on the late BER enzymes. This hypothesis is further supported by the fact that PCNA has also been shown to co-localize with DNA repair factories containing the 9-1-1 complex upon DNA damage (34). In addition the complete BER reaction involves several proteins that establish a complex network of physical and functional interactions with each other and with the 9-1-1 complex. An example of this is the recently demonstrated ‘hit and run’ mechanism that shows how Fen 1 influences Pol β (4). In this context, one can assume that the effect of the 9-1-1 complex can be abolished or masked by the influence of the other proteins present in the reaction, or that the complex might be trapped by one of the enzymes present in the reaction.
On the other hand we cannot exclude that the proposed model may differ from the in vivo situation. Another striking finding we and the others observed (41,43) using in vitro systems is that encircling of the DNA substrate by the 9-1-1 complex is not required to stimulate the activities of repair enzymes. This indicates a different mechanism than in case of the PCNA ring, which absolutely requires a loading process to further stimulate Fen 1 and Lig I activities. One explanation could be that the observed stimulatory effect results from the direct protein–protein interactions and does not involve the loading of the 9-1-1 complex in Rad17- RFC2−5 dependent manner. This is supported by the fact that APE 1, as well as Pol β, Fen 1, and the MutY DNA glycosylase also interact in vitro with the single subunits of the checkpoint clamp (this work, 39–43 for details). Still, in vivo only the loading of the 9-1-1 complex onto chromatin leads to the local increase of its concentration at the damage sites, which is very important for the functions of the 9-1-1 complex in DNA damage signaling and most probably, in DNA repair.
Moreover, the tumor suppressor p53 and its downstream target p21 are other important components of DNA damage response that are directly connected to BER (52–55). It has been speculated that at low level, p53 actively functions in BER whereas a high level of p53 supports a global DNA-damage response and, in cases of excessive genotoxic stress, apoptosis (52,53). During cellular stress p53 up-regulates the expression of p21, which in turn interacts with many other proteins involved in replication, transcription, and signal transduction. In particular it binds to PCNA causing the inactivation of PCNA mediated stimulation of Fen 1, Lig I, and Pol δ activity (54,56). Recently it has been proposed that the 9-1-1 complex may substitute for PCNA during cellular stress (57). According to this hypothesis the 9-1-1 complex might act as a platform for repair proteins when the ability of PCNA to stimulate certain components of BER is reduced due to the binding to p21. Importantly, the activity of Pol β is not affected by PCNA but is strongly enhanced by the 9-1-1 complex (39).This is in contrast to the enzymatic activity of Pol δ, which requires PCNA but is not influenced by the 9-1-1 complex (39). These properties of the two pols suggest that p21 induced by DNA damage inhibits PCNA-dependent Pol δ activity, whereas Pol β remains unaffected, thus allowing LP-BER to occur (54). On the other hand, since the 9-1-1 complex is recruited to the sites of DNA damage where it associates with various DNA damage sensors and repair proteins it may well be that it functions as a targeted response stimulator independently of the PCNA level (57).
In conclusion, our results demonstrate for the first time that the 9-1-1 complex is directly involved in LP-BER. Under in vitro conditions we showed that there is a hierarchy of stimulation by the 9-1-1 complex in the BER repairsosome and the enhancement of LP-BER activity occurs due to the specific stimulation of the two early components of repair machinery namely APE 1 and Pol β. Importantly, our data reflect a detailed biochemical study performed in vitro and, for the first time, an investigation of the effect of the 9-1-1 in a complex system containing several proteins. Although this system is simplified compared with the in vivo situation, it provides a tool to give a better insight in understanding how the 9-1-1 complex stimulates the different steps of the LP-BER process. Moreover, we showed in this report that the 9-1-1 complex interacts in vivo with APE 1, as it has previously been shown for several other LP-BER enzymes (41, 43, 45). Taken together the data presented in this manuscript and the previous studies performed on LP-BER and the 9-1-1 complex, directly connect the DNA damage response with DNA repair, suggesting that the 9-1-1 complex might act as a core component of this vital connection.
ACKNOWLEDGEMENTS
We thank M. Stucki for critical reading of the manuscript. AG, KS, MT and UH are supported by the Swiss National Science Foundation (grant 3100AO-190312), MT by a FEBS Grant, RP by the UBS “Im Auftrag eines Kunden” and EF and UH by the University of Zürich. Funding to pay the Open Access publication charge was provided by the University of Zürich.
