

Abstract
Allograft arteriosclerosis is an important characteristic of chronic graft rejection. In allograft arteriosclerosis there is a striking loss of medial smooth muscle cells (SMCs) before the development of a concentric intimal proliferative response. In this study we evaluated the role of CD8+ T lymphocytes in this medial SMC loss. Brown Norway aortic segments were transplanted into Lewis animals for 60 days (long allo-exposure) or 20 days (short allo-exposure). After 20 days allogeneic exposure aortic segments were transplanted back into syngeneic (Brown Norway) animals for 40 days. Experimental animals were treated with mAb to CD8. Apoptosis was measured by terminal dUTP nick-end labeling at 20 days and morphometry analyzed at 60 days to evaluate medial and intimal changes. Anti-CD8 treatment significantly lowered CD8+ T cell counts in peripheral blood, reduced medial SMC apoptosis at 20 days, and increased medial SMC counts at 60 days. Both short- and long-allogeneic exposure groups confirmed these findings and demonstrated that medial SMC loss is proportional to the length of allogeneic exposure. Antibody depletion of CD8+ T cells results in reduced medial SMC apoptosis and better medial SMC preservation. This supports the hypothesis that medial SMC loss occurs by apoptotic death and is driven by CD8+ T lymphocytes.
Orthotopic heart transplantation is a well-established treatment modality for end-stage heart disease. Immunosuppressive drugs have dramatically improved 1-year graft survival. However, loss of organ allografts because of chronic rejection remains a major clinical problem and has become a major limiting factor for long-term survival. It is a leading cause of late graft failure, responsible for 23% to 36% of deaths among patients that survive >1 year after heart transplantation. 1,2 Chronic rejection is characterized by a diffuse, concentric intimal proliferative response within the arteries of transplanted organs, termed allograft arteriosclerosis (AAS). 3-5 The presence of AAS can be demonstrated in up to 50% of patients 5 years after heart transplantation. 6,7 There is no current therapy for AAS other than retransplantation which has been shown to have much poorer results than the original transplant in addition to compounding the problem of donor shortage. 8 The pathogenic mechanisms of AAS remains primarily unknown.
To date, chronic rejection research has focused on allorecognition of graft endothelium with subsequent leukocyte infiltration and production of cytokines, chemokines, and growth factors. 9 In response, vascular smooth muscle cells (SMCs) are thought to transmigrate to the intimal compartment resulting in occlusive lesion formation. 10 We, and others, have observed that in arteries from both humans and animals undergoing chronic rejection the media undergoes striking thinning and loss of SMCs. 11-14 In an aortic interposition graft model, which is a well-described and reproducible model of AAS, 13,15 medial SMC loss occurred only in allografts. 16 In addition the degree of medial SMC loss and intimal proliferation observed were directly proportional to the duration of allogeneic exposure, 17,18 suggesting that medial SMC cell destruction occurs by allo-immune mechanisms and may be linked to AAS. It is also consistent with observations made in arterial injury models, that robust intimal proliferation is dependent on, and proportional to, the extent of medial SMC injury. 19 In this context, we have recently provided evidence that medial SMC loss in allografted arteries occurs by apoptotic cell death and that CD8+ T cell-derived inducers of apoptosis are up-regulated concomitantly. 12 These include granzyme B, perforin, and Fas ligand, which are the primary mediators by which cytotoxic T lymphocytes kill target cells. We therefore hypothesized that medial SMC loss occurs by CD8+ T lymphocyte-induced SMC apoptosis rather than migration. We further hypothesize that medial SMC destruction may be linked to myointimal lesion formation. In support of this hypothesis is evidence that the proliferating intimal lesion is of recipient not donor origin (Johnson P, unpublished observations). 20
In the current study we have established a role for CD8+ T lymphocytes in medial SMC loss and evaluated the correlation between this and intimal proliferation in a rat aortic interposition graft model of AAS. To achieve this, CD8+ T lymphocytes were depleted from recipient animals with a depleting mAb to the CD8 molecule (MRC OX-8). We have also used retransplantation experiments that are known to have reduced medial SMC damage and less intimal proliferation 17,18 to explore the impact of CD8 depletion on the timing and reversibility of AAS.
