


Abstract
P-TEFb regulates eukaryotic gene expression at the level of transcription elongation, and is itself controlled by the reversible association of 7SK RNA and an RNA-binding protein HEXIM1 or HEXIM2. In an effort to determine the minimal region of 7SK needed to interact with HEXIM1 in vitro, we found that an oligo comprised of nucleotides 10–48 sufficed. A bid to further narrow down the minimal region of 7SK led to a surprising finding that HEXIM1 binds to double-stranded RNA in a sequence-independent manner. Both dsRNA and 7SK (10–48), but not dsDNA, competed efficiently with full-length 7SK for HEXIM1 binding in vitro. Upon binding dsRNA, a large conformational change was observed in HEXIM1 that allowed the recruitment and inhibition of P-TEFb. Both subcellular fractionation and immunofluorescence demonstrated that, while most HEXIM1 is found in the nucleus, a significant fraction is found in the cytoplasm. Immunoprecipitation experiments demonstrated that both nuclear and cytoplasmic HEXIM1 is associated with RNA. Interestingly, the one microRNA examined (mir-16) was found in HEXIM1 immunoprecipitates, while the small nuclear RNAs, U6 and U2, were not. Our study illuminates novel properties of HEXIM1 both in vitro and in vivo, and suggests that HEXIM1 may be involved in other nuclear and cytoplasmic processes besides controlling P-TEFb.
INTRODUCTION
P-TEFb plays a key role in RNA polymerase II elongation control (1–3). It is comprised of one of two isoforms of Cdk9 (4,5) and one of three cyclins, T1, T2 (6) or K (7) in humans. One of the major targets of the kinase activity of P-TEFb is the carboxyl-terminal domain (CTD) of the largest subunit of RNA polymerase II (8), and this phosphorylation of the CTD by P-TEFb occurs during transcription elongation (9). P-TEFb also phosphorylates the negative transcription elongation factor DSIF (10), turning it into a positive elongation factor (11). P-TEFb controls gene expression by regulating the fraction of RNA polymerase II molecules that generate full-length mRNAs. In addition to its normal cellular role, P-TEFb has been shown to be recruited by viral transactivator Tat to the nascent viral transcript, TAR, near the promoter to enhance viral transcription, which is required for efficient HIV-1 replication (4,12–16).
P-TEFb is uniquely regulated by the reversible association of a small nuclear RNA, 7SK (17,18) and one of two HEXIM proteins (19–22). Glycerol gradient analyses of cell lysates indicate that two forms of P-TEFb exist in the cell. An active form of P-TEFb, free of HEXIM and 7SK, interacts with a variety of cellular factors, including NF-κB (16), c-Myc (23,24), MyoD (25) and Brd4 (26,27), to regulate gene transcription. A larger, inactive form contains 7SK and HEXIM proteins that together sequester active P-TEFb (17,19,22). When cells are treated with P-TEFb inhibitors, such as DRB, or other agents that block transcription, P-TEFb is quickly released from the large form (19). This form of P-TEFb regulation is physiologically significant, because it has been shown that all signals that trigger cardiac hypertrophy converge at the critical step of activating P-TEFb through the dissociation of 7SK and HEXIM. This activation causes increased cellular transcription and an increase in the size of cardiomyocytes (28–31). Factors that release P-TEFb from the 7SK•HEXIM1•P-TEFb complex are currently unknown.
Several studies have uncovered some of the important interactions in the 7SK•HEXIM1•P-TEFb complex. Functional domains of HEXIM1 have been identified. The N-terminal region of HEXIM1 (amino acids 1–120) is found to be self-inhibitory, preventing HEXIM1 from interacting with P-TEFb in the absence of 7SK (32,33). The region centered upon KHRR (amino acids 152–155) is involved in the binding of 7SK, and nearby sequences comprise nuclear localization signals (33,34). An adjacent region centered upon PYNT (amino acids 202–205) is involved in interaction with P-TEFb (33), and a small region centered upon Y271 is involved in inhibition of P-TEFb (32). The C-terminal region of HEXIM1 (amino acids 281–359) mediates the dimerization of HEXIM1 via a leucine zipper motif (21,32,35,36). In addition, the regions involved in interactions have been narrowed down to amino acids 1–254 of 726 of cyclin T1, all of Cdk9 and nucleotide 1–172 of 7SK (32,33). Furthermore, phosphorylation of the T-loop of Cdk9 has been implicated in the activation of P-TEFb and is required for the formation of the 7SK•HEXIM1•P-TEFb complex (32,37).
In attempts to narrow down the minimal sequence of 7SK that is needed to form the 7SK•HEXIM1•P-TEFb complex in vitro, we serendipitously discovered that HEXIM1 is a potent dsRNA-binding protein. Further analysis revealed that dsRNA induces a conformational change in HEXIM1, so that it can interact with P-TEFb. In addition, in vivo studies demonstrated that endogenous HEXIM1 can be found in both the cytoplasm and nucleus, and most HEXIM1 in cells is associated with a variety of RNAs. Interestingly, at least one microRNA was found to be associated with HEXIM1. These findings provide a mechanistic insight into the inhibition of P-TEFb by HEXIM1 and RNA, suggest a possible mechanism on how cells balance active and inactive forms of P-TEFb and indicate that HEXIM1 may play other roles besides inhibiting P-TEFb in vivo.
MATERIALS AND METHODS
EMSA
Here, 12 µl reactions were carried out in 25 mM HEPES, pH 7.6, 15% glycerol, 60 mM KCl, 0.1 mM EDTA, 5 mM DTT, 0.01% NP-40 and included 200 ng recombinant HEXIM1, recombinant P-TEFb and various RNA or DNA oligos as indicated. Reactions were incubated at room temperature for 15 min and resolved on a 6% polyacrylamide (19:1 acrylamide:bis-acrylamide ratio) gel in 0.5× Tris•glycine at 4°C for 1.5 h at 6 W. unless otherwise indicated. Proteins were visualized by silver staining.
Tryptophan fluorescence emission spectra
Tryptophan fluorescence studies were performed on a Fluorolog 3 spectrofluorimeter (Jobin Yvon, Horiba) with a 450-W xenon lamp using 4 and 8 nm excitation and emmision bandpasses, respectively. All spectra were taken under identical buffer conditions as used in EMSAs at 25°C. Emission spectra of 1 μM HEXIM1 were obtained using a constant excitation wavelength of 276 nm and, monitoring the resultant fluorescent intensity between 290 and 450 nm, emission spectra of 1 µM HEXIM1 in the presence of dsDNA or dsRNA were obtained using identical instrument conditions as HEXIM1 alone. HEXIM1 data were not corrected for contributions from the buffer or dsDNA/dsRNA, as scans of these components under conditions identical to those described above indicated a negligible contribution to the emission spectra.