Conflict of interest statement. None declared.
REFERENCES
- 1.Friedberg EC, McDaniel LD, Schultz RA. The role of endogenous and exogenous DNA damage and mutagenesis. Curr. Opin. Genet. Dev. 2004;14:5–10. doi: 10.1016/j.gde.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 3.Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase beta mediates an excision step in mammalian long patch base excision repair. J. Biol. Chem. 2000;275:4460–4466. doi: 10.1074/jbc.275.6.4460. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J. Biol. Chem. 2005;280:3665–3674. doi: 10.1074/jbc.M412922200. [DOI] [PubMed] [Google Scholar]
- 5.Aspinwall R, Rothwell DG, Roldan-Arjona T, Anselmino C, Ward CJ, Cheadle JP, Sampson JR, Lindahl T, Harris PC, Hickson ID. Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc. Natl Acad. Sci. USA. 1997;94:109–114. doi: 10.1073/pnas.94.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J. Biol. Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 8.Kim K, Biade S, Matsumoto Y. Involvement of flap endonuclease 1 in base excision DNA repair. J. Biol. Chem. 1998;273:8842–8848. doi: 10.1074/jbc.273.15.8842. [DOI] [PubMed] [Google Scholar]
- 9.Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 10.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) Embo. J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pascucci B, Stucki M, Jonsson ZO, Dogliotti E, Hubscher U. Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J. Biol. Chem. 1999;274:33696–33702. doi: 10.1074/jbc.274.47.33696. [DOI] [PubMed] [Google Scholar]
- 12.Sleeth KM, Robson RL, Dianov GL. Exchangeability of mammalian DNA ligases between base excision repair pathways. Biochemistry. 2004;43:12924–12930. doi: 10.1021/bi0492612. [DOI] [PubMed] [Google Scholar]
- 13.Stucki M, Pascucci B, Parlanti E, Fortini P, Wilson SH, Hubscher U, Dogliotti E. Mammalian base excision repair by DNA polymerases delta and epsilon. Oncogene. 1998;17:835–843. doi: 10.1038/sj.onc.1202001. [DOI] [PubMed] [Google Scholar]
- 14.Podlutsky AJ, Dianova, Podust VN, Bohr VA, Dianov GL. Human DNA polymerase beta initiates DNA synthesis during long-patch repair of reduced AP sites in DNA. Embo J. 2001;20:1477–1482. doi: 10.1093/emboj/20.6.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 16.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 17.Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 2002;14:237–245. doi: 10.1016/s0955-0674(02)00312-5. [DOI] [PubMed] [Google Scholar]
- 18.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 19.Burtelow MA, Roos-Mattjus PM, Rauen M, Babendure JR, Karnitz LM. Reconstitution and molecular analysis of the hRad9-hHus1-hRad1 (9-1-1) DNA damage responsive checkpoint complex. J. Biol. Chem. 2001;276:25903–25909. doi: 10.1074/jbc.M102946200. [DOI] [PubMed] [Google Scholar]
- 20.Shiomi Y, Shinozaki A, Nakada D, Sugimoto K, Usukura J, Obuse C, Tsurimoto T. Clamp and clamp loader structures of the human checkpoint protein complexes, Rad9-1-1 and Rad17-RFC. Genes Cells. 2002;7:861–868. doi: 10.1046/j.1365-2443.2002.00566.x. [DOI] [PubMed] [Google Scholar]
- 21.Venclovas C, Thelen MP. Structure-based predictions of Rad1, Rad9, Hus1 and Rad17 participation in sliding clamp and clamp-loading complexes. Nucleic Acids Res. 2000;28:2481–2493. doi: 10.1093/nar/28.13.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dahm K, Hubscher U. Colocalization of human Rad17 and PCNA in late S phase of the cell cycle upon replication block. Oncogene. 2002;21:7710–7719. doi: 10.1038/sj.onc.1205872. [DOI] [PubMed] [Google Scholar]
- 23.Ellison V, Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol. 2003;1:E33. doi: 10.1371/journal.pbio.0000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinho RG, Lindsay HD, Flaggs G, DeMaggio AJ, Hoekstra MF, Carr AM, Bentley NJ. Analysis of Rad3 and Chk1 protein kinases defines different checkpoint responses. Embo J. 1998;17:7239–7249. doi: 10.1093/emboj/17.24.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toueille M, Hubscher U. Regulation of the DNA replication fork: a way to fight genomic instability. Chromosoma. 2004;113:113–125. doi: 10.1007/s00412-004-0303-7. [DOI] [PubMed] [Google Scholar]