Materials and Methods
Animals
Inbred male Brown Norway (RT1.An) and Lewis (RT1.Al) rats weighing 250 to 350 g were purchased from Harlan Sprague-Dawley (Indianapolis, IN) and housed in the Medical Sciences animal care facility. All animals were housed and fed ad libitum at the Dalhousie University Animal Care Center in accordance with the guidelines of the Canadian Council of Animal Care.
Aortic Interposition Grafting
Infrarenal abdominal aortic interposition transplantation was performed as described by Mennander et al. 13 Briefly, donor Brown Norway (BN) rats were anesthetized with pentobarbital (50 mg/kg) and a midline abdominal incision was made. The infrarenal abdominal aorta was exposed and harvested for transplantation. The donor aorta was implanted orthotopically into a Lewis recipient by an end-to-end microsurgical anastomotic technique using interrupted 9-0 nylon (Sharpoint, Reading, PA) sutures. Three groups were used: 1) a long-allogeneic exposure group (n = 10); 2) a short-allogeneic exposure group (n = 10); and 3) historical syngeneic controls. In the long-allogeneic exposure group BN aortic segments were transplanted and left in Lewis recipients for a total of 60 days. In the short-allogeneic exposure group transplanted aortic segments were left in Lewis recipients for a period of 20 days at which point the same aortic segment was transplanted back into a syngeneic BN animal for an additional 40 days. In syngeneic controls a Lewis aorta was transplanted into a Lewis recipient and then retransplanted back into another Lewis recipient.
In Vivo Treatment
Antibody treatment of recipient Lewis animals was initiated 2 days before the initial transplantation in both the allogeneic exposure group and also at 20 days and 40 days after transplantation in the long-allogeneic exposure group. Two antibodies were used: an anti-CD8-depleting mAb (MRC OX-8; 2 mg injection, 9 mg/ml) and a control irrelevant mAb (B9) to pertussis toxin. The relative T-lymphocyte subset frequencies present in the peripheral blood of Lewis rats treated with OX-8 or control antibodies were analyzed by flow cytometry (see below).
Cell Isolation
Heparinized blood samples (500 μl) were collected via the tail artery from each animal before antibody injection. This was performed at day −2, 20 days, 40 days, 50 days, and 60 days. The samples were centrifuged for 15 minutes at 400 × g, the buffy coat was removed and the red cells were lysed with buffered ammonium chloride for 5 minutes at room temperature. The leukocytes were then washed three times in RPMI 1640 (ICN, Aurora, OH) supplemented with 20 mmol/L HEPES (Life Technologies, Inc., Rockville, MD). Cells were resuspended in RPMI and 0.5% bovine serum albumin (Sigma Chemical Co., St. Louis, MO) for immunofluorescent staining.
Flow Cytometric Analysis
Cells (1 × 10 5 per ml) were incubated with 9 μg/ml of anti-CD8α mAb (OX-8) or control mAb (B9) at 4°C for 30 minutes. Cells were then washed twice in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (Sigma Chemical Co.) before incubation with secondary fluorescein isothiocyanate-conjugated goat anti-mouse IgG (Cedarlane Laboratories, Hornby, ON) for 30 minutes. Cells were then washed twice and fixed in PBS containing 1% paraformaldehyde and stored at 4°C. Flow cytometry was performed on a BD FACscan (Becton Dickinson, Mountain View, CA) using Lysis II software.
Morphometric Analysis
Tissue was harvested for histology at 20 days and 60 days and immersion-fixed (4% paraformaldehyde, 0.05% v/v glutaraldehyde, 15% v/v saturated picric acid) at 4°C for 12 hours. Grafts were then paraffin-embedded and 5-μm sections were cut for histology (hematoxylin and eosin [H&E], Verhoeff elastin), for immunocytochemistry and for terminal dUTP nick-end labeling (TUNEL) studies.
Tissue sections were stained for histology using H&E (Harris hematoxylin) or Verhoeff elastin stain (hematoxylin 5%, ferric chloride 10%, Lugol’s iodine). Tissue slides from each animal were then examined by light microscopy (Nikon, Optophot, Mississauga, ON) and images digitalized and captured (JVC digital camera, TK1070U, Scarborough, ON). Morphometric analysis was carried out using Adobe Photoshop (Adobe Systems, Mountain View, CA) and NIH Image analyzer software on a Power PC (G3; Apple Computer, Cupertino, CA) to measure surface area (μm2) and cell counts per area. SMC counts and intimal surface area measurements were done at 60 days after the initial transplantation in both the long- and short-allogeneic exposure group.