Purification of P-TEFb
Baculoviruses expressing human Cdk9 and cyclin T1 (1–290) were generated using the BaculoDirect Baculovirus Expression System (Invitrogen) according to the manufacturer's instructions. Cdk9 was tagged with six histidines at its carboxyl terminus, and cyclin T1 was untagged. Sf9 cells were maintained suspended in Sf-900 II serum-free medium (SFM) (Invitrogen) at concentrations between 5 × 105 and 2 × 106 cells/ml. Here, 10 ml of a third amplification stock of each virus was added to 1 liter of cells (at 1 × 106 cells/ml) in a 2-l Erlenmeyer flask, and the cells were shaken at 100 r.p.m. at 28°C for 72 h. Cells were spun down at 1000× g, and the cell pellet was sonicated (three 10-s bursts) in 10 ml of Buffer L [10 mM Tris (pH 8.0), 150 mM NaCl, 2 mM MgCl2, 1% Triton, 0.1% of a saturated PMSF isopropanol solution, 1× EDTA-free protease inhibitor mixture (Roche Applied Science) and 10 µg/ml E-64 protease inhibitor (Roche Applied Science)]. The cell lysate was spun for 15 min at 192 000 g in a Beckman tabletop centrifuge. The supernatant was then incubated with 1 ml of nickel-nitrilotriacetic acid resin (Qiagen) for 45 min at 4°C. The resin was washed once with 10 ml of Buffer L, followed by a wash with 1 ml of 10 mM Tris (pH 8.0), 500 mM NaCl, 1% Triton, 0.1% of a saturated PMSF isopropanol solution and 25 mM imidazole and with 10 ml of 60 mM HGKEP. P-TEFb was eluted with 2.5 ml of 60 mM HGKEP with 300 mM imidazole and loaded onto a 1-ml Mono S column. P-TEFb was eluted with a 20-ml linear gradient from 0.1 to 0.5 M HGKEDP.
Kinase assay
Here, 16 µl kinase reactions containing purified recombinant P-TEFb with human DSIF as the substrate were carried out in 30 mM KCl, 20 mM HEPES pH 7.6, 7 mM MgCl2, 30 µM ATP, 1.3 µCi of [γ-32P]-ATP (Amersham), 1 µg BSA per reaction and the indicated amounts of HEXIM1 proteins. T7-transcribed 7SK RNA or RNA oligos was added last to the pre-incubation. All reactions were incubated for 10 min at 23°C prior to the addition of ATP. The kinase reactions were then incubated for 20 min at 30°C and then stopped by the addition of SDS-PAGE loading buffer. Reactions were resolved by 9% SDS-PAGE. The dried gel was subjected to autoradiography and quantified with a Packard InstantImager.
Glycerol gradient analysis
HeLaS3 cells, at 90% confluency in a T-75 flask, were scraped, spun down at 2000 r.p.m. and then lysed for 10 min on ice in 150 mM NaCl, 2 mM MgCl2, 10 mM HEPES, 1 mM EDTA, 1 mM DTT, 1% PMSF, EDTA-free complete protease inhibitor cocktail (Roche) and 0.5% NP-40, and the lysates were clarified by centrifugation for 10 min at 14 000 r.p.m. prior to fractionation on 5 ml, 5–45% glycerol gradients in the same buffer conditions used during lysis, except that NP-40 was omitted. Gradients were run at 45 000 r.p.m. for 16 h in a Beckman SW-Ti55 rotor before being fractionated.
Native gel analysis of HEXIM1 in cell extracts
Cell extracts were separated on a 6% polyacrylamide (19:1 acrylamide:bis-acrylamide ratio) gel in 0.5× Tris•glycine at 4°C for 1.5 h at 6 W, followed by transfer to BA85 PROT membrane (Whatman). HEXIM1 was then detected by western blotting analysis, as described below. For RNase treatment, 100 ng RNase A (Fermentas) was incubated with cell extracts or glycerol gradient fractions for 10 min at 30°C. Total reactions were then examined by native gel analysis, as described above.
Immunofluorescence
HeLa cells were grown on glass coverslips, fixed for 20 min in 4% paraformaldehyde/PBS. Cells were permeabilized with 0.5% Triton-X 100 in PBS for 5 min and washed in PBS. After being blocked in 2% donkey serum in PBS for 15 min, cells were incubated with affinity-purified, anti-HEXIM1 antibodies (Abcam ab28016) in blocking solution. After washing in PBS four times, cells were incubated in a 1:200 dilution of Alexa Fluor 647 donkey-anti-sheep IgG (Molecular Probes) secondary antibody for 2 h. To visualize DNA, cells were stained with 0.5 µg/ml of DAPI after secondary antibody incubation. All incubations were at room temperature. After washing in PBS, coverslips were mounted in Prolong Antifade mounting medium (Molecular Probes) and cells were visualized with a Leica DMR microscope for each fluorochrome.
Cell fractionation
HeLa cells confluent in four T-150 flasks were pelleted and washed once in ice-cold PBS, 0.1% PMSF. Cells were resuspended in 2 ml of cold buffer A [10 mM HEPES, 15 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.1% PMSF, 40 U/ml RNaseOUT (Invitrogen)] and lysed by homogenization in a 7-ml Dounce all-glass homogenizer with a tight pestle using 15 strokes. Cell lysates were spun for 5 min at 4000 r.p.m. at 4°C in a microcentrifuge to separate cytoplasm from nuclei. Nuclei were resuspended with 2 ml of cold buffer B [10 mM HEPES, 2 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, 150 mM NaCl, 0.5% NP-40, 0.1% PMSF, 40 U/ml RNaseOUT (Invitrogen)] and incubated on ice for 10 min with occasional gentle votexing. Both cytoplasm and nuclei extract were further spun for 10 min at 65 000 r.p.m. in a 100.2-Ti rotor in a Beckman tabletop centrifuge. Cleared lysates were used for western blot analysis and saved at −80°C.
Northern and western blot analysis
Total RNAs were extracted from individual glycerol gradient fractions using mirVana miRNA isolation kit (Ambion). The probe for 7SK was 5′ end labeled using T4 PNK (NEB), following manufacturer's instructions. RNAs were separated on a 6% denaturing gel and transferred to a Nytran N membrane (Whatman) using a semi-dry blotting apparatus. The membrane was blocked with ULTRAhyb (Ambion) and probed with labeled 7SK oligo, following the manufacturer's instruction. The washed membrane was exposed to film at −80°C for 30 min. The probe for 7SK was as follows (numbers in parenthesis indicate the regions to which the probes hybridize):
7SK (121–160): GGGGAUGGUCGUCCUCUUCGACCGAGCGCGCAGCUUCGGG
U6 (60–99): GGAACGCUUCACGAAUUUGCGUGUCAUCCUUGCGCAGGGG
U2 (97–136): CGGAGCAAGCUCCUAUUCCAUCUCCCUGCUCCAAAAAUCC
Mir-16-1: CGCCAAUAUUUACGUGCUGCUAAGGCACUGCUGAC
Western blots were carried out as previously described (22).