- 26.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 27.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl Acad. Sci. USA. 2003;100:13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffith JD, Lindsey-Boltz LA, Sancar A. Structures of the human Rad17-replication factor C and checkpoint Rad 9-1-1 complexes visualized by glycerol spray/low voltage microscopy. J. Biol. Chem. 2002;277:15233–15236. doi: 10.1074/jbc.C200129200. [DOI] [PubMed] [Google Scholar]
- 29.Kaur R, Kostrub CF, Enoch T. Structure-function analysis of fission yeast Hus1-Rad1-Rad9 checkpoint complex. Mol. Biol. Cell. 2001;12:3744–3758. doi: 10.1091/mbc.12.12.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindsey-Boltz LA, Bermudez VP, Hurwitz J, Sancar A. Purification and characterization of human DNA damage checkpoint Rad complexes. Proc Natl Acad Sci U S A. 2001;98:11236–11241. doi: 10.1073/pnas.201373498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bermudez VP, Lindsey-Boltz LA, Cesare AJ, Maniwa Y, Griffith JD, Hurwitz J, Sancar A. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl Acad. Sci. USA. 2003;100:1633–1638. doi: 10.1073/pnas.0437927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Majka J, Burgers PM. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc. Natl Acad. Sci. USA. 2003;100:2249–2254. doi: 10.1073/pnas.0437148100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burtelow MA, Kaufmann SH, Karnitz LM. Retention of the human Rad9 checkpoint complex in extraction-resistant nuclear complexes after DNA damage. J. Biol. Chem. 2000;275:26343–26348. doi: 10.1074/jbc.M001244200. [DOI] [PubMed] [Google Scholar]
- 34.Meister P, Poidevin M, Francesconi S, Tratner I, Zarzov P, Baldacci G. Nuclear factories for signalling and repairing DNA double strand breaks in living fission yeast. Nucleic Acids Res. 2003;31:5064–5073. doi: 10.1093/nar/gkg719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondo T, Wakayama T, Naiki T, Matsumoto K, Sugimoto K. Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science. 2001;294:867–870. doi: 10.1126/science.1063827. [DOI] [PubMed] [Google Scholar]
- 36.Roos-Mattjus P, Vroman BT, Burtelow MA, Rauen M, Eapen AK, Karnitz LM. Genotoxin-induced Rad9-Hus1-Rad1 (9-1-1) chromatin association is an early checkpoint signaling event. J. Biol. Chem. 2002;277:43809–43812. doi: 10.1074/jbc.M207272200. [DOI] [PubMed] [Google Scholar]
- 37.Giannattasio M, Lazzaro F, Longhese MP, Plevani P, Muzi-Falconi M. Physical and functional interactions between nucleotide excision repair and DNA damage checkpoint. Embo J. 2004;23:429–438. doi: 10.1038/sj.emboj.7600051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kai M, Wang TS. Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev. 2003;17:64–76. doi: 10.1101/gad.1043203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toueille M, El-Andaloussi N, Frouin I, Freire R, Funk D, Shevelev I, Friedrich-Heineken E, Villani G, Hottiger MO, Hubscher U. The human Rad9/Rad1/Hus1 damage sensor clamp interacts with DNA polymerase beta and increases its DNA substrate utilisation efficiency: implications for DNA repair. Nucleic Acids Res. 2004;32:3316–3324. doi: 10.1093/nar/gkh652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smirnova E, Toueille M, Markkanen E, Hubscher U. The human checkpoint sensor and alternative DNA clamp Rad9-Rad1-Hus1 modulates the activity of DNA ligase I, a component of the long-patch base excision repair machinery. Biochem. J. 2005;389:13–17. doi: 10.1042/BJ20050211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W, Lindsey-Boltz LA, Sancar A, Bambara RA. Mechanism of stimulation of human DNA ligase I by the Rad9-rad1-Hus1 checkpoint complex. J. Biol. Chem. 2006;281:20865–20872. doi: 10.1074/jbc.M602289200. [DOI] [PubMed] [Google Scholar]