In Situ Detection of Apoptosis by TUNEL
A commercial assay (ApopTag in situ apoptosis detection kit; Oncor, Gaithersburg, MD) using terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) was used to detect nuclear DNA fragmentation indicative of apoptosis. Tissues were fixed and embedded in paraffin as described for histology and 5-μm sections were collected onto silinated glass microscope slides. Sections were deparaffinized through a graded alcohol series into 0.1 mol/L PBS. Tissue sections were covered with 100 μg/ml RNase A (Boehringer Mannheim, Laval, Quebec) in 2× standard saline citrate buffer and incubated in a humidified chamber for 40 minutes at 37°C. Slides were washed three times in 2× standard saline citrate buffer. Endogenous peroxidase was qu-enched in fresh 0.15% hydrogen peroxide in 0.1 mol/L PBS for 25 minutes at room temperature, rinsed in 0.1 mol/L PBS, and covered with prewarmed 37°C proteinase K (Boehringer Mannheim) (20 μg/ml in TE buffer, pH 8.0). Slides were incubated at 37°C for 1 minute and then on ice in chilled 2 mg/ml glycine in 0.1 mol/L PBS for 15 minutes. After 10 minutes in −20°C ethanol:acetic acid, 2:1 slides were rinsed with 2× standard saline citrate buffer and sections were covered with equilibration buffer (ApopTag Kit) for 20 minutes. Sections were treated with TdT enzyme with digoxigenin-labeled dUTP (ApopTag Kit), as suggested by the supplier, in a humidified chamber at 37°C for 2 hours. The reaction was stopped by three 10-minute washes at 37°C in 2× standard saline citrate buffer. The sections were covered with peroxidase-labeled anti-digoxigenin antibody with 0.01% Tween-20 for 1 hour at 37°C in a humidified chamber. Slides were washed in 0.1 mol/L PBS. Color development was achieved by exposing sections to freshly prepared 0.01% hydrogen peroxide using 0.06% 3,3′-diaminobenzidine as the chromogen, for up to 20 minutes at room temperature. Sections were counterstained with methyl green, dehydrated, and mounted. Normal rat intestine was used as a positive control tissue. For negative controls, distilled water was substituted for the TdT enzyme.
Immunohistochemistry
Sections were deparaffinized; endogenous peroxidase was quenched (12% HOOH/MeOH); nonspecific staining blocked with normal horse serum; and subsequently incubated with anti-α-actin mAb (HHF-35; Enzo Diagnostics, Inc., New York, NY) or anti-CD8 mAb (MRC-OX8, Cedarlane) for 1 hour at room temperature. Sections were then washed in PBS containing 1% bovine serum albumin (Sigma Chemical Co.) three times before incubation with biotinylated secondary antibody and labeling with peroxidase avidin/biotin complex using 3.3′-diaminobenzidine as the chromogen (Vector, Burlingame, CA).
Statistics
Apoptotic cell counts, SMC counts, and surface area measurements were analyzed as continuous variables. Means were obtained from five animals in each group. Data were reported as mean and SEM. Analysis of variance was used to evaluate groups consisting of syngeneic controls, CD8-depleted allogeneic and wild-type allogeneic strain combinations. Post hoc Fishers test of least significant difference was used to assess statistical significance with P < 0.05 being the limit of significance.
Results
Depletion of CD8+ T Cells
Immunofluorescent staining of blood leukocytes was used to assess the depletion of CD8+ T cells after injection of OX-8 mAb. Approximately 20% of the blood lymphocytes were CD8+ in normal Lewis control rats. Animals receiving a single injection of OX-8 mAb had <1.5% CD8+ lymphocytes in peripheral blood after 48 hours. This depletion persisted for 20 days in the short-allogeneic exposure group (Figure 1) ▶ . In the long-allogeneic exposure group Lewis animals were additionally treated with OX-8 at 20 days and 40 days. Blood samples taken at 20 days, 40 days, 50 days, and 60 days all showed comparable depletion of CD8+ cells (<2.5% of peripheral lymphocytes staining positively for CD8).
Figure 1.

Flow cytometry analysis of blood lymphocytes (lymphocyte gated) in allo-Lewis (a) and CD8+ T cell-depleted Lewis animals 20 days after a 2-mg OX-8 injection (b). Gray area represents negative control cell population.