RESULTS
7SK (10–48) is sufficient to bind HEXIM1
While studies have been carried out to map interaction domains of HEXIM1 and P-TEFb in the large, inactive 7SK•HEXIM•P-TEFb complex, further refinement of the regions of 7SK required to form this complex are needed. Because HIV Tat protein and HEXIM1 have a nearly identical RNA-binding motif (33,34), we looked for regions of 7SK that might mimic the Tat-binding site (TAR) in the HIV 5′ UTR. Tat binds a bulged region of TAR RNA containing the sequence AUCUG. Considering that AUCUG should appear randomly only once in every 1024 nt, we were surprised to find that 7SK (330 nt) contained three AUCUG motifs in the first 70 nt. The first two reside in nucleotides 10–48 (Figure 1A). In addition, mFold analysis (Figure 1B) suggested that the structure of the first AUCUG in the context of nucleotides 10–48 resembled that predicted to occur in TAR (data not shown). These observations prompted us to test if 7SK (10–48) was able to bind HEXIM1 using a modified electrophoretic mobility shift assay (EMSA). In this assay, recombinant HEXIM1 was examined on a native gel and visualized by silver staining. As shown in Figure 1C, wild-type HEXIM1 (WT) migrated as a discrete band as expected (32) for a dimer of HEXIM1. When increasing amounts of the 7SK oligo were added, HEXIM1 shifted up the gel to a new discrete position. The shift was complete when 10 pmol of the oligo were added. Moreover, no further significant shift occurred in the presence of excess oligo up to 100 pmol. Because the reactions contained 5 pmol of HEXIM1 and a little more than 50% of the HEXIM1 was shifted by 3 pmol of the 7SK oligo, it is likely that, similar to what was found earlier with 7SK (32), a one-to-one complex of RNA to HEXIM1 dimer formed. As a control, HEXIM1 containing a substitution of ILAA for KHRR (amino acids 152–155) that does not bind to 7SK (33) was analyzed. Addition of the 7SK oligo did not result in any specific shift of the mutant HEXIM1. These results indicate that the 39-nt oligo binds to HEXIM1 and that this binding requires the same region of HEXIM1 needed for binding to 7SK.
Figure 1.

7SK (10–48) oligo interacts with HEXIM1. (A) The sequence of 7SK (10–48) oligo containing two AUCUG. (B) The predicted structure of 7SK (10–48) by mFold. (C) EMSA analysis of wild-type (WT) and ILAA mutant HEXIM1 proteins with 7SK (10–48) of indicated amount. Here, 5 pmol of WT or ILAA was used in each lane, and the 6% gel was silver stained. (D) Sequence and binding affinity of oligos tested in EMSA. (E) The predicted structures of 7SK (19–35), (57–73), (303–319) by mFold.
HEXIM1 binds double-stranded but not single-stranded nucleic acid
A set of RNA oligos were then designed to further narrow down the region of 7SK required for HEXIM1 interaction in vitro. These oligos contain either individual AUCUG regions or other regions of 7SK as negative controls (Figure 1D). The ability of these oligos to bind HEXIM1 was analyzed in EMSA (raw data not shown). To our surprise, none of these oligos changed the mobility of HEXIM1, except for one of the negative controls, 7SK (303–319), which does not have the AUCUG sequence. It showed only a weak interaction. mFold analyses of these five oligos revealed that there was a correlation not with the presence of AUCUG, but rather the stability of potential double-stranded stem structures (Figure 1D). This observation prompted us to ask if HEXIM1 actually binds to a double-stranded structure. First, we tested a set of siRNA oligos used to knock down cellular genes. They had double-stranded regions of either 19 or 25 bp with two deoxynucleotide overhangs at one or both 3′ ends and did not share any sequence similarities with each other or 7SK. Remarkably, all of them were able to shift HEXIM1 as well as the 7SK (10–48) oligo (data not shown). Again, all of them failed to shift the ILAA HEXIM1 mutant, a 7SK-binding-deficient HEXIM1 mutant (data not shown). These data suggested that HEXIM1 could associate with dsRNA, regardless of its sequence.
To eliminate the possibility that HEXIM1 required the deoxynucleotide overhangs present in those siRNA constructs, two 25-base single-stranded DNA oligos, D1 and D2, and two single-stranded RNA oligos, R1 and R2, were designed. D1 has the same sequence as R1 and is complementary to D2. Thus, we could form one dsDNA (D1/D2), one dsRNA (R1/R2) and two DNA/RNA hybrids (D1/R2 and D2/R1). When single-stranded oligos (D1, D2, R1 and R2) were used, none of them were able to shift HEXIM1 mobility significantly (Figure 2A). R1 was able to partially shift HEXIM1 at the highest concentration because it could form a semi-stable double-stranded structure (data not shown). In sharp contrast, when double-stranded oligos were used, all of them were able to shift HEXIM1. The only difference detected in this assay was that the dsDNA•HEXIM1 complex migrated differently from the dsRNA•HEXIM1 and DNA/RNA•HEXIM1 complexes (Figure 2B). The difference in mobility between dsRNA•HEXIM1 and dsDNA•HEXIM1 will be addressed later.
Figure 2.

HEXIM1 interacts with double-stranded, but not single-stranded nucleic acids. (A) EMSA analysis of HEXIM1 with single-stranded DNA (D1, D2) and single-stranded RNA (R1, R2). Each lane contains 5 pmol of wild-type HEXIM1. (B) EMSA analysis of HEXIM1 (silver stain) with double-stranded DNA (D1/D2), DNA/RNA hybrids (D1/R2, D2/R1) and double-stranded RNA (R1/R2). Each lane contains 5 pmol of wild-type HEXIM1. (C) EMSA analysis of wild-type and ILAA mutant HEXIM1proteins with labeled single-stranded RNA (ssRNA) and double-stranded RNA (dsRNA). Each lane contains 2 pmol of wild-type or ILAA mutant HEXIM1 proteins. The top panel is a silver-stained gel showing the region containing HEXIM1, the middle panel is an autoradiograph of the same region and the bottom panel is an autoradiograph (same exposure as the middle panel) of the bottom of the gel where free-labeled probes run.
Because it is possible that the mobility of HEXIM1 might not change upon binding a short oligo, the potential binding was assayed using the shift of labeled oligos. The conditions were identical to those used in preceding EMSAs, except that the R2 oligo was 5′ end labeled to low specific activity. WT and ILAA HEXIM1 (2 pmol) were incubated with increasing amounts of either R2 (ssRNA) or R1/R2 (dsRNA). HEXIM1 was visualized by silver staining. As expected, dsRNA lowered the mobility of WT HEXIM1 but not that of the ILAA RNA-binding mutant (Figure 2C, top panel). Note that the HEXIM1 shift was less dramatic than that seen earlier because the native gel was stopped very early to keep the small RNAs from running off the gel. ssRNA failed to slow the mobility of either WT or ILAA (Figure 2C, top panel). Autoradiography of the region of the gel containing the HEXIM1 proteins demonstrated that only dsRNA was associated with HEXIM1 (Figure 2C, middle panel). This binding was saturated at ∼3 pmol of dsRNA. Autoradiography of the bottom of the gel (same exposure as the middle panel) confirmed that dsRNA, but not ssRNA, was binding. The unbound labeled dsRNA was only seen after binding to HEXIM1 was saturated (Figure 2C, lower panel). We conclude that HEXIM1 binds stoichiometrically to dsRNA, but does not bind to ssRNA.
dsRNA but not dsDNA can compete efficiently with 7SK for HEXIM1 binding
Because 7SK (10–48), dsDNA and dsRNA bind the same region of HEXIM1 as 7SK does, we examined the relative binding strength of 7SK (10–48), dsDNA, dsRNA and 7SK in competition experiments. As expected, the addition of 1 pmol of 7SK lowered the mobility of 0.5 pmol of HEXIM1 (Figure 3A). When the binding reaction contained 1 pmol of 7SK and increasing amounts of dsDNA, the 7SK•HEXIM1 complex was unaffected at 0.5 and 5 pmol, and was only partially decreased at 50 pmol of dsDNA (Figure 3A). In sharp contrast, both dsRNA and 7SK (10–48) were able to compete efficiently with 7SK to form dsRNA•HEXIM1 and 7SK (10–48)•HEXIM1 complexes, respectively (Figure 3A).