- 42.Friedrich-Heineken E, Toueille M, Tannler B, Burki C, Ferrari E, Hottiger MO, Hubscher U. The two DNA clamps Rad9/Rad1/Hus1 complex and proliferating cell nuclear antigen differentially regulate flap endonuclease 1 activity. J. Mol. Biol. 2005;353:980–989. doi: 10.1016/j.jmb.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, Brandt P, Rossi ML, Lindsey-Boltz L, Podust V, Fanning E, Sancar A, Bambara RA. The human Rad9-Rad1-Hus1 checkpoint complex stimulates flap endonuclease 1. Proc. Natl Acad. Sci. USA. 2004;101:16762–16767. doi: 10.1073/pnas.0407686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang DY, Lu AL. Interaction of checkpoint proteins Hus1/Rad1/Rad9 with DNA base excision repair enzyme MutY homolog in fission yeast, Schizosaccharomyces pombe. J. Biol. Chem. 2005;280:408–417. doi: 10.1074/jbc.M406800200. [DOI] [PubMed] [Google Scholar]
- 45.Shi G, Chang DY, Cheng CC, Guan X, Venclovas C, Lu AL. Physical and functional interactions between MutY homolog (MYH) and checkpoint proteins Rad9-Rad1-Hus1. Biochem. J. 2006;400:53–62. doi: 10.1042/BJ20060774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maga G, Jonsson ZO, Stucki M, Spadari S, Hubscher U. Dual mode of interaction of DNA polymerase epsilon with proliferating cell nuclear antigen in primer binding and DNA synthesis. J. Mol. Biol. 1999;285:259–267. doi: 10.1006/jmbi.1998.2314. [DOI] [PubMed] [Google Scholar]
- 47.Beard WA, Wilson SH. Purification and domain-mapping of mammalian DNA polymerase beta. Methods Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- 48.Jonsson ZO, Hindges R, Hubscher U. Regulation of DNA replication and repair proteins through interaction with the front side of proliferating cell nuclear antigen. Embo J. 1998;17:2412–2425. doi: 10.1093/emboj/17.8.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stucki M, Jonsson ZO, Hubscher U. In eukaryotic flap endonuclease 1, the C terminus is essential for substrate binding. J. Biol. Chem. 2001;276:7843–7849. doi: 10.1074/jbc.M008829200. [DOI] [PubMed] [Google Scholar]
- 50.Budworth H, Dianova, Podust VN, Dianov GL. Repair of clustered DNA lesions. Sequence-specific inhibition of long-patch base excision repair be 8-oxoguanine. J. Biol. Chem. 2002;277:21300–21305. doi: 10.1074/jbc.M201918200. [DOI] [PubMed] [Google Scholar]
- 51.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat. Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 52.Offer H, Milyavsky M, Erez N, Matas D, Zurer I, Harris CC, Rotter V. Structural and functional involvement of p53 in BER in vitro and in vivo. Oncogene. 2001;20:581–589. doi: 10.1038/sj.onc.1204120. [DOI] [PubMed] [Google Scholar]
- 53.Offer H, Wolkowicz R, Matas D, Blumenstein S, Livneh Z, Rotter V. Direct involvement of p53 in the base excision repair pathway of the DNA repair machinery. FEBS Lett. 1999;450:197–204. doi: 10.1016/s0014-5793(99)00505-0. [DOI] [PubMed] [Google Scholar]
- 54.Tom S, Ranalli TA, Podust VN, Bambara RA. Regulatory roles of p21 and apurinic/apyrimidinic endonuclease 1 in base excision repair. J. Biol. Chem. 2001;276:48781–48789. doi: 10.1074/jbc.M109626200. [DOI] [PubMed] [Google Scholar]
- 55.Zhou J, Ahn J, Wilson SH, Prives C. A role for p53 in base excision repair. Embo J. 2001;20:914–923. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jaiswal AS, Bloom LB, Narayan S. Long-patch base excision repair of apurinic/apyrimidinic site DNA is decreased in mouse embryonic fibroblast cell lines treated with plumbagin: involvement of cyclin-dependent kinase inhibitor p21Waf-1/Cip-1. Oncogene. 2002;21:5912–5922. doi: 10.1038/sj.onc.1205789. [DOI] [PubMed] [Google Scholar]
- 57.Helt CE, Wang W, Keng PC, Bambara RA. Evidence that DNA damage detection machinery participates in DNA repair. Cell Cycle. 2005;4:529–532. doi: 10.4161/cc.4.4.1598. [DOI] [PubMed] [Google Scholar]