SMC Apoptosis at 20 Days
All apoptosis assays were performed at 20 days after transplantation. Positive nuclear labeling was localized to the media with some labeling seen in the intima and adventitia (figure not shown). The apoptotic index for medial SMC nuclei was significantly (P < 0.001; n = 5) lower in CD8-depleted Lewis recipients as compared to wild-type animals and approached that of syngeneic controls. The medial SMC apoptotic index was 1% ± 0.2 in syngeneic controls, 2.9% ± 0.5 in CD8-depleted animals (20 ± 3 apoptotic nuclei per section) compared to 9.1% ± 0.5 in wild-type animals (35 ± 3 apoptotic nuclei per section) (Figure 2) ▶ .
Figure 2.

Medial SMC apoptotic index was assessed by TUNEL assay at 20 days after transplantation in: syngeneic controls (Syn), CD8+ T cell-depleted animals (L(↓CD8)) and wild-type animals (WT). *, P < 0.02 between syngeneic and CD8+ T cell-depleted animals; **, P < 0.001 between wild-type and CD8+ T cell-depleted animals. Five animals per group were used.
Long-Allogeneic Exposure Group at 60 Days
In the long-allogeneic exposure group BN aortic segments were transplanted into a wild-type or CD8-depleted environment for the full 60 days. CD8-depleted Lewis recipients received antibody treatment before transplantation, at 20 days, and at 40 days. Histology at 60 days showed that the medial thickness and cellularity were better preserved in CD8-depleted recipients compared to wild type (Figure 3) ▶ . The number of medial SMCs in syngeneic controls was undistinguishable from a normal artery with 655 ± 45 cells/section, which was significantly higher than any allogenic strain combinations (P < 0.001). The number of medial SMCs in CD8-depleted animals were 363 ± 21 cells/section which was significantly higher (P = 0.01) than the 237 ± 27 cells/section in wild type (Figure 4) ▶ . There were a large number of infiltrating mononuclear cells that can be seen in the adventitia of both CD8-depleted and wild-type animals (Figure 3) ▶ . The intimal area was 236.2 ± 30.6 × 10 3 μm 2 in the CD8-depleted rats compared to 158.9 ± 40.9 × 10 3 μm 2 in the allo-Lewis group (P = ns) (Figure 5) ▶ . No intimal proliferation was seen in syngeneic controls.
Figure 3.

Photomicrographs of allografted aortic segment at 60 days after a long allogeneic exposure in CD8+ T cell-depleted animals (a) and wild-type animals (b). H&E, ×200. i, Intima; m, media; arrow, internal elastic lamina.
Figure 4.

Number of medial SMC nuclei per section at 60 days after a long allogeneic exposure in: syngeneic controls (Syn), CD8+ T cell-depleted animals (L(↓CD8)) and wild-type animals (WT). *, P = 0.01 between wild-type and CD8+ T cell-depleted animals. Mean number of cells in each group were obtained from five animals.
Figure 5.

Intimal area was measured in μm 2 for both the short (left) and long (right) allogeneic group at 60 days after the initial transplantation. Ten animals per group were used: five wild-type (WT) and five CD8+ T cell-depleted (L(↓CD8)) recipients.
α-Actin immunostaining for SMC actin showed positive labeling of the intimal proliferative lesion. Medial SMC immunostaining was better preserved in CD8-depleted animals and, when present, was localized mostly in proximity to the external elastic lamina (Figure 6,a and b) ▶ . Note that not all spindle-shaped cells seen in the media express α-actin. Vascular SMCs are known to loose their α-actin staining in states of proliferation or stress. 21-23 Anti-CD8 immunostaining showed a marked reduction in positive staining cells infiltrating the media of CD8-depleted animals as compared to wild-type animals (Figure 6, c and d) ▶ .
Figure 6.