Figure 3.

7SK, dsRNA and 7SK (10–48) have higher affinity than dsDNA for HEXIM1 binding. (A) Competition of 7SK•HEXIM1 interaction by dsDNA, dsRNA or 7SK (10–48) oligo analyzed by EMSA (silver-stained gel). Each lane contains 0.5 pmol of wild-type HEXIM1. (B) Competition of dsRNA•HEXIM1 interaction by dsRNA analyzed by EMSA. Each lane contains 0.5 pmol of wild-type HEXIM1 (silver-stained gel). (C) Competition of dsDNA•HEXIM1 interaction by dsDNA analyzed by EMSA. Each lane contains 0.5 pmol of wild-type HEXIM1 (silver-stained gel).
Further evidence that dsRNA has a higher affinity to HEXIM1 than dsDNA came from the direct competition between the two. Previously, we reported that the 7SK•HEXIM1 complex is relatively stable because 32P-labeled 7SK in a pre-formed 7SK•HEXIM1 complex could not be competed off by the addition of cold 7SK even at 100-fold excess (33). Because of this, an experiment was designed to look at competition for HEXIM1 binding under two conditions. DNA and RNA were mixed together before HEXIM1 was added to look at competition during binding, or DNA or RNA were added separately to allow prebinding to HEXIM1 before the other was added. When the binding reaction contained 0.5 pmol of dsRNA and increasing amounts of dsDNA were added to HEXIM1 at the same time, only dsRNA•HEXIM1 was detected (Figure 3B, dsDNA During). To examine the stability of the dsRNA•HEXIM1 complex, we preformed this complex and then added increasing amount of dsDNA. Even a 10-fold excess of dsDNA was unable to affect the dsRNA•HEXIM1 complex (Figure 3B, dsDNA After). This protocol was repeated using a constant amount of dsDNA and increasing amounts of dsRNA. Again, dsRNA was able to efficiently compete with dsDNA binding to HEXIM1, even if the dsDNA complex was preformed before dsRNA addition (Figure 3C). Taking into consideration the concentration of the various nucleic acids used, dsRNA, 7SK (10–48) and 7SK had roughly equal affinity for HEXIM1, while dsDNA had ∼1% of that affinity.
dsRNA binding induces changes in intrinsic tryptophan fluorescence of HEXIM1
The shift to lower mobility upon binding of 25-bp dsRNA (16 kDa) to HEXIM1 (80 kDa dimer) was somewhat unexpected, especially considering that the overall negative charge of the dsRNA•HEXIM1 complex was increased significantly, which would tend to increase its mobility. We reasoned that the binding of dsRNA might induce a conformational change in HEXIM1, leading to a decrease in mobility due to its more expanded shape. In an attempt to obtain further evidence for a conformational change in HEXIM1 induced by dsRNA, intrinsic fluorescence from tryptophan residues was measured. Using 1 μM HEXIM1 in a buffer identical to that used in the EMSA, the optimal excitation wavelength was found to be 276, nm and this provided a strong peak in emission at 350.7 nm (Figure 4A). An equimolar amount of dsDNA or dsRNA in the same buffer had negligible emission (data not shown). When 1 μM of dsDNA was added to the HEXIM1 solution, a 10% reduction in the emission signal was detected, and the peak in emission remained at essentially the same wavelength (350.8 nm). In contrast, the addition of 1 μM dsRNA decreased the emission signal to 50%, and shifted the peak in emission to 352.0 nm. Differences in the normalized spectra confirmed that dsRNA, but not DNA, caused a shift in the λmax as evidenced by the inversion in sign of the difference at around 350 nm (Figure 4B). We do not know why RNA additionally caused a peak at 299 nm in the difference spectrum, but DNA did not cause any change. Both the large decrease in intrinsic fluorescence, the observed changes in the difference spectrum and a shift of the emission peak are consistent with an RNA-induced change in conformation, in which one or more of the four tryptophan residues moves from a relatively hydrophobic to a more hydrophilic environment.
Figure 4.

dsRNA binding induces changes in intrinsic tryptophan fluorescence of HEXIM1. (A) Emission spectra of HEXIM1 were obtained using a constant excitation wavelength of 276 nm and monitoring the resultant fluorescent intensity between 290 and 450 nm. Emission spectra of HEXIM1 in the presence of dsDNA or dsRNA were obtained using identical instrument conditions as HEXIM1 alone. (B) After normalization of the HEXIM1, HEXIM1 + dsDNA and HEXIM1 + dsRNA spectra to 100%, the difference spectra were calculated and plotted versus wavelength.
dsRNA•HEXIM1 can recruit and inhibit P-TEFb in vitro
Next, we determined if dsRNA•HEXIM1 could interact with P-TEFb by EMSA. P-TEFb containing full-length Cdk9 and truncated cyclin T1 with amino acids 1–290 was expressed in a baculovirus system and purified. The final product had no detectable contaminants even when examined on an overloaded, silver-stained SDS gel (Figure 5A). When HEXIM1 and P-TEFb were mixed together and examined by EMSA, there was no detectable intermediate band (Figure 5B), indicating HEXIM1 does not directly interact with P-TEFb. This is in agreement with previous findings that HEXIM1 does not interact with P-TEFb without 7SK (32,33). However, when dsRNA was added, free P-TEFb disappeared, while a new band formed above the dsRNA•HEXIM1 band, demonstrating that P-TEFb was recruited to form a dsRNA•HEXIM1•P-TEFb complex (Figure 5B). Similar results were obtained with P-TEFb containing full-length cyclin T1 (data not shown). A kinase assay was then used to examine the effect of dsRNA on P-TEFb activity. As expected, HEXIM1, 7SK, dsRNA and dsDNA by themselves did not alter the kinase activity of P-TEFb. Both the 7SK•HEXIM1 and dsRNA•HEXIM1 complexes dramatically inhibited the kinase activity of P-TEFb, which is unchanged in the presence of the dsDNA•HEXIM1 complex (Figure 5C). This is in agreement with earlier results showing that both 7SK and dsRNA but not dsDNA can induce a conformational change in HEXIM1, resulting in the recruitment of P-TEFb.
Figure 5.