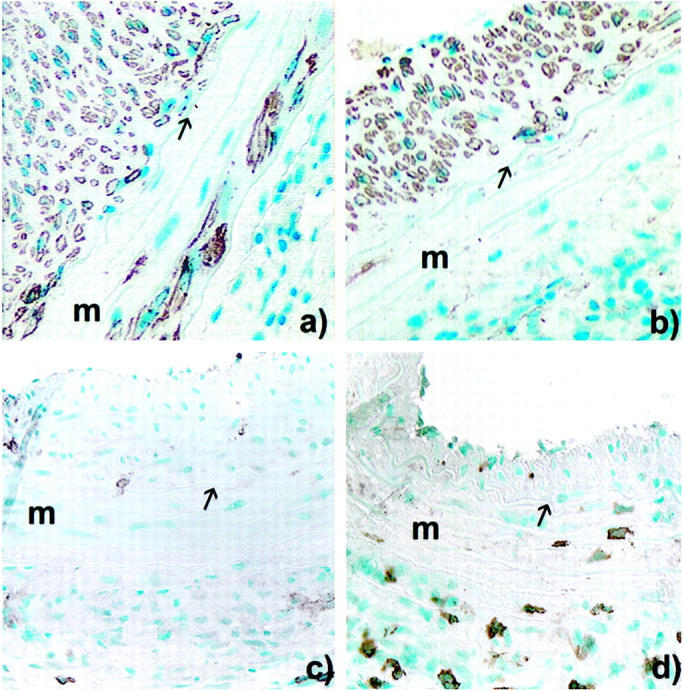
Peroxidase immunocytochemistry of allografted aortic segment at 60 days after a long allogeneic exposure in: CD8+ T cell-depleted animals (a) and wild-type animals (b) using anti-α-actin mAb (HHF35); and CD8+ depleted-animals (c) and wild-type animals (d) stained using anti-CD8 mAb (MRC-OX-8). Original magnification, ×200. m, Media, arrow, internal elastic lamina.
Short-Allogeneic Exposure Group at 60 Days
In the short-allogeneic group BN aortic segments were transplanted into wild-type or CD8-depleted Lewis animals and maintained for 20 days before being retransplanted back into syngeneic BN recipients for the remaining 40 days. Tissues were examined histologically 60 days after the initial transplantation. The medial area and cellularity were markedly higher in the CD8-depleted Lewis animal group compared to the wild-type recipients (Figure 7) ▶ . The number of medial SMCs in syngeneic controls was undistinguishable from a normal artery with 655 ± 45 cells/section, which was significantly higher than any allogenic strain combinations (P < 0.001). Medial SMC counts per section in the CD8-depleted Lewis group were 506 ± 23 cells/section and significantly higher (P = 0.01; n = 5) than 384 ± 26 cells/section in the wild-type group (Figure 8) ▶ . Intimal area was 138.5 ± 21.7 × 10 3 μm 2 in the CD8-depleted group compared to 98.8 ± 13.0 × 10 3 μm 2 in the allo-Lewis group (P = ns) (Figure 5) ▶ . No intimal proliferation was seen in syngeneic controls.
Figure 7.

Photomicrographs of allografted aortic segment at 60 days after a short allogeneic exposure in CD8+ T cell-depleted animals (a) and wild-type animals (b). H&E, ×200. i, Intima; m, media; arrow, internal elastic lamina.
Figure 8.

Number of medial SMC nuclei per section at 60 days after a short allogeneic exposure in: syngeneic controls (Syn), CD8+ T cell-depleted animals (L(↓CD8)) and wild-type animals (WT). *, P = 0.01 between wild-type and CD8+ T cell-depleted animals. The mean number of cells per group were obtained from five animals.
Retransplantation and Medial SMC Loss
The number of medial SMCs (60 days) were evaluated in terms of length of allogeneic exposure (0 days, 20 days, 60 days), and shown to significantly decrease with increasing allogeneic exposure. Counts of medial SMCs/section were 655 ± 45 cells in syngeneic controls (0 days of allogeneic exposure), 384 ± 56 cells in wild-type short allogeneic exposure (P < 0.001; 20 days of allogeneic exposure), and 237 ± 27 cells in wild-type long allogeneic exposure (P = 0.007; 60 days of allogeneic exposure).
Discussion
Clinically AAS has become a major limiting factor for long-term survival. It is the leading cause of late graft failure among patients that survive >1 year after heart transplantation. 1,2 However, the mechanisms responsible for AAS remain obscure. Experimentally AAS is at least in part an immunological phenomenon in that inbred syngeneic animals do not experience either the intimal proliferative lesion or the medial SMC loss. 16 Nonimmunological factors such as ischemic damage, however, play some role in immune activation because prolonged ischemic times will enhance AAS in allografts but not syngeneic grafts 24 indicating that nonimmune injury may augment alloreactivity but is not, in itself, sufficient do induce AAS. It is therefore clear that an understanding of the etiology of chronic rejection, including AAS, will be critical to the development of successful therapies.
In this context, we have initially targeted the development of medial SMC loss which is characteristic of AAS. We wish to determine the mechanisms involved in this loss and eventually the relationship of the SMC loss to the propagation of the intimal lesion. In this study we targeted CD8+ T cells based on our previous work which showed, by reverse transcriptase-polymerase chain reaction, an up-regulation of cytotoxic T lymphocyte (CTL) mediators of apoptosis (perforin, granzyme B, fas Ligand) in allografts undergoing chronic rejection. 12 We demonstrated here that depletion of CD8+ T lymphocytes markedly reduced medial SMC apoptosis in allografted arteries. Thus, reduced medial SMC apoptotic death, correlated with a significant preservation of medial SMC counts at later time points in CD8+ T cell-depleted animals compared to wild-type animals showing a protective effect on the media of allotransplants. Alpha-actin immunohistochemistry was used to confirm the SMC retention in the media of the CD8+ T cell-depleted animals. Parenthetically, we have found similar results in β2-microglobulin knock-out mice which have dramatically reduced levels of CD8+ T cells. 25 This, taken together with the reduction in medial SMC apoptosis in CD8+ T cell-depleted animals, provides convincing evidence that the SMC loss characteristic of AAS is mediated by CD8+ CTL-induced apoptotic cell death.
In this study, aortic segments were retransplanted into syngeneic animals after a fixed period of allogeneic exposure in wild-type or CD8+ T cell-depleted Lewis animals (short-allogeneic exposure group). Twenty days of allogeneic exposure was based on previous work that has shown that retransplantation into a syngeneic environment results in reproducible medial damage and intimal proliferation although less extensive than that in a graft maintained in an allogeneic environment. 17 Once again, CD8+ T cell-depleted animals had a significant reduction in medial SMC apoptosis and significantly higher numbers of medial SMCs when compared to wild-type animals. However, they continued to develop progressive intimal lesions. Our findings confirm that the events responsible for the generation of AAS and medial destruction occur early, and may be independent of continued allogeneic exposure or continued immune injury. These results are consistent with a previous report that an allogeneic exposure of 10 days in an aortic interposition model resulted in progressive AAS, despite back transplantation into a syngeneic environment. 18 Similar observations were made in a rat heterotopic heart model where an allogeneic exposure of 5 days or more resulted in progressive development of AAS, whereas an allogeneic exposure of <5 days resulted in complete recovery of all intimal lesions throughout time. 26 In contrast to what we would have expected from work in vascular injury models, protecting the media with CD8 depletion, did not inhibit neointimal proliferation. This finding indicates that medial SMC destruction and intimal proliferation are differentially regulated as previously suggested by Mennander et al. 16 Our findings also provide evidence that the accepted paradigm 9,10 of SMC migration from the medial compartment to the intima, under the influence of endothelial-derived growth factors, may be incorrect. Considering the brief window (hours) in which cells undergoing apoptosis can be identifiable by TUNEL assay, an index of 9% reported in this study represents a high index, and indicates that SMC loss from the allograft media could be entirely explained by apoptosis. We have shown that medial SMCs are killed by in situ immune-mediated apoptosis and could account for all of the loss of SMCs observed in AAS. 12 Moreover, there is growing indirect evidence 20,27 and some recent direct evidence (Johnson P, unpublished observations) that the proliferating intimal lesion is of recipient not donor origin, thus excluding donor SMC dedifferentiation and migration as responsible for AAS.
The role of T cell subsets in the etiology of AAS remains controversial. Some have suggested that CD8+ T cells are not involved in the progression of AAS. 28,29 Others have shown that depletion of CD8+ T cells prevents or delays the development of intimal proliferation. 30 Evidence from β2-microglobulin knock-out mice which have a gene defect which prevents the normal development of CD8+ cytotoxic T lymphocytes or antibody depletion of CD8+ T cells have yielded conflicting results depending on the model studied. 31,32 This study in the context of previously reported findings points to the complexity of the induction and expression of AAS and the fact that T cells of various phenotypes are likely to be involved in this complex process at many levels in both the regulation and propagation of this lesion.
In summary, we have demonstrated that medial SMC loss seen in allografted arteries is immune mediated and occurs by CD8+ T lymphocyte-induced apoptosis. Our findings have also shown that both medial damage and intimal proliferation occur early after transplantation, seem independent of persistent immune injury, and are differentially regulated.