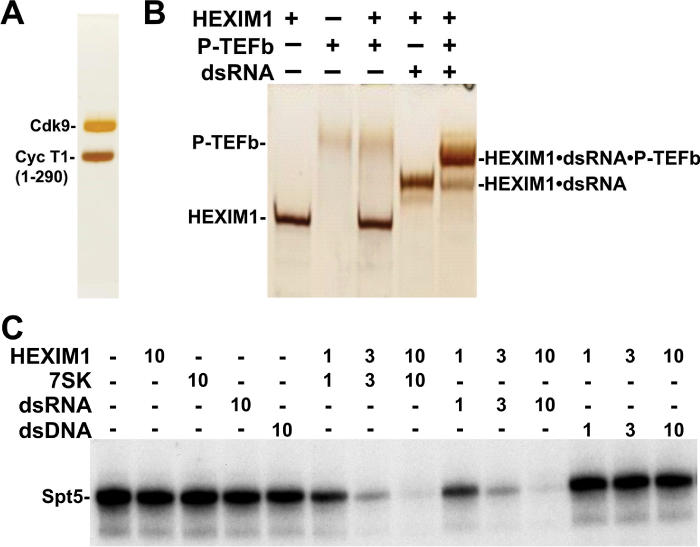
HEXIM1•dsRNA can recruit and inhibit P-TEFb. (A) Purified recombinant P-TEFb containing full-length Cdk9 and cyclin T1 (1–290) analyzed in a 9% SDS gel by silver staining. (B) EMSA analysis of HEXIM1 and P-TEFb in the absence or presence of 2 pmol of dsRNA (silver-stained gel). Here, 2 pmol of HEXIM1 and 4 pmol of P-TEFb were used. (C) The effect of 7SK, dsRNA and dsDNA on P-TEFb kinase activity in the absence or presence of HEXIM1 analyzed by P-TEFb kinase assay using human DSIF as the substrate. Label is incorporated into the large DSIF subunit (Spt5). 1× = 0.15 pmol. An autoradiograph is shown.
Most endogenous HEXIM1 associates with RNAs in vivo
Because the dsRNA•HEXIM1 interaction is independent of the sequence of dsRNA in vitro, and up to 80% of endogenous HEXIM1 is not in complex with 7SK in HeLa cells (22), we set out to determine if HEXIM1 is associated with other cellular RNAs besides 7SK. HeLa cells were lysed in the presence of a non-ionic detergent, and the lysate analyzed by glycerol gradient sedimentation. In agreement with previous studies (22), most endogenous HEXIM1 sedimented in fractions free of P-TEFb and 7SK (fractions 2–6, Figure 6A), and only a small fraction of total HEXIM1 co-migrated with P-TEFb and 7SK (fractions 8–11, Figure 6A). Glycerol gradient fractions 2–5 and 8–10 were then examined by native gel electrophoresis, followed by western blot analysis using an affinity-purified, anti-HEXIM1 antibody (Figure 6B). While most HEXIM1 in fraction 2 migrated like the recombinant HEXIM1, almost all HEXIM1 proteins in the rest of the fractions examined had reduced mobility, indicating that there were other components associated with HEXIM1. Remarkably, when individual fractions were treated with RNase A first and then examined in the same way, all HEXIM1 signals were compressed into a single band that migrated like recombinant HEXIM1 (Figure 6B). We conclude that majority of endogenous HEXIM1 is associated with RNAs.
Figure 6.

Most endogenous HEXIM1 associates with RNA. (A) HeLa cell extracts isolated in the presence of non-ionic detergent were separated on a 5–45% linear glycerol gradient sedimentation, followed by fractionation into 16 fractions. Protein components were visualized by SDS PAGE followed by western blot analysis using antibodies against the indicated proteins. Total RNA was extracted from an aliquot of each fraction, and 7SK was visualized by northern blot analysis. (B) Proteins in glycerol gradient fractions were treated with or without RNase A, separated by native gel electrophoresis first and then HEXIM1 was visualized by western blot analysis. R = recombinant HEXIM1.
The method used above to generate a HeLa cell lysate utilized non-ionic detergent, resulting in the release of HEXIM1, 7SK and most P-TEFb from the nucleus (data not shown). This raised the possibility that HEXIM1•RNA complexes observed above might be an artifact of nuclear HEXIM1 interacting with cytoplasmic RNAs. To examine this possibility, cells were lysed by homogenization in the absence of detergent, followed by differential centrifugation to separate nuclei from the cytoplasm. The nuclei were then extracted in the presence of a non-ionic detergent. As shown in Figure 7A, most if not all Cdk9 was retained in the nucleus, demonstrating successful separation of the cytoplasm from the nucleus. Surprisingly, a considerable amount of HEXIM1 was present in the cytoplasmic fraction. Both nuclear and cytoplasmic HEXIM1 proteins were then examined by native gel electrophoresis. While a small fraction of nuclear HEXIM1 migrated the same as recombinant HEXIM1, most nuclear HEXIM1 had reduced mobility compared to free HEXIM1 (Figure 7B). Also, most, if not all, cytoplasmic HEXIM1 had reduced mobility compared to recombinant HEXIM1. Again, RNase A treatment compressed virtually all HEXIM1 signals to a discrete band (Figure 7B). Consistent with the biochemical fractionation result (Figure 7A), immunofluorescence staining with affinity-purified anti-HEXIM1 antibodies demonstrated that most HEXIM1 was localized in the nucleus, while a portion of HEXIM1 was detectable in the cytoplasm (Figure 7C). Several previous studies used immunofluorescence to demonstrate that HEXIM1 was predominately nuclear (19,33,38). In those studies, cytoplasmic staining was not visible or was visible and not mentioned. The cytoplasmic localization seen here is validated by analysis of background staining (Figure 7C, secondary antibody alone) and by biochemical fractionation (Figure 7A). We conclude that the most endogenous HEXIM1 is in the nucleus and that a significant fraction is present in the cytoplasm. Importantly, most HEXIM1, regardless of location, is associated with RNA.
Figure 7.