Footnotes
Address reprint requests to Dr. Greg Hirsch, New Halifax Infirmary QEII HSC, 1796 Summer St., Rm 2269, Division of Cardiac Surgery, Halifax, Nova-Scotia, Canada, B3H 3A7. E-mail: ghirsch@is.dal.ca.
Supported by the New Brunswick Heart and Stroke Foundation Grant G-98-HI-0176.
References
- 1.Johnson DE, Alderman EL, Schroeder JS, Gao SZ, Hunt S, Decampli WM, Stinson E, Billingham M: Transplant coronary artery disease: histopathologic correlations with angiographic morphology. J Am Coll Cardiol 1991, 17:449-457 [DOI] [PubMed] [Google Scholar]
- 2.Uretsky BF, Kormos RL, Zerbe TR, Lee A, Tokarczyk T, Murali S, Reddy PS, Denys BG, Griffith BP, Hardesty RL: Cardiac events after heart transplantation: incidence and predictive value of coronary angiography. J Heart Lung Transplant 1992, 11:S45-S51 [PubMed] [Google Scholar]
- 3.Tilney NK, Whitley WD, Dimond JR, Kupiec-Weglinski JW, Adams DH: Chronic rejection, an unidentified conundrum. Transplantation 1991, 52:389-398 [DOI] [PubMed] [Google Scholar]
- 4.Hayry P: Molecular pathology of acute and chronic rejection. Transplant Proc 1994, 26:3280-3284 [PubMed] [Google Scholar]
- 5.Billingham BE: Histology of graft coronary disease. J Heart Lung Transplant 1992, 12:538-544 [PubMed] [Google Scholar]
- 6.Gao SZ, Alderman EL, Schroeder JS, Silverman JF, Hunt SA: Accelerated coronary vascular disease in the heart transplant patient: coronary arteriographic findings. J Am Coll Cardiol 1988, 12:334-340 [DOI] [PubMed] [Google Scholar]
- 7.Olivari MT, Homans DC, Wilson RF, Kubo SH, Ring WS: Coronary artery disease in cardiac transplant patients receiving triple-drug immunosuppressive therapy. Circulation 1989, 80:111-115 [PubMed] [Google Scholar]
- 8.Schnetzler B, Pavie A, Dorent R, Camproux AC, Leger P, Delcourt A, Gangjbakhch I: Heart transplantation: a 23-year single-center clinical experience. Ann Thorac Surg 1998, 65:978-983 [DOI] [PubMed] [Google Scholar]
- 9.Shirwan A: Chronic allograft rejection. Transplantation 1999, 68:715-726 [DOI] [PubMed] [Google Scholar]
- 10.Akyurek LM, Paul LC, Funa K, Larsson E, Fellstrom BC: Smooth muscle cell migration into intima and adventitia during development of transplant vasculopathy. Transplantation 1996, 62:1526-1529 [DOI] [PubMed] [Google Scholar]
- 11.Adams DH, Tilney NL, Collins JJ, Karnovsky MJ: Experimental graft arteriosclerosis. I. The Lewis-to-F344 allograft model. Transplantation 1992, 53:1115–1119 [PubMed]
- 12.Hirsch GM, Kearsey J, Burt TD, Karnovsky MJ, Lee TDG: Medial smooth muscle cell loss in arterial allografts occur by cytolytic cell induced apoptosis. Eur J Cardiothorac Surg 1998, 14:89-96 [DOI] [PubMed] [Google Scholar]
- 13.Mennander A, Tiisala S, Halttunen J, Yilmaz S, Paavonen T, Häyry P: Chronic rejection in rat aortic allografts: an experimental model for transplant arteriosclerosis. Arterioscler Thromb 1991, 11:671-680 [DOI] [PubMed] [Google Scholar]
- 14.Liu G, Butany J: Morphology of graft arteriosclerosis in cardiac transplant recipients. Hum Pathol 1992, 23:768-773 [DOI] [PubMed] [Google Scholar]
- 15.Isik FF, McDonald TO, Ferguson M, Yamanaka E, Gordon D: Transplant arteriosclerosis in rat aortic model. Am J Pathol 1992, 141:1139-1149 [PMC free article] [PubMed] [Google Scholar]
- 16.Mennander A, Paavonen T, Hayry P: Intimal thickening and medial necrosis in allograft arteriosclerosis (chronic rejection) are independently regulated. Arterioscler Thromb 1993, 13:1019-1025 [DOI] [PubMed] [Google Scholar]