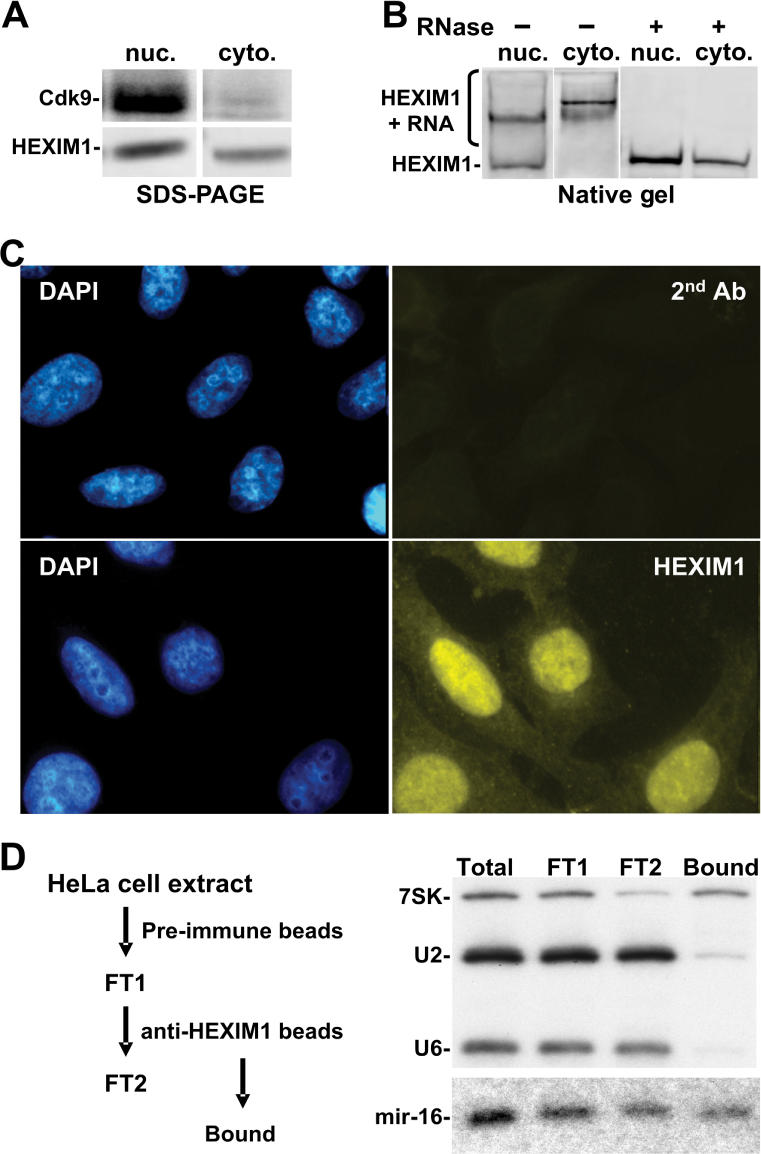
Both nuclear and cytoplasmic HEXIM1 proteins associate with RNA. (A) SDS PAGE and western blot analysis of Cdk9 and HEXIM1 present in nuclear extract (nuc) and cytosol (cyto). (B) Proteins in nuclear extract and cytosol were treated with or without RNase A, separated by native gel electrophoresis first and then HEXIM1 was visualized by western blot analysis. (C) Localization of endogenous HEXIM1 by immunofluorescence staining. The upper set of micrographs shows DAPI staining of DNA and background fluorescence due to just the secondary antibody used. The second set shows DAPI staining of DNA and the signal from affinity-purified anti-HEXIM1 antibodies using the same exposure time as that used for the secondary antibody alone. (D) Isolation of HEXIM1-bound RNAs and examination of RNA by northern blot analysis. Details are found in the Materials and methods section.
HEXIM1 associates with microRNAs in vivo
Because microRNAs have extensive double-stranded regions, we examined if HEXIM1 was associated with microRNAs in vivo. HeLa cell extracts were incubated with pre-immune serum immobilized on protein G beads. RNA was extracted from an aliquot of flowthrough, and the rest was further incubated with affinity-purified anti-HEXIM1 antibodies immobilized on protein G beads. RNA was extracted from the flowthrough. After an extensive wash, RNA was also extracted from the bound material (Figure 7D). The fractions were analyzed by northern blot using several probes. As expected, most 7SK, U2 and U6 RNAs were present in the flowthrough after incubation with pre-immune serum. In contrast, most U2 and U6 were present in the flowthrough after incubation with affinity-purified anti-HEXIM1 antibodies, while a significant portion of 7SK but not U2 or U6 was pulled down by affinity-purified anti-HEXIM1 antibodies (Figure 7D). This reinforces the observation that 7SK is a HEXIM1-binding protein in vivo, and demonstrates a successful isolation of HEXIM1-bound RNAs. Next, we examined if microRNAs could be detected in HEXIM1-bound RNAs. One microRNA, mir-16-1, was chosen because it is a relatively abundant microRNA species in HeLa cells (39). As shown in Figure 7D, about half of the total mir-16-1 was pulled down by affinity-purified anti-HEXIM1 antibodies. It is possible that the fraction of mir-16-1 associated with HEXIM1 is a double-stranded intermediate and that the unbound mir-16-1 is the mature single-stranded microRNA that would be associated with a RISC complex and unavailable to bind to HEXIM1. Although it might be expected that pre-mir-16-1 RNA (89 nt) would bind to HEXIM1 based on its content of double-stranded regions, it was not detected in this study. This could have been due to its low abundance or the presence of other, more specific mir-16-1-binding proteins.
DISCUSSION
In this article, we demonstrated that HEXIM1 is a dsRNA-binding protein, and this interaction is independent of RNA composition. Although dsDNA could interact with HEXIM1, dsRNA exhibited at least 100-fold higher affinity. We also provided evidence that dsRNA, but not dsDNA, binding induced a large conformational change in HEXIM1, which allows it to recruit P-TEFb. Significantly, P-TEFb kinase activity was inhibited in this complex. Furthermore, we showed that endogenous HEXIM1 localized in both cytoplasm and the nucleus, and the majority of HEXIM1 in both locations was generally associated with RNA.
Several previous studies have analyzed the potential secondary structure of 7SK and the regions that support association with HEXIM1. In an extensive analysis of 7SK sequences required for association of HEXIM1 and P-TEFb in HeLa cells, Egloff et al. found that many mutations within the first 100 nt of 7SK resulted in the loss of HEXIM1 binding (40). Their mutations were based on a structure proposed in a previous study (41) that mapped chemical and nuclease sensitivities of 7SK RNA and 7SK RNPs pulled out of cells. In that study, most of the results supported the proposed structure, but several sites of moderate modification (for example, U28, U30 and U66) were inexplicably found embedded in stems, not loops, as predicted from the known function of the modification reagents used (41). If mFold is used to predict the secondary structure of the first 100 nt of 7SK, two different structures are predicted that have similar thermodynamic stability (Supplementary Figure 1). One (ΔG = −40.06 kcal/mole) is very similar to that proposed by Wasserman and Steitz, and the other (ΔG = −40.37 kcal/mole) is completely different. In the slightly more stable second structure, U28, U30 and U66 are present in two prominent loops instead of stems. The complete chemical and nuclease sensitivity data would be accommodated, if the isolated 7SK RNP contained a mixture of the two structures. The structure predicted for 7SK 10–48 used in our study is present in the alternative structure predicted for the entire 1–100 nt of 7SK (compare Figure 1 with Supplementary Figure 1). Our results clearly demonstrate that HEXIM1 can bind to 7SK 10–48 or other short dsRNAs in vitro. However, determination of what 7SK structure is present when HEXIM1 and P-TEFb are associated in vivo will require chemical modification analysis of the RNA in 7SK RNPs pulled down with antibodies to HEXIM1 or P-TEFb.
Here, we demonstrated that dsRNA can functionally replace 7SK in vitro, and that most HEXIM1 is associated with RNAs in vivo, yet in cells, 7SK is the only identified RNA that allows formation of a complex with both HEXIM1 and P-TEFb. A small stem and loop near the 3′ end of 7SK has been shown to be essential for P-TEFb association in vivo (40). However, our results indicate that that region of 7SK is not needed in vitro and, in fact, that no region of 7SK is needed for the dsRNA-dependent conformational change that results in recruitment and inhibition of P-TEFb. Because the two studies measured HEXIM1 and P-TEFb binding under very different conditions, the results are not contradictory. However, they suggest an interesting possibility. There could be a modification in HEXIM1, for example in its P-TEFb interaction domain, which prevents it from interacting with P-TEFb even when bound with RNAs. This modification would not be present on the recombinant HEXIM1 utilized for the in vitro experiments here, but could be present on the endogenous HEXIM1. A factor associated with the 3′ end of 7SK, but not other RNAs, could be responsible for removing this negative modification from HEXIM1, thereby allowing only 7SK•HEXIM1 but not other RNA•HEXIM1 complexes to recruit P-TEFb.
The finding that HEXIM1 is a dsRNA-binding protein is surprising because 7SK has been previously regarded as the only RNA associated with HEXIM1. Although dsDNA could interact with HEXIM1 (Figure 2A), this interaction can be readily displaced by dsRNA (Figure 3C). On the contrary, dsRNA•HEXIM1 interaction is very stable and cannot be disrupted by dsDNA (Figure 3B). Our results demonstrate that HEXIM1 prefers to bind A-form nucleic acids, and also suggest that dsDNA•HEXIM1 interaction may not be physiologically relevant. Our unpublished data indicated that the minimal length of dsRNA for HEXIM1 interaction is 12 bp. However, there are no uninterrupted helices of >10 nt in 7SK and 7SK (10–48) (Figure 1B). Taking into consideration that HEXIM1 is a dimer with two RNA-interaction regions (32,35) and HEXIM1 monomer does not bind RNA (data not shown), each HEXIM1 RNA-interaction region may bind a short stretch of dsRNA, and the distance between the two RNA-interaction regions has to be at least the length of a 12-bp dsRNA. It is not clear if structures in between the two short stretches of dsRNA regions contribute significantly to the dsRNA•HEXIM1 interaction.
We provide two lines of evidence to support the previous hypothesis that RNA binding induces a conformational change in N-terminal self-inhibitory domain of HEXIM1 (32,33). The first line of evidence came from the observation that dsRNA reduced the mobility of HEXIM1 dramatically. There are two major determinants for a protein's mobility on a native gel, i.e. its overall negative charges and its shape. More negative charges and a compact conformation would give rise to a faster migration than a less negative charged and extended conformation. Considering that the overall negative charge of the dsRNA•HEXIM1 complex is increased significantly, one would expect that the mobility of HEXIM1 should increase. Instead, dsRNA•HEXIM1 showed slower mobility than HEXIM1, suggesting that it adapts a more expanded conformation. In fact, HEXIM2 (21,22,35), a HEXIM1 homolog that has a much shorter N-terminal domain, but otherwise shares high sequence identity with HEXIM1, showed increased mobility upon dsRNA binding (data not shown). The second line of evidence came from a large decrease in intrinsic tryptophan fluorescence and a slight shift of the emission peak upon dsRNA, but not dsDNA binding. Although a decrease in intrinsic fluorescence could be caused by the quenching of tryptophan fluorescence by nucleic acids, only dsRNA, but not dsDNA, reduced the intrinsic fluorescence of HEXIM1 dramatically. In addition, only dsRNA, but not dsDNA, caused a shift of the emission wavelength. This is consistent with the observation, obtained from the native gel analysis, that only dsRNA but not dsDNA, reduces the mobility of HEXIM1 dramatically. Taking all the evidence into account, it is likely that, upon dsRNA binding, there is a conformational change in HEXIM1 that exposes the P-TEFb-binding site.
Another interesting observation is that after RNase A treatment, both cytoplasmic and nuclear HEXIM1 migrated the same as the recombinant HEXIM1 protein on a native gel. Recombinant HEXIM1 has been shown to be a dimer (32) under the same condition. Therefore, endogenous HEXIM1 is likely to be a dimer as well. The calculated molecular weight for a HEXIM1 dimer is 80 kDa. However, biophysical studies estimated that endogenous HEXIM1, free of 7SK and P-TEFb, has a size of 95–130 kDa (35). Our observation that most endogenous HEXIM1 has RNAs associated could explain this apparent discrepancy.
dsRNA-binding proteins are involved in a variety of cellular processes such as RNA editing, RNAi, viral defense and transcription (42). Unlike most dsRNA-binding proteins, HEXIM1 lacks the characteristic 50–100 amino acid dsRNA-binding motif (dsRBM). The RNA-binding ability of HEXIM1 largely depends on KHRR (amino acids 152–155). On the other hand, like most, if not all, characterized dsRNA-binding proteins, HEXIM1 binds to any dsRNA, regardless of its primary sequence in vitro. Given this, a dsRNA substrate of one dsRNA-binding protein could easily be bound by other dsRNA proteins in the cell, resulting in crosstalk between different dsRNA-mediated pathways. However, some dsRNA-binding proteins do recognize specific dsRNA substrates in cells, and how this specificity is achieved remains elusive (42–44). Our results do not at present demonstrate conclusively that HEXIM1 is involved in cellular processes other than controlling P-TEFb. However, future studies including the identification of other HEXIM1-bound RNAs may shed light on the function of the HEXIM1•RNA complexes found in the cytoplasm and the nucleus.
SUPPLEMENTARY DATA
Supplementary Data is available at NAR Online.
ACKNOWLEDGEMENTS
This work was supported by NIH grants GM35500 (D.H.P.) and GM57001 (M.A.S.) and the American Heart Foundation fellowship 0510040Z (Q.L.). Funding to pay the Open Access publication charge was provided by NIH grant GM35500.
Conflict of interest statement. None declared.