- 17.Hare GM, Lee TD, Jordan JL, Hirsch GM: Medial cell apoptosis precedes the development of intimal hyperplasia after transient allogeneic exposure in a rat aortic allograft model of chronic rejection. Transplant Proc 1999, 31:1384-1385 [DOI] [PubMed] [Google Scholar]
- 18.Mennander A, Hayry P: Reversibility of allograft arteriosclerosis after retransplantation to donor strain. Transplantation 1996, 62:526-529 [DOI] [PubMed] [Google Scholar]
- 19.Reidy MA: Biology of disease: a reassessment of endothelial injury and arterial lesion formation. Lab Invest 1985, 53:513-520 [PubMed] [Google Scholar]
- 20.Plissonnier D, Nochy D, Poncet P, Mandet C, Hinglais N, Bariety J, Michel JB: Sequential immunological targeting of chronic experimental arterial allografts. Transplantation 1995, 60:414-424 [DOI] [PubMed] [Google Scholar]
- 21.Kocher O, Gabbiani F, Gabbiani G, Reidy MA, Cokay MS, Peters H, Huttner I: Phenotypic features of smooth muscle cells during the evolution of experimental carotid artery intimal thickening. Lab Invest 1991, 65:459-470 [PubMed] [Google Scholar]
- 22.Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, Gabbiani G: A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol 1986, 103:2787-2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clowes AW, Clowes MM, Reidy MA: Kinetics of cellular proliferation after arterial injury. III. Endothelial and smooth muscle growth in chronically denuded vessels. Lab Invest 1986, 54:295–303 [PubMed]
- 24.Knight RJ, Dikman S, Martinelli GP: Cold ischemic injury accelerates the progression to chronic rejection in a rat cardiac allograft model. Transplantation 1997, 64:1102-1107 [DOI] [PubMed] [Google Scholar]
- 25.O’Neill JL, Lee TDG, Hirsch GM: CD8+ cytotoxic T lymphocytes are involved in inducing medial cell dropout via apoptosis in mouse aortic allografts. Transplant Proc 1999, 31:865-866 [DOI] [PubMed] [Google Scholar]
- 26.Izutani H, Miyagawa S, Shirakura R, Matsumiya G, Nakata S, Shimazaki Y, Matsuda H: Evidence that graft coronary arteriosclerosis begins in early phase after transplantation and progress without chronic immunoreaction. Transplantation 1995, 60:1073-1079 [DOI] [PubMed] [Google Scholar]
- 27.Aziz S, McDonald TO, Gohra H: Transplant arterial vasculopathy: evidence for a dual pattern of endothelial injury and the source of smooth muscle cells in lesions of intimal hyperplasia. J Heart Lung Transplant 1995, 14:5123-5136 [PubMed] [Google Scholar]
- 28.Izutani H, Miyagawa S, Mikata S, Shirakura R, Matsuda H: Essential initial immunostimulation in graft coronary arteriosclerosis induction detected by retransplantation technique in rats: the participation of T cell subsets. Transplant Immunol 1997, 5:11-15 [DOI] [PubMed] [Google Scholar]
- 29.Forbes RDC, Zheng SX, Gomersall M, Al-Saffar M, Guttmann RD: Evidence that recipient CD8+ T cell depletion does not alter development of chronic vascular rejection in a rat heart allograft model. Transplantation 1994, 57:1238-1246 [DOI] [PubMed] [Google Scholar]
- 30.Allan JS, Choo JK, Vesga L, Arns JS, Pins MR, Sachs DH, Madsen JC: Cardiac allograft vasculopathy is abrogated by anti-CD8 monoclonal antibody therapy. Ann Thorac Surg 1997, 64:1019-1025 [DOI] [PubMed] [Google Scholar]
- 31.Shi C, Lee WS, He Q, Zhang D, Fletcher DL, Newell JB: Immunologic basis of transplant-associated arteriosclerosis. Proc Natl Acad Sci USA 1996, 93:4051-4056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z, Cobbold S, Metcalfe S, Waldman H: Tolerance in the mouse to major histocompatibility complex-mismatched heart allografts, and to rat xenografts, using monoclonal antibodies to CD4 and CD8. Eur J Immunol 1992, 22:805-810 [DOI] [PubMed] [Google Scholar]