REFERENCES
- 1.Zhou Q, Yik JH. The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol. Mol. Biol. Rev. 2006;70:646–659. doi: 10.1128/MMBR.00011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 3.Marshall RM, Grana X. Mechanisms controlling CDK9 activity. Front. Biosci. 2006;11:2598–2613. doi: 10.2741/1994. [DOI] [PubMed] [Google Scholar]
- 4.Zhu Y, Pe'ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shore SM, Byers SA, Maury W, Price DH. Identification of a novel isoform of Cdk9. Gene. 2003;307:175–182. doi: 10.1016/s0378-1119(03)00466-9. [DOI] [PubMed] [Google Scholar]
- 6.Peng J, Marshall NF, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J. Biol. Chem. 1998;273:13855–13860. doi: 10.1074/jbc.273.22.13855. [DOI] [PubMed] [Google Scholar]
- 7.Fu TJ, Peng J, Lee G, Price DH, Flores O. Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. J. Biol. Chem. 1999;274:34527–34530. doi: 10.1074/jbc.274.49.34527. [DOI] [PubMed] [Google Scholar]
- 8.Marshall NF, Peng J, Xie Z, Price DH. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 1996;271:27176–27183. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- 9.Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995;270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 10.Wada T, Takagi T, Yamaguchi Y, Ferdous A, Imai T, Hirose S, Sugimoto S, Yano K, Hartzog GA, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998;12:343–356. doi: 10.1101/gad.12.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol. Cell. 2006;21:227–237. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 12.Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997;11:2633–2644. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Q, Chen D, Pierstorff E, Luo K. Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J. 1998;17:3681–3691. doi: 10.1093/emboj/17.13.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garber ME, Wei P, Jones KA. HIV-1 Tat interacts with cyclin T1 to direct the P-TEFb CTD kinase complex to TAR RNA. Cold Spring Harb. Symp. Quant. Biol. 1998;63:371–380. doi: 10.1101/sqb.1998.63.371. [DOI] [PubMed] [Google Scholar]
- 15.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 16.Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell. 2001;8:327–337. doi: 10.1016/s1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Zhu Q, Luo K, Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- 19.Michels AA, Nguyen VT, Fraldi A, Labas V, Edwards M, Bonnet F, Lania L, Bensaude O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol. Cell. Biol. 2003;23:4859–4869. doi: 10.1128/MCB.23.14.4859-4869.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 21.Yik JH, Chen R, Pezda AC, Zhou Q. Compensatory contributions of HEXIM1 and HEXIM2 in maintaining the balance of active and inactive positive transcription elongation factor b complexes for control of transcription. J. Biol. Chem. 2005;280:16368–16376. doi: 10.1074/jbc.M500912200. [DOI] [PubMed] [Google Scholar]
- 22.Byers SA, Price JP, Cooper JJ, Li Q, Price DH. HEXIM2, a HEXIM1-related protein, regulates positive transcription elongation factor b through association with 7SK. J. Biol. Chem. 2005;280:16360–16367. doi: 10.1074/jbc.M500424200. [DOI] [PubMed] [Google Scholar]
- 23.Eberhardy SR, Farnham PJ. Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J. Biol. Chem. 2002;277:40156–40162. doi: 10.1074/jbc.M207441200. [DOI] [PubMed] [Google Scholar]
- 24.Kanazawa S, Soucek L, Evan G, Okamoto T, Peterlin BM. c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene. 2003;22:5707–5711. doi: 10.1038/sj.onc.1206800. [DOI] [PubMed] [Google Scholar]
- 25.Simone C, Stiegler P, Bagella L, Pucci B, Bellan C, De Falco G, De Luca A, Guanti G, Puri PL, et al. Activation of MyoD-dependent transcription by cdk9/cyclin T2. Oncogene. 2002;21:4137–4148. doi: 10.1038/sj.onc.1205493. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 27.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 28.Sano M, Schneider MD. Cyclin-dependent kinase-9: an RNAPII kinase at the nexus of cardiac growth and death cascades. Circ. Res. 2004;95:867–876. doi: 10.1161/01.RES.0000146675.88354.04. [DOI] [PubMed] [Google Scholar]
- 29.Sano M, Schneider MD. Cyclins that don't cycle – cyclin T/cyclin-dependent kinase-9 determines cardiac muscle cell size. Cell Cycle. 2003;2:99–104. [PubMed] [Google Scholar]
- 30.Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, Michael LH, DeMayo FJ, Schneider MD. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat. Med. 2002;8:1310–1317. doi: 10.1038/nm778. [DOI] [PubMed] [Google Scholar]
- 31.Kulkarni PA, Sano M, Schneider MD. Phosphorylation of RNA polymerase II in cardiac hypertrophy: cell enlargement signals converge on cyclin T/Cdk9. Recent Prog. Horm. Res. 2004;59:125–139. doi: 10.1210/rp.59.1.125. [DOI] [PubMed] [Google Scholar]
- 32.Li Q, Price JP, Byers SA, Cheng D, Peng J, Price DH. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem. 2005;280:28819–28826. doi: 10.1074/jbc.M502712200. [DOI] [PubMed] [Google Scholar]
- 33.Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, Sedore SC, Price JP, Price DH, et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004;23:2608–2619. doi: 10.1038/sj.emboj.7600275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yik JH, Chen R, Pezda AC, Samford CS, Zhou Q. A human immunodeficiency virus type 1 Tat-like arginine-rich RNA-binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA-mediated inactivation of P-TEFb. Mol. Cell. Biol. 2004;24:5094–5105. doi: 10.1128/MCB.24.12.5094-5105.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dulac C, Michels AA, Fraldi A, Bonnet F, Nguyen VT, Napolitano G, Lania L, Bensaude O. Transcription-dependent association of multiple positive transcription elongation factor units to a HEXIM multimer. J. Biol. Chem. 2005;280:30619–30629. doi: 10.1074/jbc.M502471200. [DOI] [PubMed] [Google Scholar]
- 36.Blazek D, Barboric M, Kohoutek J, Oven I, Peterlin BM. Oligomerization of HEXIM1 via 7SK snRNA and coiled-coil region directs the inhibition of P-TEFb. Nucleic Acids Res. 2005;33:7000–7010. doi: 10.1093/nar/gki997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen R, Yang Z, Zhou Q. Phosphorylated positive transcription elongation factor b (P-TEFb) is tagged for inhibition through association with 7SK snRNA. J. Biol. Chem. 2004;279:4153–4160. doi: 10.1074/jbc.M310044200. [DOI] [PubMed] [Google Scholar]
- 38.Shimizu N, Ouchida R, Yoshikawa N, Hisada T, Watanabe H, Okamoto K, Kusuhara M, Handa H, Morimoto C, et al. HEXIM1 forms a transcriptionally abortive complex with glucocorticoid receptor without involving 7SK RNA and positive transcription elongation factor b. Proc. Natl Acad. Sci. USA. 2005;102:8555–8560. doi: 10.1073/pnas.0409863102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson PT, Baldwin DA, Scearce LM, Oberholtzer JC, Tobias JW, Mourelatos Z. Microarray-based, high-throughput gene expression profiling of microRNAs. Nat. Methods. 2004;1:155–161. doi: 10.1038/nmeth717. [DOI] [PubMed] [Google Scholar]
- 40.Egloff S, Van Herreweghe E, Kiss T. Regulation of polymerase II transcription by 7SK snRNA: two distinct RNA elements direct P-TEFb and HEXIM1 binding. Mol. Cell. Biol. 2006;26:630–642. doi: 10.1128/MCB.26.2.630-642.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wassarman DA, Steitz JA. Structural analyses of the 7SK ribonucleoprotein (RNP), the most abundant human small RNP of unknown function. Mol. Cell. Biol. 1991;11:3432–3445. doi: 10.1128/mcb.11.7.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian B, Bevilacqua PC, Diegelman-Parente A, Mathews MB. The double-stranded-RNA-binding motif: interference and much more. Nat. Rev. Mol. Cell. Biol. 2004;5:1013–1023. doi: 10.1038/nrm1528. [DOI] [PubMed] [Google Scholar]
- 43.Chang KY, Ramos A. The double-stranded RNA-binding motif, a versatile macromolecular docking platform. FEBS J. 2005;272:2109–2117. doi: 10.1111/j.1742-4658.2005.04652.x. [DOI] [PubMed] [Google Scholar]
- 44.Fierro-Monti I, Mathews MB. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem. Sci. 2000;25:241–246. doi: 10.1016/s0968-0004(00)01580-2. [DOI] [PubMed] [Google Scholar]